

Abstract
Background
PLS is a rare autosomal recessive disorder characterized by early onset periodontopathia and palmar plantar keratosis. PLS is caused by mutations in the cathepsin C (CTSC) gene. Dipeptidyl-peptidase I encoded by the CTSC gene removes dipeptides from the amino-terminus of protein substrates and mainly plays an immune and inflammatory role. Several mutations have been reported in this gene in patients from several ethnic groups. We report here mutation analysis of the CTSC gene in three Indian families with PLS.
Methods
Peripheral blood samples were obtained from individuals belonging to three Indian families with PLS for genomic DNA isolation. Exon-specific intronic primers were used to amplify DNA samples from individuals. PCR products were subsequently sequenced to detect mutations. PCR-SCCP and ASOH analyses were used to determine if mutations were present in normal control individuals.
Results
All patients from three families had a classic PLS phenotype, which included palmoplantar keratosis and early-onset severe periodontitis. Sequence analysis of the CTSC gene showed three novel nonsense mutations (viz., p.Q49X, p.Q69X and p.Y304X) in homozygous state in affected individuals from these Indian families.
Conclusions
This study reported three novel nonsense mutations in three Indian families. These novel nonsense mutations are predicted to produce truncated dipeptidyl-peptidase I causing PLS phenotype in these families. A review of the literature along with three novel mutations reported here showed that the total number of mutations in the CTSC gene described to date is 41 with 17 mutations being located in exon 7.
Background
With an incidence of 1 to 4/million, Papillon-Lefèvre Syndrome (PLS, MIM 245000) is a rare autosomal recessive disorder with an onset usually by two to three years of age [1,2]. The disease is mainly characterized by severe early onset periodontitis, leading to premature tooth shedding of both the deciduous and permanent teeth, alveolar bone destruction (such that by 15 years of age the patients are usually edentulous), and hyperkeratosis characteristically involving the palms and soles and sometimes the knees, elbows, knuckles, and back [1]. PLS can manifest itself as early as 2 months of age with the appearance of hyperkeratotic lesions of the hands and feet [3]. Other clinical features not often reported are an increased susceptibility to infections (especially furunculosis and pyoderma, pyogenic liver abscess etc.,) with recurrent fevers in childhood and calcification of the dura. Males and females are equally affected with no racial predominance [3]. Approximately one-third of the families show consanguinity [2].
PLS is a genetically homogeneous disorder with a locus mapped on chromosome 11q14-q21 [4-6]. Toomes et al. [7] have subsequently reported that loss-of-function mutations in the cathepsin C gene result in the PLS phenotype. The cathepsin C gene encodes a cysteine lysosomal protease also known as dipeptidyl-peptidase I which functions by removing dipeptides from the amino terminus of the protein substrate. It also has endopeptidase activity. It is highly expressed in various tissues such as the cells of the immune system (polymorphic nuclear leukocytes, alveolar macrophages and their precursors), and in the lungs, kidneys and other epithelial tissues [8]. Its main functions are thought to be protein degradation and pro-enzyme activation in addition to its immunological role [8]. The CTSC gene is over 46 kb long and consists of seven exons and six introns [7]. Several mutations have been reported in the CTSC gene in individuals from diverse ethnic groups. In addition, mutation in this gene was also detected in a Jewish-Hindu family with Haim-Munk syndrome [9] and prepubertal periodontitis [10]. We report here mutation analysis of the CTSC gene in three Indian families with members suffering from PLS. We also present a summary of cathepsin C mutations reported to date in PLS patients.
Methods
Patients
We have ascertained three families from the southern part of India, IISC-PLS1, IISC-PLS2 and IISC-PLS3 with PLS phenotype. Families IISC-PLS1 and IISC-PLS3 are non-consanguineous, whereas family IISC-PLS2 shows consanguinity. A total of 11 individuals including five patients were recruited for the study from these three families. All family members were examined in detail by clinicians and the patients with PLS were diagnosed according to previously established criteria [1,2,11]. This research followed the tenets of the Declaration of Helsinki. Informed consent was obtained from the individuals included in this study.
PCR amplification and mutation analysis
Genomic DNA samples were isolated from peripheral blood samples of all available members of three families using a Wizard® Genomic DNA Extraction kit (Promega Inc., USA). Genomic DNA from one affected member from each family was initially PCR amplified for all seven exons spanning the CTSC gene using pairs of exon-specific intronic primers. The primer sequences are described in Toomes et al. [7]. PCR amplification was carried out in a 25 μl reaction volume containing ~100 ng genomic DNA, 60 ng of each primer, 200 μmol of each dNTP, 1 unit Taq DNA polymerase (Bangalore Genei®, India) in a standard PCR buffer supplied by the manufacturer. Amplification was performed in a Minicycler™ (MJ Research® Inc., USA) under the following conditions: an initial denaturation at 95°C for 2 min was followed by 35 cycles at 94°C for 30 s, 62°C for 30 s, 72°C for 1 min and a final extension of 5 min at 72°C. PCR amplified products were purified using a Wizard® PCR Preps DNA Purification System (Promega Inc., USA) and subjected to direct sequencing using an fmol® DNA Cycle Sequencing System kit (Promega Inc., USA) according to the manufacturer's instruction. Once a nucleotide change was detected in a patient, other available members from the family were also sequenced to determine if the change was also present in them. PCR-SSCP and allele-specific oligonucleotide hybridization (ASOH) analyses were used to determine if a nucleotide change was present in ethnically matched normal controls as described in Kumar et al. [12] and Cormand et al. [13] respectively.
Results
All patients had a history of the onset of the disorder by the age of 2 to 3 years. Examination of these patients revealed that they all had severe palmar plantar keratoderma, and severe periodontitis (except individual II-2 from family IISC-PLS3) leading to shedding of most of their teeth. Patient II-2 from family IISC-PLS3 had a subsidence of periodontitis, as he was edentulous, having lost all his teeth by the age of 12 years. Patients IV-3 and IV-4 from family IISC-PLS2 also had hyperkeratosis of the knuckles, knees and elbows in addition to palmar plantar keratoderma and severe periodontitis. Fig. 1 illustrates clinical manifestations of PLS. Individual II-2 from family IISC-PLS3 was known to have frequent episodes of infections especially of the skin and recurrent fevers in childhood, suggesting an increased susceptibility to microbial infections. Heterozygous parents from families IISC-PLS1 and IISC-PLS2 and heterozygous siblings (IV-1 and IV-5) from family IISC-PLS2 were unaffected.
Figure 1.
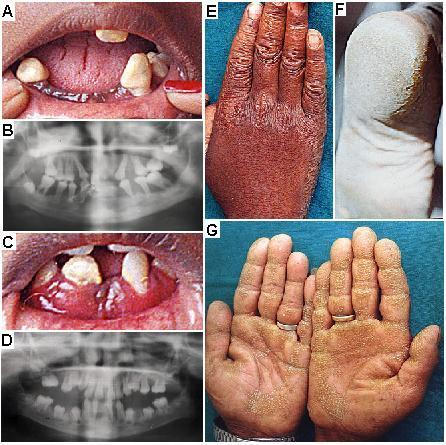
Clinical features of PLS. (A) Periodontitis affecting secondary dentition in the patient IV-3 at 16 yrs of age from the family IISC-PLS2. (B) Orthopantomogram of oral cavity of patient IV-3 at 11 years of age from the family IISC-PLS2 with severe periodontitis. All teeth in this patient are permanent and there is an extensive loss of alveolar bone both in maxillary and mandibular arches. (C) Periodontitis in the patient IV-4 at 12 years of age from the family IISC-PLS2. (D) Orthopantomogram of oral cavity of patient IV-4 at 12 years of age from the family IISC-PLS2 with severe periodontitis. All teeth in this patient are permanent and there is an extensive loss of alveolar bone both in maxillary and mandibular arch. (E) Knuckle hyperkeratosis in the patient IV-4 of the family IISC-PLS2. (F) Plantar hyperkeratosis in the patient IV-3 of the family IISC-PLS2. (G) Palmar hyperkeratosis (punctate type) in the patient II-2 of the family IISC-PLS3.
We initially screened PCR products of all seven exons of the CTSC gene for mutation(s) in one patient from each family using DNA sequencing technique (Fig. 2). Once the mutation was detected, it was confirmed in other members of the family. Sequence analysis of the exon 1 PCR product from the patient III-1 of the family IISC-PLS1 showed a nucleotide change in a homozygous state at position 145 from C>T (c.145C>T) which introduced a premature stop codon in the exon 1 (p.Q49X) (Fig. 2A). This change was also present in affected sibling III-2 in a homozygous state, whereas both parents were heterozygous for this change (Fig. 2A). PCR-SSCP analysis showed that this change was not present in 60 normal chromosomes (data not shown).
Figure 2.
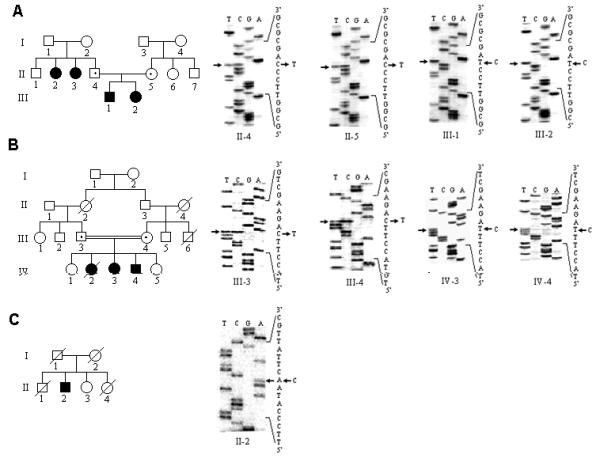
Three novel nonsense mutations in the CTSC gene. The numbering of the wild type sequence is based upon the cDNA sequence of the CTSC gene (GenBank accession # NM-001814). (A) Pedigree diagram of the family IISC-PLS1. DNA samples from individuals II-4, II-5, III-1 and III-2 were analysed from this family. Direct sequencing of the PCR products from both heterozygous parents II-4 and II-5 and homozygous patients III-1 and III-2 are shown on the right. Arrows indicate the145C>T nucleotide change. (B) Pedigree diagram of the family IISC-PLS2. DNA samples from individuals III-3, III-4, IV-1, IV-3, IV-4 and IV-5 were analysed from this family. Direct sequencing of the PCR products from both heterozygous parents III-3 and III-4, and homozygous patients IV-3 and IV-4 are shown on the right. Arrows indicate the 205C>T nucleotide change. (C) Pedigree diagram of the family IISC-PLS3. DNA sample from only individual II-2 was analysed from this family. Direct sequencing of the PCR product from the patient II-2 is shown on the right. Arrow indicates the 912 C>A change.
Sequence analysis of the exon 2 PCR-product from the patient IV-3 of the family IISC-PLS2 showed a nucleotide change in a homozygous state at position 205 from C>T (c.205C>T) which introduced a premature stop codon in the exon 2 (p.Q69X) (Fig. 2B). As expected, affected sibling IV-4 from this family was also homozygous for this change, whereas both parents (Fig. 2B) and both unaffected siblings (IV-1 and IV-5) were heterozygous for this change (data not shown). Since PCR-SSCP analysis failed to demonstrate this change, we used ASOH analysis to determine if this change was present in normal controls. ASOH analysis showed that this change was not present in 60 normal chromosomes (data not shown).
Sequence analysis of the exon 7 PCR product from the patient II-2 of the family IISC-PLS3 (Fig. 2C) showed a nucleotide change in a homozygous state at position 912 from C>A (c.912C>A) which introduced a premature stop codon in the exon 7 (p.Y304X). PCR-SSCP analysis showed that this change was not present in 60 normal chromosomes (data not shown).
Discussion
All three nucleotide changes reported here fulfilled the criteria of a mutation [12] as these changes were not present in the controls and result in the truncation of the CTSC encoded enzyme dipeptidyl-peptidase I with the introduction of premature stop codons. The c.145C>T mutation in the family IISC-PLS1 would be expected to produce an abnormally short enzyme dipeptidyl-peptidase I of 49 amino acid length in contrast to a normal enzyme of 463 amino acids [8]. Similarly, the c.205C>T change in the family IISC-PLS2 would be expected to produce a truncated enzyme with 69 amino acids and the c.912C>A mutation in the family IISC-PLS3 would be expected to produce a truncated enzyme with 304 amino acids [8]. These nonsense mutations would reduce any residual dipeptidyl peptidase I activity through nonsense mutation-mediated decay of the transcript, which might result in very low steady-state levels of CTSC messages leading to the disease phenotype in patients reported in the present study. In addition, this protein is lysosomal and may not be transported to the organelle if truncated, leading to the disease phenotype. It has been suggested that dipeptidyl peptidase I is involved in a wide variety of immune and inflammatory responses that include the activation of phagocytic cells and T-lymphocytes, leading to the final elimination of pathogens [7]. Therefore, inactivation of the dipeptidyl-peptidase I due to mutations would result in the blocking of these responses leading to infection of the gums and surrounding tissues by pathogens, ultimately leading to tooth loss. Alternatively, dipeptidyl-peptidase I might influence periodontal disease progression through its role in epithelial differentiation or desquamation. Since the sulcular and junctional epithelium represent the first line of defense against pathogens, their aberrant differentiation due to mutant CTSC gene may alter the mechanical barrier to periodontal pathogens [7].
A review of the literature on the number of total mutations reported in the CTSC gene shows a total of 38 mutations [[3,7,14-22], http://www.genetics.pitt.edu/CTSC/] with 27 being novel and 11 recurrent (Table 1). The total number of known mutations in the CTSC gene is now 41. Of these, 11 mutations are recurrent (Table 1). A majority of mutations (a total of 17 mutations) are located in exon 7 (Table 1). Of the known mutations, 11 are nonsense, 20 are missense, 2 are insertions, 7 are deletions and 1 is a splice site mutation (Table 1). This suggests that although most of the mutations (a total of 17) are located in exon 7, mutations are scattered across the coding region of the CTSC gene, suggesting that mutation analysis of the cathepsin C gene will require evaluation of the complete gene. Missense mutation c.815G>C (p.R272P) has been found in nine families including four families from Saudi Arabia. The haplotype analysis has shown that these four Saudi Arabian families have inherited this mutation from a common ancestor [17]. Similarly, Zhang et al. [17] have demonstrated that a recurrent nonsense mutation c.856C>T (p.Q286X) found in three families from Turkey has a common ancestral origin. A nonsense mutation c.1286G>A (p.W429X) found in four families from Turkey has also been found to have a common ancestral origin by haplotype analysis [17]. On the other hand, it is not clear if five recurrent mutations- c.96T>G (p.Y32X) found in three families, splice site mutation IVS3_1G>A found in two families, c.901G>A (p.G301S) found in three families, c.1015C>T (p.R339C) found in four families and c.1040A>G (p.Y347C) found in two families- have originated from common ancestors or these mutation sites represent mutation hotspots.
Table 1.
Disease-causing mutations in the cathepsin C gene detected so far.
| Sl# | Exon | Type | Mutation | Effect | Origin / Disease | No. of families |
| 1 | 1 | Nonsense | c.72C>A (p.C24X) | Premature stop | Moroccan 19 / PLS | 1 |
| 2 | 1 | Nonsense | c.96T>G (p.Y32X) | Premature stop | Mexican 20, §Caucasian 20, French 19 / PLS | 3 |
| 3 | 1 | Nonsense | c.145C>T (p.Q49X) | Premature stop | Indian A / PLS | 1 |
| 4 | 1 | Missense | c.116G>C (p.W39S) | AHCR** | Puerto Rican 16 / PLS | 1 |
| 5 | 2 | Nonsense | c.205C>T (p.Q69X) | Premature stop | Indian A / PLS | 1 |
| 6 | 2 | Deletion | c.199_222del (p. 67_74) | Frameshift | * Chinese 15 / PLS | 1 |
| 7 | 3 | Missense | c.380A>C (p. H127P) | AHCR** | French 19 / PLS | 1 |
| 8 | 3 | Missense | c.415G>A (p.G139R) | AHCR** | §S Caucasian 20 / PLS | 1 |
| 9 | 3 | Insertion | c.445_446insATGT (p.V149fsX157) | Frameshift & premature stop | Indian 15 / PLS | 1 |
| 10 | Splice site (Intron 3) | IVS3_1G>A | Altered splicing | Egyptian 7, Jordanian 22 / PLS | 2 | |
| 11 | 4 | Nonsense | c.545G>A (p.W185X) | Premature stop | Brazilian 21 / PLS | 1 |
| 12 | 4 | Missense | c.587T>C (p.L196P) | AHCR** | Brazilian 3 / PLS | 1 |
| 13 | 4 | Insertion | 622_623insC (p.H208fsX223) | Frameshift & Premature stop | Turkish 15 / PLS | 1 |
| 14 | 4 | Nonsense | c.628C>T (p.R210X) | Premature stop | Lebanese7, Algerian 19 / PLS | 2 |
| 15 | 5 | Nonsense | c.704G>A (p.W235X) | Premature stop | Iranian 15 / PLS | 1 |
| 16 | 5 | Missense | c.706G>T (p.D236Y) | AHCR** | φ Spanish 18 / PLS | 1 |
| 17 | 5 | Deletion | c.711del14 | Frameshift & Premature stop | Algerian 19 / PLS | 1 |
| 18 | 5 | Missense | c.745G>T (p.V249F) | AHCR** | Indian-Pakistani 7 / PLS | 1 |
| 19 | 5 | Nonsense | c.748C>T (p.R250X) | Premature stop | Turkish 15 / PLS | 1 |
| 20 | 5 | Missense | c.755A>T (p.Q252L) | AHCR** | Egyptian 7 / PLS | 1 |
| 21 | 6 | Missense | c.815G>C (p.R272P) | AHCR** | Lebanese7, Turkish 15, §Caucasian20, (4) Saudi 17, Holland19, French 19 / PLS | 9 |
| 22 | 6 | Nonsense | c.856C>T (p.Q286X) | Premature stop | Turkish 9,14,17 / PLS | 3 |
| 23 | 6 | Missense | c.857A>G (p.Q286R) | AHCR** | Indian 9, Spanish18 / HMS, PLS | 2 |
| 24 | 6 | Missense | c.872G>A (p.C291Y) | AHCR** | φ Spanish 18 /PLS | 1 |
| 25 | 7 | Missense | c.898G>A (p.G300S) | AHCR** | Φ Vietnamese 15 / PLS | 1 |
| 26 | 7 | Missense | c.899G>A (p.G300D) | AHCR** | Saudi 17 / PLS | 1 |
| 27 | 7 | Missense | c.901G>A (p.G301S) | AHCR** | Indian-Pakistani 7, Iranian 15, Japanese 16 / PLS | 3 |
| 28 | 7 | Missense | c.902G>T (p.G301V) | AHCR** | Iranian 15 / PLS | 1 |
| 29 | 7 | Missense | c.910T>A (p.Y304N) | AHCR** | Panamanian 15 / PLS | 1 |
| 30 | 7 | Nonsense | c.912C>A (p.Y304X) | Premature stop | Indian A / PLS | 1 |
| 31 | 7 | Missense | c.956A>G (p.E319G) | AHCR** | Iranian 15 / PLS | 1 |
| 32 | 7 | Deletion | c.984del7 | Frameshift & Premature stop | French 19 / PLS | 1 |
| 33 | 7 | Missense | c.1015C>T (p.R339C) | AHCR** | Egyptian 7,15, Turkish10, Martinique 19 / PLS | 4 |
| 34 | 7 | Deletion | c.1028_1029delCT (p.S343X) | Frameshift & Premature stop | Turkish 14 / PLS | 1 |
| 35 | 7 | Missense | c.1040A>G (p.Y347C) | AHCR** | Indian-Pakistani7, Jordanian 10 / PLS, PPP | 2 |
| 36 | 7 | Deletion | c.1047delA (p.G349fsX359) | Frameshift & Premature stop | Turkish 14 / PLS | 1 |
| 37 | 7 | Deletion | c.1056delT | Frameshift & Premature stop | French 19 / PLS | 1 |
| 38 | 7 | Deletion | c.1141delC (p.L381fsX393) | Frameshift & Premature stop | §S Caucasian20, French 19 / PLS | 2 |
| 39 | 7 | Nonsense | c.1286G>A (p.W429X) | Premature stop | Turkish 14,15 / PLS | 4 |
| 40 | 7 | Missense | c.1287G>C (p. W429C) | AHCR** | French19 / PLS | 1 |
| 41 | 7 | Missense | c.1360A>G (p.E447G) | AHCR** | Φ Vietnamese 15 / PLS | 1 |
[A] Novel mutations identified in this study; [20] Zhang et al. 2002; [16] Nakano et al. 2001; [15] Hart et al. 2000c; [17] Zhang et al. 2001; [18] Allende et al. 2001; [19] Lefevre et al. 2001; [7]Toomes et al. 1999; [3] Cury et al. 2002; [21] Hart et al. 2002; [10] Hart et al. 2000b; [14] Hart et al. 1999; [22] Nusier et al. 2002; [9] Hart et al. 2000a. * Proband was a compound heterozygote for the 199–222 del and 458C>T mutations ** Alteration of highly conserved residue. §Probands were compound heterozygote for the 96T>G and 815G>C mutations. §S Proband was a compound heterozygote for the 415G>A and 1141delC mutations. φ Proband was a compound heterozygote for the 706G>T and 872G>A mutations. Φ Proband was a compound heterozygote for the 898G>A and 1360A>G mutations.
An interesting feature of the cathepsin C gene is that mutations in this gene also result in two other closely related conditions: the Haim-Munk Syndrome [9], and prepubertal periodontitis [10]. A common clinical manifestation in all three syndromes is severe early-onset periodontitis. Haim-Munk syndrome (HMS) is an ethnically specific disorder described only in Jews of South Indian origin (the so called "Cochin Jews"). The clinical phenotypes of HMS overlap with PLS and prepubertal periodontitis (PPP) and include congenital keratosis palmoplantaris, onychogryposis, periodontitis, pes planus, arachnodactyly, and acroosteolysis. Thus HMS, PPP and PLS seem to be allelic variants. A common mutation c.1040A>G has been shown to cause two distinct phenotypes, PLS and PPP, suggesting that other factors such as genetic or environmental, play a role in the ultimate phenotype [15]. A missense mutation c.857A>G (p.Q286R) in the CTSC gene has been found to cause HMS [9]. This mutation has also been detected in a homozygous state in a Spanish PLS patient, suggesting that the HMS and PLS are clinical variants of the same homozygous cathepsin C gene mutation [18]. It is possible that a part of the clinical manifestations in HMS patients (viz., hyperkeratosis and periodontitis) is caused by a mutation in the CTSC gene where as other features of HMS (viz., onychogryposis, pes planus, arachnodactyly, and acroosteolysis) are caused by mutations in another hitherto undescribed gene. However, this possibility remains to be proven.
Hart et al. al. [15] have carried out genotype-phenotype correlation using 22 probands on whom genotype and phenotype data were available. The categories for genotype were mutations in the pro-region and mutations in the mature enzyme [15]. The categories for phenotypes were the presence or absence of transgressions of the hyperkeratosis lesions on the knees and elbows [15]. No correlation was found as affected subjects with transgressions of dermal lesions onto knees or elbows or both had mutations in both the pro- and mature regions of the enzyme [15]. Our analysis on 41 probands suggested a similar finding. Our analysis also did not find any correlation between the types of mutations (missense, nonsense, insertion, deletion and splice site) and the presence or absence of the lesions on the knees or elbows or both as observed previously by Hart et al. [15].
Conclusions
In this study, we have reported three novel nonsense mutations responsible for PLS phenotype in three Indian families. In addition, our review of the literature showed that the total number of mutations described to date in the cathepsin C gene, along with the ones reported during the present study, reaches to 41 including a common mutation reported in HMS patients.
Competing interests
None declared
Authors' contributions
VS carried out the molecular genetic studies. MM coordinated specimen collection and wrote the first draft of the manuscript. PVSP, PS, GS and SCS ascertained the families. NT provided primers and their PCR conditions. AK conceived the study, carried out the molecular genetic studies along with VS and drafted the final version of the manuscript
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
We thank the reviewers, Drs. Douglas Peter C. Dickinson and Jouni Uitto, for their valuable suggestions to improve the manuscript.
Contributor Information
Veeriah Selvaraju, Email: selvacell@rediffmail.com.
Manjunath Markandaya, Email: mmanju@mrdg.iisc.ernet.in.
Pullabatla Venkata Siva Prasad, Email: prasaderm@hotmail.com.
Parthasarathy Sathyan, Email: sathyan.p@cbe.aravind.org.
Gomathy Sethuraman, Email: kgsethu@yahoo.com.
Satish Chandra Srivastava, Email: srivastavadr@yahoo.co.in.
Nalin Thakker, Email: nthakker@man.ac.uk.
Arun Kumar, Email: karun@mrdg.iisc.ernet.in.
References
- Papillon MN, Lefevre B. Two cases of familial symmetric palmoplanter keratosis (Maleda's disease) in a brother and his sister. Alterations in both cases (in French) Bull Soc Francaise Dermatologie Syphiligraphie. 1924;31:81–84. [Google Scholar]
- Gorlin RJ, Sedano H, Anderson VF. The syndrome of palmo-plantar hyperkeratosis and premature periodontal destruction of the teeth. A clinical and genetic analysis of the Papillon-Lefevre syndrome. J Pediatr. 1964;65:895–908. doi: 10.1016/s0022-3476(64)80014-7. [DOI] [PubMed] [Google Scholar]
- Cury VF, Costa JE, Gomez RS, Boson WL, Loures CG, Marco LD. A novel mutation of the cathepsin C gene in Papillon-Lefevre syndrome. J Periodontol. 2002;73:307–312. doi: 10.1902/jop.2002.73.3.307. [DOI] [PubMed] [Google Scholar]
- Fischer J, Blanchet-Bardon C, Prud'homme J-F, Pavek S, Steijlen PM, Dubertret L, Weissenbach J. Mapping of Papillon-Lefevre syndrome to the chromosome 11q14 region. Eur J Hum Genet. 1997;51:156–160. [PubMed] [Google Scholar]
- Laass MW, Hennies HC, Preis S, Stevens HP, Jung M, Leigh IM, Wienker TF, Reis A. Localization of a gene for Papillon-Lefevre syndrome to chromosome 11q14-q21 by homozygosity mapping. Hum Genet. 1997;101:376–382. doi: 10.1007/s004390050645. [DOI] [PubMed] [Google Scholar]
- Hart TC, Bowden DW, Ghaffar KA, Wang W, Cutler CW, Cebeci I, Efeoglu A, Firatli E. Sublocalization of the Papillon-Lefevre syndrome on chromosome 11q14-q21. Am J Med Genet. 1998;79:134–139. doi: 10.1002/(SICI)1096-8628(19980901)79:2<134::AID-AJMG9>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Toomes C, James J, Wood AJ, CL Wu, McCormick D, Lench N, Hewitt C, Moynihan L, Roberts E, Woods CG, Markham A, Wong M, Widmer R, Ghaffar KA, Pemberton M, Hussein IR, Temtamy SA, Davies R, Read AP, Sloan P, Dixon MJ, Thakker NS. Loss-of function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat Genet. 1999;23:421–424. doi: 10.1038/70525. [DOI] [PubMed] [Google Scholar]
- Rao NV, Rao GV, Hoidal JR. Human dipeptidyl-peptidase I. Gene characterization, localization, and expression. J Biol Chem. 1997;272:10260–10265. doi: 10.1074/jbc.272.15.10260. [DOI] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Michalec MD, Zhang Y, Firatli E, Van Dyke TE, Stabholz A, Zlotogorski A, Shapira L, Soskolne WA. Haim-Munk syndrome and Papillon-Lefevre syndrome are allelic mutations in cathepsin C. J Med Genet. 2000;37:88–94. doi: 10.1136/jmg.37.2.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Michalec MD, Zhang Y, Marazita ML, Cooper M, Yassin OM, Nusier M, Walker S. Localization of a gene for prepubertal periodontitis to chromosome 11q14 and identification of a cathepsin C gene mutation. J Med Genet. 2000;37:95–101. doi: 10.1136/jmg.37.2.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haneke E. The Papillon-Lefevre syndrome: keratosis palmoplantaris with periodontopathy: Report of a case and review of the cases in the literature. Hum Genet. 1979;51:1–35. doi: 10.1007/BF00278288. [DOI] [PubMed] [Google Scholar]
- Kumar A, Kandt RS, Wolpert C, Roses AD, Pericak-Vance MA, Gilbert JR. Mutation analysis of the TSC2 gene in an African-American family. Hum Mol Genet. 1995;4:2295–2298. doi: 10.1093/hmg/4.12.2295. [DOI] [PubMed] [Google Scholar]
- Cormand B, Vilageliu L, Burguera JM, Balcells S, Gonzalez-Duarte R, Grinberg D, Chabas A. Gaucher disease in Spanish patients: analysis of eight mutations. Hum Mutat. 1995;5:303–309. doi: 10.1002/humu.1380050406. [DOI] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Bowden DW, Michalec MD, Callison SA, Walker SJ, Zhang Y, Firatli E. Mutations of the cathepsin C gene are responsible for Papillon-Lefèvre syndrome. J Med Genet. 1999;36:881–887. [PMC free article] [PubMed] [Google Scholar]
- Hart PS, Zhang Y, Firatli E, Uygur C, Lotfazar M, Michalec MD, Marks JJ, Lu X, Coates BJ, Seow WK, Marshall R, Williams D, Reed JB, Wright JT, Hart TC. Identification of cathepsin C mutations in ethnically diverse Papillon-Lefevre syndrome patients. J Med Genet. 2000;37:927–932. doi: 10.1136/jmg.37.12.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A, Nomura K, Nakano H, Ono Y, LaForgia S, Pulkkinen L, Hashimoto I, Uitto J. Papillon-Lefevre syndrome: mutations and polymorphisms in the cathepsin C gene. J Invest Dermatol. 2001;116:339–343. doi: 10.1046/j.1523-1747.2001.01244.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lundgren T, Renvert S, Tatakis DN, Firatli E, Uygur C, Hart PS, Gorry MC, Marks JJ, Hart TC. Evidence of a founder effect for four cathepsin gene mutations in Papillon-Lefevre syndrome patients. J Med Genet. 2001;38:96–101. doi: 10.1136/jmg.38.2.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allende LM, Garcia-Perez MA, Moreno A, Corell A, Carasol M, Martinez-Canut P, Arnaiz-Villena A. Cathepsin C gene: First compound heterozygous patient with Papillon-Lefevre syndrome and a novel symptomless mutation. Hum Mutat. 2001;17:152–153. doi: 10.1002/1098-1004(200102)17:2<152::AID-HUMU10>3.3.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Lefevre C, Blanchet-Bardn C, Jobard F, Bouadjar B, Stalder JF, Cure S, Hoffmann A, Prud'homme JF, Fischer J. Novel point mutations, deletions and polymorphisms in the cathepsin C gene in nine families from Europe and North Africa with Papillon-Lefevre syndrome. J Invest Dermatol. 2001;117:1657–1661. doi: 10.1046/j.0022-202x.2001.01595.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Hart PS, Moretti AJ, Bouwsma OJ, Fisher EM, Dudlicek L, Pettenati MJ, Hart TC. Biochemical and mutational analyses of the cathepsin C gene (CTSC) in three North American families with Papillon-Lefevre syndrome. Hum Mutat. 2002;20:75. doi: 10.1002/humu.9040. [DOI] [PubMed] [Google Scholar]
- Hart PS, Pallos D, Zhang Y, Sanchez J, Kavamura I, Brunoni D, Hart TC. Identification of a novel cathepsin C mutation (p.W185X) in a Brazilian kindred with Papillon-Lefevre syndrome. Mol Genet Metab. 2002;76:145–147. doi: 10.1016/S1096-7192(02)00031-8. [DOI] [PubMed] [Google Scholar]
- Nusier M, Zhang Y, Yassin O, Hart TC, Hart PS. Demonstration of altered splicing with the IVS3-1G> A mutation of the cathepsin C. Mol Genet Metab. 2002;75:280–283. doi: 10.1006/mgme.2002.3304. [DOI] [PubMed] [Google Scholar]