

Abstract
Expression of the porin genes of Escherichia coli is regulated in part by the osmolarity of the growth medium. The process is controlled by the histidine kinase EnvZ and the response regulator OmpR. We have previously shown that phosphorylation of OmpR increases its affinity for the upstream regulatory regions of ompF and ompC. We now report that, in the presence of DNA, there is a dramatic stimulation in the level of phospho-OmpR. This effect is independent of the source of phosphorylation, i.e., stimulation of phosphorylation is observed with a small phosphorylating agent such as acetyl phosphate or with protein-catalyzed phosphorylation by the kinase EnvZ. The dephosphorylation rate of phospho-OmpR is affected only slightly by the presence of DNA; thus, the increased level is largely caused by an increased rate of phosphorylation. Stimulation of phosphorylation requires specific binding of DNA by OmpR. Occupancy of the DNA binding domain exposes a trypsin cleavage site in the linker, which connects the phosphorylation domain with the DNA binding domain. Our results indicate that when DNA binds in the C terminus, it enhances phosphorylation in the N terminus, and the linker undergoes a conformational change. A generalized mechanism involving a four-state model for response regulators is proposed.
Keywords: winged helix–turn–helix, response regulator, porin regulation, two-component regulatory system, transcriptional activator
All organisms must communicate with their environment to survive. Two-component regulatory systems have emerged as a paradigm for adaptive responses. In its simplest form, a two-component system contains a sensor, a histidine kinase, and a response regulator, often a transcriptional activator. Changes in the environment result in phosphorylation of the sensor followed by transphosphorylation onto the response regulator. Adaptive responses controlled by two-component regulatory systems are diverse and include chemotaxis, fruit ripening, sporulation, and virulence gene expression in numerous pathogens (see ref. 1).
The outer membrane proteins OmpF and OmpC in Escherichia coli are regulated in response to changes in the osmolarity of the medium (see ref. 2 for a recent review). At low osmolarity, OmpF predominates; at high osmolarity, ompF is repressed, and OmpC is the major porin in the outer membrane (3). This process is controlled by the EnvZ/OmpR two-component regulatory system (4). EnvZ is a histidine kinase located in the inner membrane, OmpR is a cytoplasmic DNA binding protein (5). The two proteins communicate via a series of phosphorylation and phosphotransfer reactions. EnvZ senses the osmotic environment and is autophosphorylated from intracellular ATP at His-243 (6, 7). EnvZ-P phosphorylates OmpR at Asp-55 (8), and phospho-OmpR (OmpR-P) binds to the upstream sites of the porin genes to regulate their expression (9–12). EnvZ also stimulates the dephosphorylation of OmpR-P, thereby controlling the concentration of cellular OmpR-P (11, 13). Phosphorylation of OmpR results in an increase in its affinity for DNA (9, 14), and recent studies indicate that this effect is 10- to 30-fold, depending on the binding site (15).
There is accumulating evidence that OmpR also plays a global regulatory role, because it regulates genes in addition to the porin genes ompF and ompC. A specific mutation in envZ, V241G, results in a pleiotropic phenotype in which OmpR regulates additional genes outside of its normal repertoire, including phoA and phoE of the Pho regulon and lamB and malT of the maltose regulon (16). OmpR modulates expression of genes in the flagellar operon flhDC (17); a fatty acid receptor gene, fadL (18); and the tripeptide permease TppB (19). OmpR also activates transcription of the mcb operon in both exponential and stationary phases (20). Recently, a mutation in ompR was implicated in the production of a biofilm phenotype in E. coli, resulting from curli fimbrial expression (21); ompR also plays a role in curli fimbrial expression in Salmonella typhimurium (22). Mutations in ompR that result in attenuated virulence in mice have been reported in S. typhimurium (23) and Yersinia enterocolitica (24). OmpR also affects Salmonella-induced filament formation (triggered by intracellular S. typhimurium) (25), the ability of Salmonella to escape from macrophages (26), and cell-to-cell spreading and epithelial-cell killing in Shigella flexneri (27, 28). In Salmonella typhi, a cryptic porin ompS1, is regulated by OmpR (29). Thus, in a wide variety of organisms, OmpR plays a central regulatory role in controlling gene expression.
OmpR is a bifunctional protein, containing an N-terminal phosphorylation domain (30) structurally similar to the chemotaxis protein CheY (31, 32). The C-terminal domain contains a winged helix–turn–helix motif that binds DNA (33–35). The two domains are connected to one another via an exposed Q linker (36) that is sensitive to proteolysis (37). In this study, we report that DNA binding in the C-terminal region of OmpR stimulates phosphorylation at the N terminus. The effect of DNA binding is to change the conformation around the active site of phosphorylation at Asp-55. Evidence is also presented that the exposure of trypsin cleavage sites in the linker region is altered in conjunction with either phosphorylation in the N terminus or DNA binding in the C terminus, suggesting that the linker conformation changes as a result.
Materials and Methods
Materials.
Acetyl phosphate, ATP, trypsin, and trypsin inhibitor were purchased from Sigma. Oligonucleotides were prepared by the Microbiology Core Facility, Oregon Health Sciences University. One of the sequences of the oligonucleotides used in this study was (5′–3′) pstS (cctctctgtcataaaactgtcat), and the others have been described in ref. 15. Hitrap Desalt columns, NAP-5, and the Probe Quant columns were purchased from Amersham Pharmacia. N-terminal amino acid sequencing was performed by the Emory University Microsequencing Facility, Atlanta, GA. PyMPO {1-(2-maleimidylethyl)-4-[5-(4-methoxyphenyl)oxazol-2-yl]pyridinium methane sulfonate} was from Molecular Probes.
Purification of the OmpR Protein.
OmpR was purified according to the method of Jo et al. (38) with the modifications described in ref. 15 and stored at 4°C in 20 mM Tris, pH 7.4/5% (vol/vol) glycerol/0.1 mM EDTA/0.1 mM DTT. The protein was freshly dialyzed in the appropriate buffer before use.
Reverse-Phase HPLC of OmpR.
Separation of OmpR-P from OmpR was performed as described (15).
OmpR Phosphorylation and Dephosphorylation Reactions.
OmpR was phosphorylated with acetyl phosphate as described (37). Phosphorylation of OmpR with a truncated form of the kinase, EnvZ115 (6), was for 15 min, the peak of an experimentally determined time course, with conditions as described (39). EnvZ115 does not localize to the inner membrane but contains all of the biochemical activities associated with the kinase (39). For phosphorylation with MBP-EnvZ (14), varying OmpR and MBP-EnvZ concentrations were incubated in a buffer containing (in mM) 5 ATP/50 Tris⋅HCl, pH 7.6/50 KCl/20 MgCl2 for 3 h at room temperature (RT). For the dephosphorylation studies, OmpR was phosphorylated with acetyl phosphate for 3 h as before. A sample of each phosphorylation reaction was chromatographed on C4 by using reverse-phase HPLC, representing 100% at the zero time point. Acetyl phosphate was removed by passage of the reaction over a NAP-5 gel filtration column. The removal of acetyl phosphate was confirmed in an assay for acetyl phosphate (40). The absorbance of the fractions at 280 nm was measured, and the OmpR-containing fractions were pooled. At the times indicated, 50-μl samples were removed from the reaction and chromatographed on C4 reverse-phase HPLC; the faster eluting peak corresponded to OmpR-P, as previously determined by mass spectrometry (15).
DNA Binding Assays.
Electrophoretic mobility-shift assays were performed at RT by incubating 100 pmol of 32P-labeled C1 oligonucleotide with 200 pmol of OmpR or OmpR-P for 3 h at RT. The binding reactions were performed in 25 μl of 50 mM Na2HPO4, pH 8/20 mM MgCl2 and separated on a 6% acrylamide Tris/borate/EDTA gel.
Fluorescent Labeling of OmpR with PyMPO.
OmpR (1 ml; 20 μM) was reacted with a 50-fold molar excess of PyMPO in 40 mM Imidazole/1 mM EDTA, pH 7.6 for 30 min at RT. PyMPO was resuspended in DMSO at approximately 10 mM; the concentration was determined spectrophotometrically. The products of the labeling reaction were separated by SDS/12% PAGE.
Limited Proteolysis with Trypsin.
A 36-bp double-stranded oligonucleotide corresponding to a C1 binding site (15) was incubated at RT with 50 μg of OmpR for 30 min. The trypsin digest and the subsequent protein transfer were conducted as described (37). The protein bands were excised from the blot, and the N-terminal sequence was determined by automated Edman degradation.
Results
Previously, we used fluorescence anisotropy to measure the apparent affinities of OmpR and OmpR-P to the ompF and ompC regulatory regions. Our findings indicated that phosphorylation of OmpR by acetyl phosphate had a 10- to 30-fold effect on its affinity for DNA (15). It is predicted that the converse should also be true; thus, we investigated the effects of DNA binding on the OmpR phosphorylation reaction.
DNA Binding Stimulates OmpR Phosphorylation.
We developed a means of separating OmpR from OmpR-P by using reverse-phase HPLC on a C4 column (15), which enabled us to quantitate the extent of phosphorylation in our DNA binding assay. To determine the effect of DNA binding on phosphorylation, we added the oligonucleotide C1 to the phosphorylation reaction. It was the highest affinity OmpR binding site that we had previously measured (15). We observed that DNA binding stimulated OmpR phosphorylation by acetyl phosphate (Fig. 1). In the absence of C1 DNA, only 32% of the OmpR protein was phosphorylated after 120 min (Fig. 1A), whereas, in the presence of C1 DNA, 94% of the OmpR protein was phosphorylated (Fig. 1B).
Figure 1.

The effect of C1 DNA on OmpR phosphorylation by acetyl phosphate. OmpR (7.5 μM) was phosphorylated in a buffer described in Materials and Methods with 25 mM acetyl phosphate in 300 μl for 120 min in the absence (A) or presence (B) of 15 μM C1 DNA, and the products were isolated on C4 reverse-phase HPLC as described (15). In A, 32% of the OmpR is phosphorylated compared with 94% in B. The percentage of the protein that is phosphorylated is determined by combining the areas of the two peaks (OmpR-P + OmpR). The area under each individual peak, representing the fraction of either OmpR-P or OmpR, is expressed as a percentage of the total protein.
Time Course of Stimulation of OmpR Phosphorylation by C1 DNA and the Effect of C1 DNA on OmpR-P Dephosphorylation.
In Fig. 2A, a time course of OmpR phosphorylation by acetyl phosphate in the absence and presence of C1 DNA is shown. The presence of C1 dramatically stimulates the steady-state level of OmpR phosphorylation (Fig. 2A, upper curve). The time required to phosphorylate 50% of the OmpR protein is 10 min in the presence of C1 DNA and 240 min in the absence of DNA. To determine whether the effect of DNA was caused by a stabilization of OmpR-P by slowing dephosphorylation, we also examined the effect of C1 DNA on the rate of OmpR-P dephosphorylation after removal of acetyl phosphate by gel filtration (Fig. 2B). The dephosphorylation rates differ by only 2-fold. The presence of C1 DNA does not greatly stabilize the phosphoprotein, and OmpR-P dephosphorylates at nearly the same rate whether DNA is present or absent. Thus, the presence of C1 DNA stimulates the rate of phosphorylation of OmpR. The most straightforward interpretation of this result is that DNA binding in the C-terminal domain of OmpR alters the conformational state of the active site of phosphorylation in the N-terminal domain and makes it a better substrate for phosphorylation. Alternatively, OmpR may exist in two conformations: one that is readily phosphorylated, the other that is phosphorylated only very slowly. Binding to DNA shifts the distribution, favoring the form that is more readily phosphorylated.
Figure 2.

(A) Time course of OmpR phosphorylation. OmpR (7.5 μM) was incubated with 25 mM acetyl phosphate in the presence (triangles) or absence (circles) of a 2-fold molar excess of C1 DNA. At 5, 15, 30, 60, 90, 120, 150, and 180 min, 50-μl samples were withdrawn from the phosphorylation reaction and injected onto a C4 column and separated by reverse-phase HPLC. OmpR-P is plotted as a percentage of total protein versus time. (B) Time course of OmpR-P dephosphorylation. OmpR is phosphorylated as described for A, and after 180 min, the acetyl phosphate is removed by gel filtration. OmpR-P is monitored via C4 reverse-phase HPLC, and this value represents 100% at time zero. OmpR-P is plotted in the presence (triangles) and absence (circles) of C1 DNA at the times indicated.
Stimulation of OmpR Phosphorylation by DNA Requires Binding of Specific DNA.
We determined whether the presence of a specific DNA binding site is required to stimulate phosphorylation or whether nonspecific interactions are involved (Table 1). In the presence of an F1 or C1 binding site, stimulation of OmpR phosphorylation occurs. An F1 or C1 half-site is unable to stimulate phosphorylation. Nonspecific DNA, such as a pstS oligonucleotide containing a PhoB binding site (to which OmpR does not bind), does not stimulate phosphorylation (Table 1). Stimulation of phosphorylation is observed only with the high-affinity sites F1 and C1 or in compound sites that contained the high-affinity sites. Low-affinity sites do not stimulate phosphorylation. The upstream binding site F4 has been difficult to characterize; however, recently, we have been successful in measuring saturable binding of both OmpR and OmpR-P to F4 by using fluorescence anisotropy (V. Tran and L.J.K., unpublished results). Under the conditions in which DNA binding stimulates phosphorylation, the DNA is indeed bound, as indicated by a shifted band in an electrophoretic mobility-shift assay (Fig. 3).
Table 1.
The effect of DNA on OmpR phosphorylation by acetyl phosphate
| OmpR-P, percentage of total | Fold stimulation | |
|---|---|---|
| No DNA added | 45 | |
| F1 | 84 | 1.9 |
| F1 half site | 42 | 0 |
| F2 | 59 | 1.3 |
| F3 | 45 | 0 |
| F4 | 85 | 1.9 |
| F1–F2–F3 | 83 | 1.8 |
| C1 | 86 | 1.9 |
| C1 half site | 41 | 0 |
| C2 | 36 | 0 |
| C3 | 33 | 0 |
| C1–C2–C3 | 84 | 1.9 |
| pstS | 42 | 0 |
The role of individual binding sites in stimulating OmpR phosphorylation by acetyl phosphate. In the first column, the oligonucleotides present during the phosphorylation reaction are listed. In the second column, OmpR-P is expressed as a percentage of the total OmpR protein present in the reaction, as determined by C4 reverse-phase HPLC. The third column shows the stimulation caused by the presence of DNA. Phosphorylation was for 3 h as described in Materials and Methods, and the oligonucleotides were present at a 2-fold molar excess in the presence of 10 μM OmpR.
Figure 3.
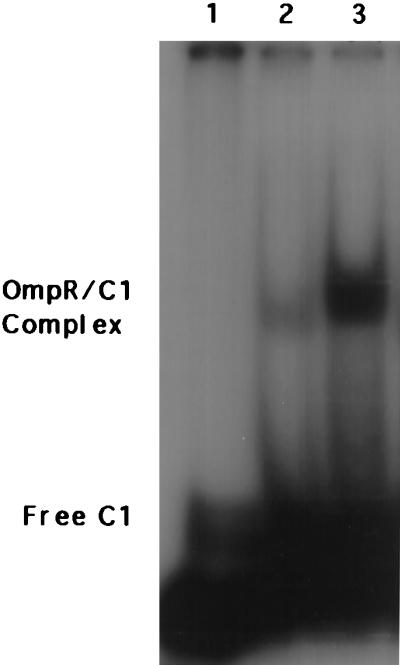
Electrophoretic mobility-shift assay. The free radiolabeled C1 probe in the absence of OmpR protein is shown in lane 1 (indicated by Free C1 to the left of the lane). OmpR (8 μM) was incubated with 4 μM C1 for 3 h at RT in the absence (lane 2) or presence (lane 3) of 25 mM acetyl phosphate, and the shifted complex is indicated by OmpR/C1 Complex to the left of the gel.
OmpR Phosphorylation in the Presence of DNA Inhibits PyMPO Labeling.
Because DNA binding alters the phosphorylation site, it is likely that there are more extensive changes that occur throughout the OmpR protein as a consequence of DNA binding. It might also affect the exposure of the lone cysteine in OmpR located near the site of phosphorylation. We examined the reactivity of Cys-67 with the fluorescent maleimide PyMPO in response to phosphorylation and the presence of C1 DNA (Fig. 4). In the presence of C1, there is a stimulation of OmpR labeling by PyMPO (Fig. 4, compare lane 2 to lane 1). When OmpR is phosphorylated before labeling, there is a decrease in PyMPO labeling (Fig. 4, lane 3). When the concentration of OmpR-P is increased by binding to C1 DNA, significantly less OmpR is labeled (Fig. 4, lane 4). Labeling with PyMPO before incubation of OmpR with acetyl phosphate decreased phosphorylation to 27% as determined by C4 HPLC (data not shown).
Figure 4.

Fluorescent labeling of OmpR with PyMPO. OmpR (9.5 μM) is reacted with a 50-fold excess of PyMPO for 30 min at RT in 40 mM Imidazole/1 mM EDTA, pH 7.6 (lane 1). Lane 2 shows a stimulation of PyMPO labeling in the presence of C1. OmpR (9.5 μM) is incubated with acetyl phosphate for 3 h at RT in the absence (lane 3) or presence (lane 4) of C1 DNA and then labeled with PyMPO as before. The products of the labeling reaction are separated on SDS/12% PAGE. The visible fluorescent band is the PyMPO-labeled OmpR protein.
DNA Binding Stimulates OmpR Phosphorylation by EnvZ.
We determined whether stimulation of OmpR phosphorylation is observed when phosphorylating with EnvZ or whether it is merely a result of phosphorylating with small-molecule phosphorylating agents such as acetyl phosphate. We performed the phosphorylation reaction by using the kinase EnvZ-P, generated via phosphorylation from ATP. For these experiments, we used either a fusion protein, MBP-EnvZ (14), or an N-terminal truncated form of EnvZ, EnvZ115 (6, 39). Clearly, DNA binding stimulates OmpR phosphorylation by both the phosphodonor acetyl phosphate and the kinase EnvZ-P (Table 2). At low concentrations of the kinase or with acetyl phosphate, there is approximately a 2-fold stimulation of phosphorylation in the presence of DNA. Higher concentrations of the kinase further stimulate OmpR phosphorylation when DNA is present.
Table 2.
Stimulation of OmpR phosphorylation by C1 DNA: Comparison of phosphodonors
| Phosphodonor | OmpR-P, percentage of total
|
||
|---|---|---|---|
| −C1 | +C1 | Fold stimulation | |
| Acetyl phosphate | 49 | 84 | 1.7 |
| MBP-EnvZ | 10 | 21 | 2.1 |
| 11 | 78 | 7.1 | |
| EnvZ115 | 14 | 37 | 2.6 |
| 12 | 56 | 4.7 | |
OmpR (3.3 μM) is phosphorylated by acetyl phosphate (top row); by 8.8 μM OmpR and 1 μM MBP-EnvZ (second row); by 3.3 μM OmpR and 1 μM MBP-EnvZ (third row); by 5 μM OmpR with 30 μM of the truncated EnvZ protein EnvZ115 (fourth row); or by 44 μM EnvZ115 (bottom row), as described in Materials and Methods. OmpR-P is expressed as a percentage of the total OmpR protein present in the reaction, as determined by reverse-phase HPLC on a C4 column in the absence of C1 DNA (−C1) and in the presence of a 2-fold molar excess of C1 DNA (+C1). The degree of stimulation by C1 DNA is listed in the third column. The values shown in the table represent the means of duplicate samples, and the variation among duplicates was less than 10%.
C-Terminal DNA Binding Causes a Conformational Change in the Linker Region.
We used limited proteolysis with trypsin to identify conformational changes in OmpR resulting from phosphorylation (37). Phosphorylation of OmpR protects a cleavage site in the linker region. We observed three N-terminal proteolytic fragments of OmpR (labeled I, II, and III in Fig. 5; see also ref. 37). When OmpR was phosphorylated, only N-terminal fragments I and II were observed. The protected cleavage site mapped to the C terminus of the linker region (37). Because DNA binding stimulates OmpR phosphorylation, it was of interest to examine the trypsin cleavage pattern of OmpR in the presence of DNA (Fig. 5, lanes 3 and 4). Digesting OmpR with trypsin in the presence of C1 DNA produces a new fragment (labeled QANEL) that is not observed in the absence of DNA (Fig. 5, lanes 1 and 2). This fragment is the result of cleavage at Arg-122. It maps to the N terminus of the linker region. The appearance of the QANEL fragment was also observed in the presence of F1 DNA (data not shown). Thus, when the C-terminal domain of OmpR binds to DNA, it exposes a trypsin cleavage site in the N terminus of the linker.
Figure 5.

Limited proteolytic digestion of OmpR in the presence of C1 DNA. The conditions for proteolysis of OmpR are described in Materials and Methods. The ompC DNA was present at a 2:1 (mol/mol) ratio to OmpR. The digestion time was 30 min at RT. The products of digestion were isolated by SDS/15% PAGE and transferred to a poly(vinylidene difluoride) membrane. The band identified as QANEL was determined by N-terminal sequencing of the stained band excised after transfer to Immobilon. The sizes of the molecular mass markers (in kilodaltons) are indicated to the left of the figure. See Results for a description of fragments I, II, and III.
Discussion
DNA binding in the C-terminal domain of OmpR stimulates OmpR phosphorylation in the N terminus. The stimulation is caused largely by a stimulation in the OmpR phosphorylation rate, and only a minor effect of DNA is observed on OmpR-P dephosphorylation (Figs. 1 and 2). Stimulation of OmpR phosphorylation by DNA requires specific DNA; a PhoB binding site does not stimulate, nor do F1 or C1 half sites (Table 1). The DNA that stimulates phosphorylation is bound to OmpR (Fig. 3).
The simplest interpretation of our results is that DNA binding in the C terminus changes the conformation of OmpR to make the N terminus a better substrate for phosphorylation. This change could result from a localized effect that would merely alter the reactivity of the phosphorylated residue, Asp-55. Alternatively, DNA binding could drive a conformational change affecting the phosphorylation site and result in a more global change in the OmpR molecule. Because we observe stimulation of OmpR phosphorylation by both acetyl phosphate as well as the kinase EnvZ, the latter interpretation seems to be the more likely one.
In the presence of binding sites for which unphosphorylated OmpR can bind with reasonably high affinity (Kd < 200 nM or so), OmpR binds, and phosphorylation is stimulated. These include F1, C1, F4, and the complex sites F1–F2–F3 and C1–C2–C3 (Table 1). At sites such as F2, F3, C2, or C3, unphosphorylated OmpR does not bind, and OmpR-P binds only with low affinity (Kd = ≈300 nM; ref. 15). When only these low-affinity sites are present, OmpR must first become phosphorylated and then bind, and the presence of a low-affinity binding site does not stimulate OmpR phosphorylation further.
Fluorescent labeling of OmpR at Cys-67 inhibits phosphorylation at Asp-55, and phosphorylation at Asp-55 eliminates labeling at Cys-67. This exclusion is especially apparent in the presence of DNA, which results in a high level of OmpR-P and no labeling at Cys-67 (Fig. 4). Labeling with PyMPO at Cys-67 results in an uncoupling of phosphorylation from DNA binding. Furthermore, it suggests that the conformational change associated with phosphorylation is transmitted in part to the DNA binding domain via α-helix 3 (where Cys-67 is located).
OmpR is a bifunctional protein consisting of an N-terminal phosphorylation domain and a C-terminal DNA binding domain, joined by a flexible linker that is protease sensitive. The linker seems to be sensitive to both the phosphorylation status and the DNA binding domain occupancy. When OmpR is phosphorylated, a trypsin cleavage site in the linker is protected (Fig. 5; ref. 37), and when DNA is bound, a new cleavage site is exposed (Fig. 5). It may be that the linker undergoes a major structural change such as a helix–to–coil transition in response to either phosphorylation or DNA binding. Because the linker represents only 6% of the total protein, this mechanistically important structural change would be difficult to detect by low-resolution structural methods such as circular dichroism, but it is readily resolved by proteolytic analysis. It is unclear whether the linker functions as a signal transducer, actively communicating between the phosphorylation domain and the DNA binding domain in response to those signals. It may have only a passive role, changing its conformation when the two domains shift their relative interfaces. Alternatively, OmpR protein–protein interactions resulting from phosphorylation or DNA binding may result in an intermolecular reaction, causing the linker conformation to change.
In the crystal structure of another two-domain response regulator, NarL, the linker region is not discernible, suggesting that it is disordered. Access to the recognition helix of the C-terminal DNA binding domain is sterically blocked by the N-terminal phosphorylation domain (41). Phosphorylation in the N terminus of NarL must alter the exposure of the DNA binding domain to allow DNA binding. This inhibition does not occur with OmpR. In our previous study with limited proteolysis, we observed that the exposure of trypsin cleavage sites in the DNA binding domain was not affected by OmpR phosphorylation (37). Deletions within the 16-aa linker region of NarL inhibit the ability of NarL to activate a narG∷lacZ fusion (M. Jarvis and R. P. Gunsalus, personal communication). Activation was more sensitive than repression (of a frdA∷lacZ fusion) to deletions in the NarL linker. In order for the C-terminal recognition helix to bind to DNA (helix 9), it must rotate away from the N-terminal domain. It seems likely that the linker region is involved in this process.
With the two-domain response regulator CheB, phosphorylation in the N terminus stimulates methylesterase activity in the C terminus. In the absence of phosphorylation, the methylesterase is inhibited (42). The two domains are connected by an 18-residue linker that, except for a short helical turn of 4 residues, does not exhibit secondary structure (43). Mutations that enhance methylesterase activity in the absence of phosphorylation have been isolated, and seven of eight of the substitutions map to this linker region (44).
Clearly, OmpR reacts differently from the response regulators NarL and CheB. In the absence of phosphorylation, the C-terminal domains of CheB and NarL are inhibited. In the absence of phosphorylation, OmpR binds DNA but with lower affinity (15). The C-terminal domain of OmpR is not inhibited by its N-terminal domain. These differences among two domain response regulators may indicate interesting differences in their linker regions. Perhaps the OmpR linker is more flexible compared with CheB and NarL, permitting low-affinity DNA binding in the absence of phosphorylation.
We can represent OmpR as an equilibrium mixture of four distinct states (Fig. 6). Phosphorylation shifts OmpR into the active conformation (Fig. 6B) that binds DNA with high affinity (Fig. 6D). In the absence of phosphorylation, OmpR binds to DNA with low affinity (Fig. 6C). In this work, we show that when OmpR is bound to DNA, it stimulates phosphorylation (the C-to-D transition). DNA binding lowers the energy required for phosphorylation and vice versa. Genetic approaches have resulted in the isolation of mutations that can be interpreted as affecting these equilibria (see legend to Fig. 6).
Figure 6.

Four-states model for OmpR. (A and C) The unphosphorylated forms of OmpR. (B and D) The phosphorylated forms of OmpR. (A and B) The forms of OmpR that are not bound to DNA. (C and D) The DNA-bound forms of OmpR. Relevant mutants are shown next to the arrows denoting the equilibria that seem to be affected by the substitution. The OmpR mutant K105R (C) binds to DNA with an affinity similar to unphosphorylated OmpR, but the mutant is not stably phosphorylated (15). DNA-binding mutants shown are E96A and R115S (48). They can be phosphorylated by EnvZ-P; however, they do not bind ompF and ompC, and they have an F−C− phenotype. The DNA binding mutants T162I, R182L, S200F, T224I, and G229S (49) and the double-mutant M211V/V212A (50) are defective in binding. Their ability to be phosphorylated has not been determined but is presumed to be unaffected by the mutation or mutations. The V203M mutant is affected in binding at ompC and at some ompF sites, and its phenotype is FcC− (51–53). Surprisingly, phosphorylation by acetyl phosphate of this mutant is inhibited (V. Tran and L.J.K., unpublished results), and it is therefore shown locked in the A/C position. The constitutively active mutants Y102C (54), T83A, and G94S (55) bind DNA with high affinity in the absence of phosphorylation. The activation mutants S181P (49), P179L, A196V, E198K, R42C/H (56), and E193K (52) bind but cannot activate transcription and are presumed to be defective in their ability to interact with RNA polymerase. Note that the linker is represented as being in a different conformation in each of the four states. N = N terminus; C = C terminus, P = phosphoryl group.
The equilibria among four states may be a general mechanism of response regulators, whether they are two-domain proteins or not. Silversmith and Bourret (45) have identified four states of CheY based on genetically separable steps in the activation and inactivation of clockwise signaling. In each case, the four states correspond to the combinations of phosphorylated and unphosphorylated, active and inactive conformations. With CheY, the active conformation results in clockwise signaling; in the case of OmpR, the active conformation binds DNA. When we compare the four OmpR states with those of CheY, interesting differences emerge. CheY mutants K109R and T87I are phosphorylated but fail to cause clockwise flagellar rotation (B). The analogous OmpR mutant K105R (C) binds to DNA with an affinity similar to unphosphorylated OmpR, but the mutant is not stably phosphorylated (15). It seems that in this mutant, the A ⇌ B and the C ⇌ D equilibria are both pushed to the left, i.e., toward dephosphorylation. The CheY mutant D13K is like the constitutively active mutants Y102C, etc., because it puts the protein in the active conformation in the absence of phosphorylation (i.e., it affects the A ⇌ C equilibrium; refs. 46 and 47). The OmpR mutant T83I has not been characterized.
Previously, we have shown that OmpR phosphorylation stimulates DNA binding (A → B → D; ref. 15). In the present work, we show that DNA binding greatly stimulates OmpR phosphorylation (C → D ≫ A → B). Thus, under our conditions, the favored pathway is A → C → D. We have also shown that dephosphorylation of OmpR-P is not affected greatly by DNA binding (i.e., B → A ≅ D → C). Analysis of the factors affecting the equilibria among the states of this four-state model and the effects of mutants on them will lead to a more systematic description of the role of response regulators in cellular signaling.
Because phosphorylation of OmpR stimulates DNA binding, it follows that DNA binding should stimulate phosphorylation. We have shown that this principle holds for the response regulator OmpR, and it is anticipated to be a general mechanism for other response regulators that are also transcription factors. At a cellular concentration of 1 μM OmpR and binding affinities for unphosphorylated OmpR of 85–300 nM (15), the possibility exists that unphosphorylated OmpR is bound to the ompF and ompC regulatory regions in vivo. Our results suggest that response regulators may be phosphorylated while bound to their target DNA.
Acknowledgments
We thank Charlotte Head for HPLC expertise and OmpR purification; Michele Igo for the generous gift of MBP-EnvZ; Becky Kapphahn for purification; our laboratory members; and Steve Mansoor, Tom Dunham, Dave Farrens, Richard Brennan, and Sydney Kustu for helpful discussions. L.J.K. thanks Jack H. Kaplan for comments on the manuscript, scientific input, and encouragement. L.J.K. is deeply indebted to R. Bourret for helpful discussions and to R. Bourret and Ruth Silversmith for their comments on the manuscript before submission. This work was supported by National Science Foundation Grant MCB-9513275.
Abbreviations
- OmpR-P
phospho-OmpR
- PyMPO
1-(2-maleimidylethyl)-4-(5-(4-methoxyphenyl)oxazol-2-yl)pyridinium methane sulfonate
- RT
room temperature
References
- 1.Hoch J A, Silhavy T J. Two-Component Signal Transduction. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 2.Pratt L A, Silhavy T J. In: Two-Component Signal Transduction. Hoch J A, Silhavy T J, editors. Washington, DC: Am. Soc. Microbiol.; 1995. pp. 105–127. [Google Scholar]
- 3.van Alphen W, Lugtenberg B. J Bacteriol. 1977;131:623–630. doi: 10.1128/jb.131.2.623-630.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hall M, Silhavy T J. J Mol Biol. 1981;151:1–15. doi: 10.1016/0022-2836(81)90218-7. [DOI] [PubMed] [Google Scholar]
- 5.Forst S, Comeau D, Norioka S, Inouye M. J Biol Chem. 1987;262:16433–16438. [PubMed] [Google Scholar]
- 6.Igo M M, Silhavy T J. J Bacteriol. 1988;170:5971–5973. doi: 10.1128/jb.170.12.5971-5973.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts D L, Bennett D W, Forst S A. J Biol Chem. 1994;269:8728–8733. [PubMed] [Google Scholar]
- 8.Delgado J, Forst S, Harlocker S, Inouye M. Mol Microbiol. 1993;10:1037–1047. doi: 10.1111/j.1365-2958.1993.tb00974.x. [DOI] [PubMed] [Google Scholar]
- 9.Aiba H, Nakasai F, Mizushima S, Mizuno T. J Biochem. 1989;106:5–7. doi: 10.1093/oxfordjournals.jbchem.a122817. [DOI] [PubMed] [Google Scholar]
- 10.Aiba H, Mizuno T, Mizushima S. J Biol Chem. 1989;264:8563–8567. [PubMed] [Google Scholar]
- 11.Igo M M, Ninfa A J, Stock J B, Silhavy T J. Genes Dev. 1989;3:1725–1734. doi: 10.1101/gad.3.11.1725. [DOI] [PubMed] [Google Scholar]
- 12.Igo M M, Silhavy T J. Genes Dev. 1989;3:598–605. doi: 10.1101/gad.3.5.598. [DOI] [PubMed] [Google Scholar]
- 13.Aiba H, Nakasai F, Mizushima S, Mizuno T. J Biol Chem. 1989;264:14090–14094. [PubMed] [Google Scholar]
- 14.Huang K J, Igo M M. J Mol Biol. 1996;262:615–628. doi: 10.1006/jmbi.1996.0540. [DOI] [PubMed] [Google Scholar]
- 15.Head C G, Tardy A, Kenney L J. J Mol Biol. 1998;281:857–870. doi: 10.1006/jmbi.1998.1985. [DOI] [PubMed] [Google Scholar]
- 16.Verhoef C, Lugtenberg B, van Boxtel R, de Graaff P, Verheij H. Mol Gen Genet. 1979;169:137–146. doi: 10.1007/BF00271664. [DOI] [PubMed] [Google Scholar]
- 17.Shin S, Park C. J Bacteriol. 1995;177:4696–4702. doi: 10.1128/jb.177.16.4696-4702.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higashitani A, Nishimura Y, Hara H, Aiba H, Mizuno T, Horiuchi K. Mol Gen Genet. 1993;240:339–347. doi: 10.1007/BF00280384. [DOI] [PubMed] [Google Scholar]
- 19.Gibson M M, Ellis E M, Graeme-Cook K A, Higgins C F. Mol Gen Genet. 1987;207:120–129. doi: 10.1007/BF00331499. [DOI] [PubMed] [Google Scholar]
- 20.Mao W, Siegele D A. Mol Microbiol. 1998;27:415–424. doi: 10.1046/j.1365-2958.1998.00690.x. [DOI] [PubMed] [Google Scholar]
- 21.Vidal O, Longin R, Prigent-Combaret C, Dorel C, Hooreman M, Lejeune P. J Bacteriol. 1998;180:2442–2449. doi: 10.1128/jb.180.9.2442-2449.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romling U, Sierralta W D, Eriksson K, Normark S. Mol Microbiol. 1998;28:249–264. doi: 10.1046/j.1365-2958.1998.00791.x. [DOI] [PubMed] [Google Scholar]
- 23.Dorman C J, Chatfield S, Higgins C F, Hayward C, Dougan G. Infect Immun. 1989;57:2136–2140. doi: 10.1128/iai.57.7.2136-2140.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorrell N, Li S R, Everest P H, Dougan G, Wren B W. FEMS Microbiol Lett. 1998;165:145–151. doi: 10.1111/j.1574-6968.1998.tb13139.x. [DOI] [PubMed] [Google Scholar]
- 25.Mills S D, Ruschkowski S R, Stein M A, Finlay B B. Infect Immun. 1998;66:1806–1811. doi: 10.1128/iai.66.4.1806-1811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindgren S W, Stojiljkovic I, Heffron F. Proc Natl Acad Sci USA. 1996;93:4197–4201. doi: 10.1073/pnas.93.9.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernardini M L, Fontaine A, Sansonetti P J. J Bacteriol. 1990;172:6274–6281. doi: 10.1128/jb.172.11.6274-6281.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernardini M L, Sanna M G, Fontaine A, Sansonetti P J. Infect Immun. 1993;61:3625–3635. doi: 10.1128/iai.61.9.3625-3635.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez-Mora M, Oropeza R, Puente J L, Calva E. Gene. 1995;158:67–72. doi: 10.1016/0378-1119(95)00171-2. [DOI] [PubMed] [Google Scholar]
- 30.Kato M, Aiba H, Tate S, Nishimura Y, Mizuno T. FEBS Lett. 1989;249:168–172. doi: 10.1016/0014-5793(89)80617-9. [DOI] [PubMed] [Google Scholar]
- 31.Stock A M, Mottonen J M, Stock J B, Schutt C E. Nature (London) 1989;337:745–749. doi: 10.1038/337745a0. [DOI] [PubMed] [Google Scholar]
- 32.Volz K, Matsumura P. J Biol Chem. 1991;266:15511–15519. doi: 10.2210/pdb3chy/pdb. [DOI] [PubMed] [Google Scholar]
- 33.Tate S, Kato M, Nishimura Y, Arata Y, Mizuno T. FEBS Lett. 1988;242:27–30. doi: 10.1016/0014-5793(88)80978-5. [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Hackert E, Stock A M. Structure. 1997;5:109–124. doi: 10.1016/s0969-2126(97)00170-6. [DOI] [PubMed] [Google Scholar]
- 35.Kondo H, Nakagawa A, Nishihira J, Nishimura Y, Mizuno T, Tanaka I. Nat Struct Biol. 1997;4:28–31. doi: 10.1038/nsb0197-28. [DOI] [PubMed] [Google Scholar]
- 36.Wootton J C, Drummond M H. Protein Eng. 1989;2:535–543. doi: 10.1093/protein/2.7.535. [DOI] [PubMed] [Google Scholar]
- 37.Kenney L J, Bauer M D, Silhavy T J. Proc Natl Acad Sci USA. 1995;92:8866–8870. doi: 10.1073/pnas.92.19.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jo Y-L, Nara F, Ichihara S, Mizuno T, Mizushima S. J Biol Chem. 1986;261:15252–15256. [PubMed] [Google Scholar]
- 39.Kenney L J. Arch Biochem Biophys. 1997;346:303–311. doi: 10.1006/abbi.1997.0315. [DOI] [PubMed] [Google Scholar]
- 40.Skarstedt M T, Silverstein E. J Biol Chem. 1976;251:6775–6783. [PubMed] [Google Scholar]
- 41.Baikalov I, Schroder I, Kaczor-Grzeskowiak M, Grzeskowiak K, Gunsalus R P, Dickerson R E. Biochemistry. 1996;35:11053–11061. doi: 10.1021/bi960919o. [DOI] [PubMed] [Google Scholar]
- 42.Lupas A, Stock J. J Biol Chem. 1989;264:17337–17342. [PubMed] [Google Scholar]
- 43.Djordjevic S, Goudreau P N, Xu Q, Stock A M, West A H. Proc Natl Acad Sci USA. 1998;95:1381–1386. doi: 10.1073/pnas.95.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anand G S, Goudreau P N, Stock A M. Biochemistry. 1998;37:14038–14047. doi: 10.1021/bi980865d. [DOI] [PubMed] [Google Scholar]
- 45.Silversmith R E, Bourret R B. Trends Microbiol. 1999;17:16–22. doi: 10.1016/s0966-842x(98)01409-7. [DOI] [PubMed] [Google Scholar]
- 46.Bourret R B, Hess J F, Simon M I. Proc Natl Acad Sci USA. 1990;87:41–45. doi: 10.1073/pnas.87.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bourret R B, Drake S K, Chervitz S A, Simon M I, Falke J J. J Biol Chem. 1993;268:13089–13096. [PMC free article] [PubMed] [Google Scholar]
- 48.Nakashima K, Kanamaru K, Aiba H, Mizuno T. FEMS Microbiol Lett. 1991;66:43–47. doi: 10.1016/0378-1097(91)90418-a. [DOI] [PubMed] [Google Scholar]
- 49.Kato N, Tsuzuki M, Aiba H, Mizuno T. Mol Gen Genet. 1995;248:399–406. doi: 10.1007/BF02191639. [DOI] [PubMed] [Google Scholar]
- 50.Aiba H, Kato N, Tsuzuki M, Mizuno T. FEBS Lett. 1994;351:303–307. doi: 10.1016/0014-5793(94)00846-9. [DOI] [PubMed] [Google Scholar]
- 51.Mizuno T, Kato M, Jo Y L, Mizushima S. J Biol Chem. 1988;263:1008–1012. [PubMed] [Google Scholar]
- 52.Russo F D, Slauch J M, Silhavy T J. J Mol Biol. 1993;231:261–273. doi: 10.1006/jmbi.1993.1281. [DOI] [PubMed] [Google Scholar]
- 53.Forst S, Kalve I, Durski W. FEMS Microbiol Lett. 1995;131:147–151. doi: 10.1111/j.1574-6968.1995.tb07769.x. [DOI] [PubMed] [Google Scholar]
- 54.Kanamaru K, Mizuno T. J Biochem. 1992;111:425–430. doi: 10.1093/oxfordjournals.jbchem.a123773. [DOI] [PubMed] [Google Scholar]
- 55.Brissette R E, Tsung K L, Inouye M. J Bacteriol. 1991;173:3749–3755. doi: 10.1128/jb.173.12.3749-3755.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pratt L A, Silhavy T J. J Mol Biol. 1994;243:579–594. doi: 10.1016/0022-2836(94)90033-7. [DOI] [PubMed] [Google Scholar]