

Abstract
Background
The engineering of fusion proteins has become increasingly important and most recently has formed the basis of many biosensors, protein purification systems, and classes of new drugs. Currently, most fusion proteins consist of three or fewer domains, however, more sophisticated designs could easily involve three or more domains. Using traditional subcloning strategies, this requires micromanagement of restriction enzymes sites that results in complex workaround solutions, if any at all.
Results
Therefore, to aid in the efficient construction of fusion proteins involving multiple domains, we have created a new expression vector that allows us to rapidly generate a library of cassettes. Cassettes have a standard vector structure based on four specific restriction endonuclease sites and using a subtle property of blunt or compatible cohesive end restriction enzymes, they can be fused in any order and number of times. Furthermore, the insertion of PCR products into our expression vector or the recombination of cassettes can be dramatically simplified by screening for the presence or absence of fluorescence.
Conclusions
Finally, the utility of this new strategy was demonstrated by the creation of basic cassettes for protein targeting to subcellular organelles and for protein purification using multiple affinity tags.
Background
Conceptually, a fusion protein is constructed by joining two different domains to produce a new chimeric protein, which retains the properties of the individual domains. For instance, a tumor necrosis factor (TNF) inhibitor was created from the fusion of the TNF receptor to the Fc domain of human immunoglobin G as it retains the ability to bind TNF and to be targeted by the immune system [1]. By using the principle of fluorescence resonance energy transfer, protein biosensors can be created from multiple domain fusions with fluorescent proteins to image cellular events such as Ca2+ signaling, phosphorylation, and caspase proteolytic cleavage [2]. In practice, such fusion proteins are created by inserting PCR products of the individual domains into an expression vector at the available restriction endonuclease sites. Previously, the flexibility of design is compromised as the choice of insertion sites limits the possible locations for future fusions into the same expression vector. In turn, many initially unplanned but simple extensions to existing fusion proteins cannot be constructed because available sites are exhausted or incompatible. As this issue is exacerbated when constructing multiple domain fusion proteins, we have created a new expression vector for subcloning using a cassette-based strategy. A basic cassette contains the sequence of an individual domain that can be recombined with other cassettes irrespective of order, without the progressively more complex management of sites. Therefore, the creation of a cassette library of commonly used domains facilitates the rapid prototyping of multiple domain fusion proteins that can perform numerous functions.
Results and discussion
Standard vector structure of the cassette
Our cassettes must have a standard vector structure where a domain(s) is flanked by cut sites 1 and 2a at the 5' end and 2b and 3 at the 3' end (Figure 1a). Site 1 and 3 can be selected arbitrarily, but site 2a and 2b must be derived from different restriction enzymes producing blunt or compatible cohesive ends. For example, there are two ways to create the AB fusion cassette from cassette A and B (likewise for the BA fusion cassette): ligate the insert from cassette B (site 2a and 3) to the host cassette A (site 2b and 3) (Figure 1b) or ligate the insert from cassette A (site 1 and 2b) to the host cassette B (cut site 1 and 2a) (Figure 1c). Since the ligation point of site 2a and 2b produces the recognition site of neither, it cannot be cut with either restriction enzymes. Therefore, the AB fusion cassette has the same standard vector structure and can be used for further fusion following the same concept. Note that if no more than one of the four enzyme sites are found inside the domain sequence of the cassette, it is still possible to create any fusion because cassettes can be fused on either the 5' or 3' end.
Figure 1.

The cloning methodology. (A) Design of cassette A and B. Creation of the AB fusion cassette by (A) a C-terminal fusion to cassette A and (B) a N-terminal fusion to cassette B. (D) Schematic diagram of the pCfvtx vector. pTriEx1.1-Hygro (Novagen) was chosen as the base vector because it allows expression in both prokaryotic and eukaryotic cells.
pCfvtx embodies the standard vector structure and allows fluorescence screening
Our new expression vector, pCfvtx, Cassette Fused with Venus [3] in the p Trie X1.1-Hygro vector (Novagen), allows for rapid subcloning of basic and fusion cassettes by screening positive colonies using fluorescence (Figure 1d). This vector fixes site 1 and 3 as NcoI and XhoI, respectively, but there are many choices for site 2a and 2b: StuI and SmaI or NheI and SpeI or BamHI and BglII. Since standardization of these specific sites is required to create a basic cassette, they must be added to the domain of interest by PCR and then inserted into the vector. pCfvtx was constructed with a stop codon flanked by two multiple cloning sites (MCS1 and MCS2) upstream of Venus [3], a mutant variant of green fluorescent protein (GFP). When a fragment is subcloned into the vector between MCS1 and MCS2, the stop codon is removed and therefore, a fluorescent cassette is created since it is fused with Venus. As the leak expression of the fusion protein is enhanced by the presence of the T7lac promoter and the absence of the lacI repressor gene [4], positive colonies will be fluorescence on bacterial culture plates (Figure 3a). To create a non-fluorescent cassette, Venus can be removed by cutting with PmeI, performing a self-ligation and then screening for the absence of fluorescence.
Figure 3.
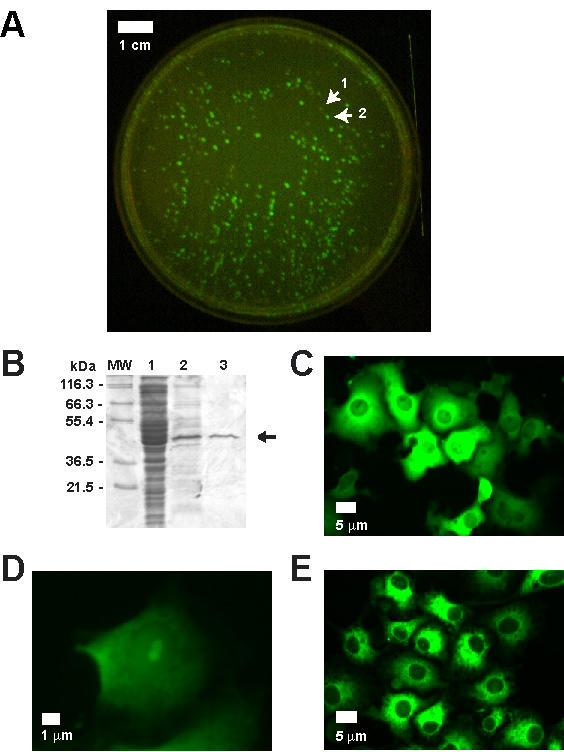
Fluorescence screening, protein purification and subcellular localization. (A) Fluorescence screening in bacterial colonies using the leak expression of the target protein fused with Venus. A non-fluorescent colony is identified by arrow 1; a fluorescent colony, by arrow 2. (B) The purification of the 6xHis-Venus-GST construct (identified by the arrow). Lane MW is the molecular weight marker; lane 1, the cell lysate; lane 2, the elusion from the Ni-NTA column; lane 3, the elusion from the GST column. All vectors were transfected into COS-7 cells for fluorescence imaging. The 10× magnification of (C) the cytoplasmic distribution of the 6xHis-Venus-GST and (E) the endoplasmic distribution of Venus N-terminally fused with the interleukin-4 leader sequence and C-terminally fused with the KDEL retention signal. (D) The 40× magnification of the nucleolar distribution of Venus N-terminally fused with the HIV Tat domain.
Fluorescence screening with Venus
It should be noted that Venus is the fastest folding and brightest GFP mutant to date [3]. Accordingly, the positive colonies will become fluorescent immediately, whereas other GFP variants may require several days. Second, these fluorescent colonies ensure that the inserted fragment is in-frame and without nonsense mutations. Also, the C-terminal fusion of GFP to target proteins is an effective assay for protein solubility and fold stability – the more fluorescent the fusion protein, the more soluble and well-folded the inserted fragment [5,6]. Lastly, any desired fusion cassette can be designed, such that at each intermediate step, a positive colony is selected by the presence or absence of fluorescence (Figure 2). As only one fluorescent or non-fluorescent colony is needed and the random gain or loss of this property is improbable, fluorescence is a robust reporter that tolerates much of the inefficiency in the subcloning process. In sum, through the use of fluorescence, subcloning is performed rapidly and precisely such that it is possible to efficiently create many fusion cassettes in parallel.
Figure 2.

Flow diagram of our fluorescent cassette-based strategy to construct the AB fusion cassette. First, the fluorescent cassettes A and B are created by insertion of the respective domains into the pCfvtx vector. Then, cassette A and B is created by the excision of Venus by PmeI restriction and then self-ligation. The path of the thick arrows highlight the fusion steps required for creating the AB fusion cassette. The fluorescent cassette B is ligated to cassette A to create a fluorescent cassette AB. The final AB fusion cassette is created by the excision of Venus as described previously.
Protein purification cassettes
To demonstrate the utility of our cassette-based strategy, we first applied it to protein expression/purification systems, which often involve either an N-terminal or C-terminal fusion of the target protein with an affinity tag. Cassettes were made using two popular tags – 6xHis (Qiagen) and Glutathione S-transferase (GST) tag (Pharmacia) [7]. The N-terminal fusion of the 6xHis tag to Venus allows binding to Ni-NTA (nickel-nitrilotriacetic acid) agarose beads, however, a simple elution yields an impure sample (Figure 3b). The additional C-terminal fusion of GST to Venus allows binding of the previous elution to GST sepharose beads. Since the affinity tags flank the target protein and it is unlikely that a protein will non-specifically bind to both affinity beads, only full-length fusion proteins will be eluted from the GST beads. Note that the newly created 6xHis-Venus-GST fusion cassette is itself a useful affinity tag that additionally could be used to estimate protein expression greater than ~1 nM as fluorescence intensity from the Venus domain is linearly proportional to target protein concentration. Finally, the flexibility of our cassette-based strategy opens new opportunities for the design of tandem affinity purification (TAP) tags [8], which were useful in protein complex purification in the yeast proteome [9]. The customization of TAP tags is desirable as the same affinity tag may not be suitable for all organisms [10].
Protein subcellular targeting cassettes
The creation of protein biosensors has allowed the observation of signaling events in single cells [11-13]. Such events are often isolated to subcellular organelles such as the nucleus or endoplasmic reticulum and therefore, the ability to easily localize biosensors to these sites is important. The localization of proteins to specific organelles relies on vital cellular mechanisms that recognize leader sequences and signal peptides [14]. If a protein (such as the 6xHis-Venus-GST protein) is expressed in the cell without any localization peptides, it will be found inside the cytoplasm (Figure 3c). To localize a target protein to the nucleolus, a cassette was created containing the protein transduction domain of human immunodeficiency virus (HIV) Tat [15]. When this cassette was N-terminally fused to Venus and transfected into COS-7 cells, fluorescence was most intense in the nucleolus (Figure 3d). To localize to the lumen of the endoplasmic reticulum, a cassette was created containing the leader sequence from interleukin-4 and another cassette was created with the KDEL retention signal. When these cassettes were fused N- and C-terminally to Venus, it localized to the endoplasmic reticulum (Figure 3e). In summary, the creation of these cassettes allows the flexibility of localizing any cassette in our library to those organelles.
Conclusions
Using the pCfvtx vector as a starting point, basic cassettes are subcloned from target genes or domains of interest by PCR. These basic cassettes can then be recombined in any order and number of times to create fusion cassettes of multiple domain proteins. Each step of the subcloning process of cassettes is rapidly and reliably screened by the presence or absence of fluorescence. In contrast to the common β-Gal screen [16], our fluorescence approach may potentially have applications in high-throughput structural genomics by identifying in-frame fragments with favorable folding and solubility properties. Unlike fluorescence, the subcloning of a target sequence using the β-Gal screen disrupts expression of the lacZ α-peptide, so a subsequent fusion cannot use the same screening process. Finally, the use of our fluorescent cassette-based strategy offers significant long-term advantages in protein engineering as each new cassette enriches the functionality of the growing library of cassettes (Table 1). Thus, future designs can efficiently build on previous work to create progressively more complex and sophisticated fusion proteins which are capable of performing a wide range of functions.
Table 1.
List of cassettes
| Cassette | Function |
| General | |
| pCfvtx | Fundamental vector for rapidly inserting PCR products using a fluorescence assay to create basic cassettes |
| pVentx | Venus for acquisition of fluorescence |
| Protein purification | |
| pHistx | 6xHis affinity tag |
| pGsttx | GST affinity tag |
| pHisventx | 6xHis affinity tag that also allows protein expression estimation by fluorescence |
| pHisvengsttx | A double affinity tag using 6xHis and GST for improved purity and protein expression estimation by fluorescence |
| Subcellular organelle targeting | |
| pTattx | HIV TAT protein tranduction domain for targeting to the nucleolus or peptide-mediated delivery of proteins to the nucleus |
| pIl4tx | Interleukin-4 signal peptide for secretion of target protein (alone) or for localization to the ER (with KDEL retention) |
| pKdeltx | KDEL retention signal for localization the ER (with signal peptide) |
| Organelle markers | |
| pTatvtx | A fluorescent marker for the nucleolus |
| pIl4venkdeltx | A fluorescent marker for the ER |
Methods
Fluorescence screening
Vectors were transformed into E. coli strain DH5α and plated on LB (Luria Broth) agarose with 100 μg/mL ampicilin. The culture plates were then incubated overnight at 37°C. Venus fluorescence was observed on the culture plate using the Lighttools Illuminatool Tunable Lighting System equipped with a 535 nm viewing filter and 488 nm/10 nm filter cup.
Construction of the pCfvtx vector
To create the pInsvtx intermediate vector, Venus was PCR amplified from the pVenus vector [3] using primers Insv-sense (5'-CATGCCATGGGCCTGACTAGTAGGCCTGCTAGCCTGTTTAAACTGGTGAGCAAGGGCGAGGAGCTG-3') and Insv-antisense (5'-CCGCTCGAGTTACAGTTTAAACAGGGCGGCGGTCACGAACTCCA-3'). The Insv-sense contained NcoI, SpeI, NheI and PmeI sites, while Insv-antisense contained PmeI and XhoI sites. The fragment was subcloned into the pTriEx1.1-Hygro vector (Novagen) at the NcoI and XhoI sites by selecting a fluorescent colony. The pInstx intermediate vector was created by self-ligating after cutting at the PmeI site of pInsvtx and a non-fluorescent colony was selected. Finally, to create the pCfvtx vector, a fragment containing the multiple cloning sites (SpeI, BamHI, StuI, BglII, SmaI, NheI) sandwiching a stop codon was created using 5'-end phosphorylated primers Mcs-sense (5'-CTAGTGGATCCAGGCCTTAAAGATCTCCCGGGG-3') and Mcs-anti-sense (5'-CTAGCCCCGGGAGATCTTTAAGGCCTGGATCCA-3'). Mcs-sense and Mcs-antisense were self-hybridized and subcloned into the pInsvtx vector at the SpeI and NheI sites. A non-fluorescent colony was selected.
Construction of the subcellular targeting vectors
To create the pVentx vector, Venus was amplified using primers Ven-sense (5'-CATGCCATGGGCCTGACTAGTGTGAGCAAGGGCGAGGAGCTG-3') and Ven-antisense (5'-CCGCTCGAGTTAGCCGCTAGCGGCGGCGGTCACGAACTCCA-3'). The Ven-sense contained NcoI and SpeI, sites, while Ven-antisense contained XhoI and NheI sites. The fragment was subcloned into the pInstx vector at the NcoI and XhoI sites by selecting a fluorescent colony. To create pTatvtx, 5'-end phosphorylated primers Tat-sense (5'-CATGGGCCTGACTAGTTACGGCAGGAAGAAGAGGAGGCAGAGGAGGAGGGGGG-3') and Tat-anti-sense (5'-CTAGCCCCCCTCCTCCTCTGCCTCCTCTTCTTCCTGCCGTAACTAGTCAGGCC-3') were self-hybridized and subcloned into the pCfvtx vector at the NcoI and NheI sites. Similarly, to create the pIl4vtx vector, 5'-end phosphorylated primers il4-sense (5'-CATGGGCCTGACTAGTCAGCTGCTGCCGCCCCTGTTCTTCCTGCTGGCCTGCG-3') and il4-anti-sense (5'-CTAGCGCAGGCCAGCAGGAAGAACAGGGGCGGCAGCAGCTGACTAGTCAGGCC-3') were self-hybridized and subcloned into the pCfvtx vector at the NcoI and NheI sites. The pIl4tx was created by self-ligating after cutting at the PmeI site of pIl4vtx. To create the pVkdeltx intermediate vector, Venus was amplified using primers Ven-sense and kdel-antisense (5'-CCGCTCGAGTTACAGCTCGTCCTTACTAGTGGCGGCGGTCACGAACTCCA-3'). The kdel-antisense contained XhoI and SpeI sites. The fragment was subcloned into the pInstx vector at the SpeI and XhoI sites. The pKdeltx was subcloned by cutting and self-ligating at the SpeI site. To create pVenkdeltx vector, pVentx was cut with NcoI and NheI and the fragment was subcloned into pKdeltx at NcoI and SpeI sites. To create pIl4venkdeltx, pVenkdeltx was cut with SpeI and XhoI and the fragment was subcloned into pIl4tx at the NheI and XhoI sites.
Construction of the 6xHis-Venus-GST cassette (pHisvengsttx vector)
To create pGstvtx, GST was amplified from pGEX2T (Invitrogen) using primers gst-sense (5'-GACTAGTATGTCCCCTATACTAGGTTATTG-3') and gst-antisense (5'-GAAGATCTATCCGATTTTGGAGGATGGTCG-3'). The gst-sense and gst-antisense contained SpeI and BglII sites, respectively. The fragment was subcloned into the pCfvtx vector at the SpeI and BglII sites. pGsttx was created by cutting and self-ligation at the PmeI site. To create the pHisvtx vector, 5'-end phosphorylated primers his-sense (5'-CATGGGCCTGACTAGTGGCAGCAGCCACCACCACCACCACCACAGCAGCGGCG-3') and his-anti-sense (5'-CTAGCGCCGCTGCTGTGGTGGTGGTGGTGGTGGCTGCTGCCACTAGTCAGGCC-3') were self-hybridized and subcloned into the pCfvtx vector at the NcoI and NheI sites. pHistx was created by cutting and self-ligation at the PmeI site. To create pHisventx vector, pVentx was cut with SpeI and XhoI and the fragment was subcloned into the pHistx at NheI and XhoI sites. To create pHisvengsttx, pHisventx was cut with NcoI and NheI and the fragment was subcloned into pGsttx at the NcoI and SpeI sites.
Transfection and imaging of tissue cultures
COS-7 cells were transfected with GeneJuice (Novagen). Between 2 and 5 days after transfection, cells were imaged at 22°C on an Olympus IX70 microscope with a CCD camera (MicroMax 1300YHS) controlled by MetaMorph 4.5r2 software (Universal Imaging).
Authors' Contribution
KT developed the subcloning strategy, performed all the subcloning and prepared the manuscript. AK provided technical mentorship. MI provided funding and supervision for the work. All authors have read and approved the final manuscript.
Acknowledgments
Acknowledgements
The vector pVenus was generously provided by A. Miyawaki. We thank K. Tong for technical assistance. This work was supported by grants to KT from the Canadian Institutes of Health Research (CIHR) and to MI from the Cancer Research Society, Inc. and the Institute for Cancer Research of the CIHR.
Contributor Information
Kevin Truong, Email: ktruong@uhnres.utoronto.ca.
Ahmad Khorchid, Email: khorchid@uhnres.utoronto.ca.
Mitsuhiko Ikura, Email: mikura@uhnres.utoronto.ca.
References
- Mannik M, Wener M. Treatment of rheumatoid arthritis with a tumor necrosis factor receptor- Fc fusion protein. N Engl J Med. 1997;337:1560–1. [PubMed] [Google Scholar]
- Truong K, Ikura M. The use of FRET imaging microscopy to detect protein-protein interactions and protein conformational changes in vivo. Curr Opin Struct Biol. 2001;11:573–8. doi: 10.1016/S0959-440X(00)00249-9. [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Pedelacq JD, Piltch E, Liong EC, Berendzen J, Kim CY, Rho BS, Park MS, Terwilliger TC, Waldo GS. Engineering soluble proteins for structural genomics. Nat Biotechnol. 2002;20:927–32. doi: 10.1038/nbt732. [DOI] [PubMed] [Google Scholar]
- Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17:691–5. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- Sheibani N. Prokaryotic gene fusion expression systems and their use in structural and functional studies of proteins. Prep Biochem Biotechnol. 1999;29:77–90. doi: 10.1080/10826069908544695. [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–2. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–7. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Forler D, Kocher T, Rode M, Gentzel M, Izaurralde E, Wilm M. An efficient protein complex purification method for functional proteomics in higher eukaryotes. Nat Biotechnol. 2003;21:89–92. doi: 10.1038/nbt773. [DOI] [PubMed] [Google Scholar]
- Truong K, Sawano A, Mizuno H, Hama H, Tong KI, Mal TK, Miyawaki A, Ikura M. FRET-based in vivo Ca2+ imaging by a new calmodulin-GFP fusion molecule. Nat Struct Biol. 2001;8:1069–73. doi: 10.1038/nsb728. [DOI] [PubMed] [Google Scholar]
- Sato M, Ozawa T, Inukai K, Asano T, Umezawa Y. Fluorescent indicators for imaging protein phosphorylation in single living cells. Nat Biotechnol. 2002;20:287–94. doi: 10.1038/nbt0302-287. [DOI] [PubMed] [Google Scholar]
- Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906–18. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- Stroud RM, Walter P. Signal sequence recognition and protein targeting. Curr Opin Struct Biol. 1999;9:754–9. doi: 10.1016/S0959-440X(99)00040-8. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Hruska KA, Dowdy SF. Protein transduction: unrestricted delivery into all cells? Trends Cell Biol. 2000;10:290–5. doi: 10.1016/S0962-8924(00)01771-2. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. New York: Cold Spring Harbor Laboratory Press. 2 1989. Molecular cloning : a laboratory manual. [Google Scholar]