

Abstract
Background
Components of the insulin signaling pathway are important regulators of growth. The FOXO (forkhead box, sub-group "O") transcription factors regulate cellular processes under conditions of low levels of insulin signaling. Studies in mammalian cell culture show that activation of FOXO transcription factors causes cell death or cell cycle arrest. The Caenorhabiditis elegans homologue of FOXO, Daf-16, is required for the formation of dauer larvae in response to nutritional stress. In addition, FOXO factors have been implicated in stress resistance and longevity.
Results
We have identified the Drosophila melanogaster homologue of FOXO (dFOXO), which is conserved in amino acid sequence compared with the mammalian FOXO homologues and Daf-16. Expression of dFOXO during early larval development causes inhibition of larval growth and alterations in feeding behavior. Inhibition of larval growth is reversible upon discontinuation of dFOXO expression. Expression of dFOXO during the third larval instar or at low levels during development leads to the generation of adults that are reduced in size. Analysis of the wings and eyes of these small flies indicates that the reduction in size is due to decreases in cell size and cell number. Overexpression of dFOXO in the developing eye leads to a characteristic phenotype with reductions in cell size and cell number. This phenotype can be rescued by co-expression of upstream insulin signaling components, dPI3K and dAkt, however, this rescue is not seen when FOXO is mutated to a constitutively active form.
Conclusions
dFOXO is conserved in both sequence and regulatory mechanisms when compared with other FOXO homologues. The establishment of Drosophila as a model for the study of FOXO transcription factors should prove beneficial to determining the biological role of these signaling molecules. The alterations in larval development seen upon overexpression of dFOXO closely mimic the phenotypic effects of starvation, suggesting a role for dFOXO in the response to nutritional adversity. This work has implications in the understanding of cancer and insulin related disorders, such as diabetes and obesity.
Background
The biological control of the size of an organism is one of the most elusive concepts in biology. What mechanisms determine the size differences between species? What genetic and environmental factors contribute to variations of size within a species? How does an individual regulate the size of its organs to maintain proportion with the rest of the body? Although much remains unanswered, it is clear that the size of an individual is directly related to the number of cells it has, and the size of these cells [1-3]. Thus, the final size of an organism is determined by the number of cell divisions that occur during development, and the amount of growth these cells undergo. When considering the size difference between two organisms, such as a mouse and a human, it is obvious that the main cause of the size difference is the total number of cells [2]. Intuitively, this may lead to the conclusion that the size of an organism is related to the rate of cell proliferation during development. However, experimental evidence shows that there are more subtle controls involved [4,5]. For example, increasing or decreasing cell proliferation in the Drosophila imaginal discs does not alter the final size, but instead produces discs with either an increased number of small cells or a decreased number of large cells [4,5]. These studies indicate that there must be a genetically predetermined total cell mass and a mechanism for sensing this critical size.
Studies in Drosophila demonstrate that the evolutionarily conserved insulin signaling pathway is involved in the control of body size, through alterations of cell size and cell number [1]. Seven Drosophila insulin-like peptides (Dilps) have been identified that are able to promote organism growth when expressed ubiquitously during development [6,7]. The Dilps activate cell signaling through the Drosophila insulin receptor (dInr), a receptor tyrosine kinase, which can promote growth when overexpressed in the developing eye [6,8]. Loss of function mutations in dInr are lethal during embryogenesis [8]. However, reduction of dInr levels through combination of weak heteroallelic mutations [9], or through partial loss of function mutations[6], reduces growth and leads to the development of small adults that have reduced cell size and number. In mammals, the insulin receptor promotes signaling through adaptor proteins, the insulin receptor substrates (IRS) 1–4, which are required to activate phosphoinositide-3-kinase (PI3K) [10,11]. PI3K is a lipid kinase that phosphorylates inositide lipids on the inner surface of the cell membrane, leading to the activation of the serine/threonine kinase Akt. Once activated, Akt phosphorylates many substrates that are involved in the regulation of metabolism, cell death/survival, and cell proliferation. Negative regulation of insulin signaling occurs through the tumor suppressor, PTEN. PTEN removes phosphates from inositide lipids, thus acting in opposition to PI3K. This signaling mechanism appears to be conserved in Drosophila, and the Drosophila homologues of IRS 1–4 (chico), PI3K (dPI3K), Akt (dAkt) and PTEN (dPTEN) have all been individually implicated in the regulation of cell size, and cell number [1]. Flies that are homozygous for a null mutation in chico are smaller than normal due to a reduction in cell size and cell number [12]. Null mutations in dAkt are lethal [13], however, rescue of dAkt mutants through ectopic expression of dAkt during embryogenesis results in a small fly phenotype [14] similar to that seen with chico mutants and through reduction of dInr activity. Clearly, components of the insulin signaling pathway act to control body and organ size through regulation of cell size and cell number during development.
In addition to developmentally predetermined size control, many cells and organisms can alter their size according to environmental stimuli, such as nutrient limitation. When Drosophila larvae are raised under nutrient limited conditions the adults are smaller than well-fed flies[15,16] This phenomena appears to be phenocopied in the generation of small adults through inhibition of Drosophila insulin signaling [6,9,12,14]. Interestingly, expression of Dilps 3, 5, and 7 has been linked to the availability of nutrients [7]. These Dilps are produced in neurosecretory cells in the larval brain where they are released into the circulatory system [7]. These studies indicate that nutritional signals may regulate body size by modulating the levels of Dilps 3, 5, and 7 in the body.
Newly hatched Drosophila larvae require a nutritional signal to initiate the cell cycle in mitotic tissues [17]. Well-fed larvae increase their body mass very rapidly due to replication of cells in mitotic tissues. In contrast, larvae hatched into conditions of amino acid starvation live in a state of developmental arrest for several days until nutrients become available to initiate the cell cycle[16,17]. Dominant negative inhibition of dPI3K in developing Drosophila larvae has been shown to phenocopy the effects of amino acid starvation [18]. Expression of dPI3K in subsets of cells in the imaginal discs of starved larvae allows these cells to divide in the absence of nutritional signals [18]. Expression of dPI3K in the fat bodies of starved larvae significantly reduces their survival, thus conferring starvation sensitivity in these larvae [18]. This suggests that Drosophila insulin signaling may play a protective role in the response to starvation.
An insulin-like signaling pathway involved in the response to nutrient limitation also exists in the nematode Caenorhabiditis elegans. When C. elegans are raised under conditions of nutrient limitation, they enter an alternate developmental stage called the dauer larvae. The dauer stage is characterized by arrest of growth at a sexually immature stage along with altered metabolism to increase the storage of fat [19]. Mutations in components of the insulin signaling pathway in C. elegans lead to dauer larvae formation and increased life span [20-24]. A null mutation in the C. elegans gene, Daf-16, negates dauer formation and the life expanding effect of these mutations [21,25,26]. Thus, in C. elegans, Daf-16 is necessary for dauer formation and seems to be the primary effector molecule under conditions of low levels of insulin signaling.
Daf-16 is the C. elegans homologue of a highly conserved group of Akt phosphorylatable forkhead transcription factors, the FOXO (forkhead box, subgroup "O") transcription factors. These transcription factors were first discovered as proto-oncogenes, which were disrupted as a result of chromosomal translocations leading to acute myeloid leukemia and rabdomyosarcoma[27,28]. Three versions of FOXO have been identified in humans (FOXO1, FOXO3a, and FOXO4; formerly known as FKHR, FKHR-L1, and AFX) and mice (Foxo1, Foxo3, and Foxo4), and additional homologues have been identified in zebrafish and chickens[29]. The FOXO transcription factors share a highly conserved forkhead box DNA binding domain in the N-terminal half of the protein, and three highly conserved Akt phosphorylation sites. Mammalian cell culture studies have shown that in the absence of Akt signaling, FOXO is able to activate gene transcription and cause cell death, cell cycle arrest, or cell senescence [30,31]. In the presence of activated Akt, FOXO becomes phosphorylated and is sequestered in the cytoplasm through facilitation of 14-3-3 binding [32-35], and/or disruption of a nuclear localization signal[34,36]. The down-regulation of FOXO in this manner is, possibly, one of the most important consequences of Akt mediated signaling.
Based on evidence from studies in C. elegans and mammalian cell culture, it appears that FOXO transcription factors are a critical mediator of cellular processes under conditions of low levels of insulin signaling. To investigate this further, we have identified and characterized the Drosophila melanogaster version of FOXO. We show that Drosophila FOXO (dFOXO) retains the conserved domains seen in other organisms and is involved in the regulation of growth. Of special interest is that dFOXO appears to have an effect upon feeding behavior, and may be a key player in the response of Drosophila larvae to nutritional stress.
Results
dFOXO retains the functional domains found in Daf-16 and the mammalian FOXO homologues
The dFOXO gene consists of 10 exons and is spread out over approximately 31 kb in polytene chromosome section 88A within the genomic scaffolding region, AE003703, of the Berkeley Drosophila Genome Project (BDGP) (Figure 1A). dFOXO encodes a theoretical protein of 463 amino acids (Figure 1B). Analysis of the complete Drosophila genome for additional dFOXO homologues revealed none.
Figure 1.

dFOXO encodes a protein that retains important functional domains found in other FOXO homologues. (A) Schematic representation of the dFOXO cDNA clone LD05569 and its location in the genomic scaffolding, region AE003703, of the BDGP sequence. (B) ClustalW alignment of the proposed dFOXO amino acid sequence with that of mammalian homologues (FOXO1a, FOXO3a, and FOXO4) and Daf-16a1. Highlighted are: the T1, S1, and S2 Akt target sequences (yellow shading); the potential DYRK1a/mnb phosphorylation site (arrow, and grey shading); and the forkhead box DNA binding domain (black box). "*" indicates nucleotides that are identical in all sequences in the alignment, ":" indicates conserved substitutions, according to the chemical nature of the amino acids, and "." indicates semi-conserved substitutions. Colors indicate the chemical nature of the amino acid; Red = small hydrophobic (including aromatic), Blue = Acidic, Magenta = Basic, and Green = basic amino acids with hydroxyl groups and/or amine groups.
Alignment of dFOXO with the human homologues of FOXO and Daf-16a1 using ClustalW [37] (Figure 1B) revealed that although the overall identity of amino acids is not high, the identity in the forkhead box DNA binding domain is between 74 and 86 percent. The Akt phosphorylation sites are also well conserved in their relative position in the protein, and in sequence. The T1 site is located at T24 in dFOXO, the S1 site at S160, and the S2 site at S239. These sites align well with the human FOXO homologues in the ClustalW alignment, however the Daf-16 S1, and S2 sites are slightly out of line (Fig 1B). All three of the potential Akt phosphorylation sites in dFOXO fit the Akt consensus target sequence (RxRxxS/T).
Other notable features found in FOXO homologues include a DYRK1a phosphorylation site, a 14-3-3 binding site, a nuclear localization signal (NLS), a nuclear export signal (NES), and Ral dependent phosphorylation sites. A DYRK1a phosphorylation site was confirmed experimentally in FOXO1 at S329 [38]. This serine residue is conserved in human FOXO3a (S324), FOXO4 (S267), Daf-16a1 (S317), and dFOXO (S248) (Figure 1B). In addition, the sequence surrounding this site in dFOXO (LS248PI) is identical to that in FOXO1. The high conservation of this sequence indicates that dFOXO may be phosphorylated at this site by the Drosophila homologue of DYRK1a, minibrain (mnb).
Binding to 14-3-3 proteins is thought to be an important part of FOXO sequestration [30,31]. 14-3-3 proteins normally bind to a consensus site containing a phosphoserine residue, either RSxSPxP, or RxxxSPxP [39]. In the case of dFOXO, the sequence surrounding the T1 Akt phosphorylation site fits the former perfectly, aside from the substitution of a threonine for a serine. It has been shown experimentally that 14-3-3 does bind to this site in FOXO1 [40], FOXO3a [33], and Daf-16[32], hence, it is likely that this region functions as a 14-3-3 binding site in Drosophila.
The current model for FOXO deactivation suggests that a NES exists which causes constitutive localization of FOXO in the cytoplasm in the absence of a functional NLS [31]. A non-conventional NLS was identified in human FOXO4 from amino acids 180–221 [36]. The corresponding sequence in dFOXO (amino acids 147–194) is 38% identical and 66% similar in amino acid content (Figure 1B). This similarity suggests that this region may act as an NLS in dFOXO as well. A leucine rich NES has been identified in FOXO1 (368 MENLLDNLNL 377) and the conservation of this sequence is quite high FOXO3a, FOXO4, and Daf-16[30] (Figure 1B). The corresponding region in dFOXO retains two of the important leucine residues (281 LTGTMADELTL 291). However, the remaining sequence is more divergent, and may or may not act as an NES in Drosophila.
FOXO4 has previously been shown to be phosphorylated in a Ral-dependent manner at threonines 447 and 451[41]. However, these sites do not appear to be conserved in the other human FOXO homologues, Daf-16, or dFOXO (Figure 1B), indicating that Ral dependent phosphorylation of FOXO may be specific to FOXO4.
Interestingly, the carboxy-terminal three amino acids are conserved between dFOXO and FOXO1 (VSG). Also, FOXO3a contains a similar sequence in the final three amino acids (VPG). In view of this conservation, it is possible that this tail plays a functional role in FOXO regulation.
dFOXO expression during development phenocopies starvation and alters feeding behavior
Drosophila larvae feed continuously for about 5 days after egg laying (AEL). During this time the appetite and growth rate of the larvae is enormous. If young larvae are deprived of food, they do not grow and tend to disperse randomly[16,17,42]. When the food supply is replenished, the larvae immediately move towards it and continue eating until they are close to pupation. If the food supply is depleted, the larvae will disperse again[42]. We utilized the UAS/Gal4 ectopic expression system [43] to overexpress dFOXO in the developing larvae under the control of the ActGal4 driver[44]. This resulted in complete developmental arrest of the larvae, which remained as first instar for up to 7 days (Figure 2A), similar to the life expectancy of starved larvae [16-18]. This trend was also seen using a constitutively active version of Murine Foxo1 (mFoxo1) containing an alanine substitution at the T1 (T24A), and S1 (S253A) Akt phosphorylation sites (mFoxo1-AA) [45] (Figure 2A). In addition, larvae expressing dFOXO and mFoxo1-AA were often found to be wandering far from their food supply. We monitored feeding behavior by assessing the number of larvae away from their food at 48 and 72 hours after egg laying (AEL). Larvae expressing dFOXO and mFoxo1-AA showed a 3–4 fold increase in wandering over larvae expressing Gal4 alone (Figure 2B). Thus, dFOXO expression drastically alters feeding behavior and is able to induce a starvation type response in larvae which have an adequate food supply.
Figure 2.

Expression of dFOXO in first instar larvae phenocopies starvation and effects feeding behavior. Expression of dFOXO and mFOXO1-AA early in larval development using the (A) ActGal4 and (C) hsGal4 driver lines leads to developmental arrest similar to that seen in starved larvae. Developmentally arrested larvae are capable of surviving for up to seven days after egg laying (AEL). (B) Expression of dFOXO (red bars) and mFOXO1-AA (green bars) leads to alterations in feeding behavior when compared to controls (grey bars). The percentage of wandering larvae is significantly greater in larvae expressing dFOXO and mFOXO1-AA at 48 hours and 72 hours AEL (p = 0.05). Expression of dPI3K-DN (blue bars) did not increase larval wandering. (D) Developmental arrest is reversible upon removal of dFOXO expression (red bars), but not upon removal of mFOXO1-AA expression (green bars). Grey bars represent the controls. Each bar reflects the average of three separate trials, with 50 larvae per trial. Genotypes are; (A-top, B-grey bars) w; ActGal4/+, (A-middle, B-red bars) w; ActGal4/+; UAS-dFOXO/+, (A-bottom, B-green bars) w, UAS-mFoxo1-AA/w; ActGal4/+, (C-top, D-grey bars) w; hsGal4/+, (C-middle, D-red bars), w; hsGal4/UAS-dFOXO, (C-bottom, D-green bars) w, UAS-mFoxo1-AA/w; hsGal4/+, (B-blue bars) w; ActGal4/UAS-dPI3K-DN.
In Drosophila, PI3K consists of an adaptor subunit, dp60, and a catalytic subunit, dp110. Unexpectedly, expression of an inhibitory or "dominant negative" version of dp110 (UAS-dPI3K-DN)[46] under the control of the ActGal4 did not lead to increased larval wandering (Figure 2B). Expression of this construct also did not appear to inhibit larval growth, whereas other negative regulators of insulin signaling do [18]. It is possible that the level of expression of this construct is not high enough under the control of the ActGal4 driver to have a complete dominant negative effect.
Starved larvae which are developmentally arrested are able to resume growth upon acquisition of food [17]. We examined if larvae that were expressing dFOXO could resume growth upon termination of dFOXO expression. To do this we utilized the hsGal4 driver [47]. dFOXO was expressed in the larvae by heat shock treatment (HST) for 10 minutes every 24 hours. This treatment was sufficient to inhibit growth while allowing controls to survive to adulthood with a 48 hour delay in the time to pupation (Figure 2C). When dFOXO expression was discontinued after 2, 4, and 6 days of HST, developmentally arrested larvae were able to recover with decreased levels of survival as time progressed (Figure 2D). Significant lethality was observed in controls as well suggesting that low survival rate was partially due to the expression of Gal4, which can induce apoptosis [48], or the HST itself (Figure 2D). Nevertheless, developmental arrest caused by dFOXO is clearly reversible as these individuals could be returned to their normal path of development.
dFOXO performs an analogous function to C. elegans, Daf-16
The formation of dauer larvae in C. elegans is a developmental response to nutrient limitation [19]. The dauer larvae provides a temporary defense mechanism allowing the nematode to persevere until nutrients are available, at which point development can continue. Interestingly, constitutive activation of Daf-16 by mutation of its Akt phosphorylation sites to alanine residues causes obligatory dauer larvae formation[49]. We found a similar result in the Drosophila larvae using the constitutively active mFoxo1-AA [45]. This construct had an effect similar to that of dFOXO when expressed under the control of ActGal4 (Figure 2A), and hsGal4 (Figure 2C). Upon removal from HST, larvae expressing mFoxo1-AA did not resume growth but remained in a state of developmental arrest until death (Figure 2D). Although a few larvae did survive to adulthood after 2 days of HST, none of the larvae were able to continue development after 4, or 6 days of HST (Figure 2D). Out of 450 larvae examined at all time points, only 10 expressing mFoxo1-AA survived, when compared to 110 and 180 for larvae expressing dFOXO, and Gal4 alone, respectively. Presumably this occurs because Akt is unable to deactivate mFoxo1-AA, allowing it to continue functioning long after expression is induced. Taken together, this data suggests that dFOXO is evolutionarily conserved in function, possibly playing a role in the response to nutritional adversity, as seen in the formation of dauer larvae in C. elegans.
dFOXO inhibits growth through alterations in cell size and cell number
Expression of dFOXO in the third instar larvae caused significant lethality, however, rare flies that did survive were much smaller than control flies (Figure 3A), showing a phenotype similar to that caused by mutations in chico [12], dAkt [14] and dInr [6,9]. Expression of dFOXO under the control of the ubiquitous low level Gal4 drivers, armadillo-Gal4, and hsGal4 (raised at 25°C with no heat shock) had very little effect on growth (data not shown). In contrast, increasing expression of dFOXO using the hsGal4 driver in flies raised at 29°C lead to the development of small adults, which were approximately half the weight of control flies (Figures 3B and 3D). Analysis of the wings of these flies showed that the wing area was reduced by nearly one third and that this reduction was due to a decrease in both cell size and cell number (Figures 3C and 3D). SEM analysis of the eyes revealed reductions in both ommatidia number and ommatidia area, which reflect cell number and cell size, respectively (Figures 3E and 3F). These results implicate dFOXO in the control of body size through alterations in cell size and cell number.
Figure 3.
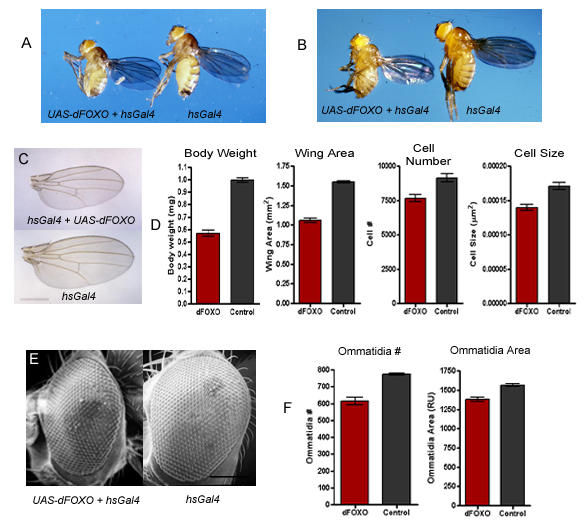
dFOXO reduces growth through alterations in cell size and cell number (A) Expression of UAS-dFOXO in the third larval instar produces small flies (left) when compared to controls (right). w; hsGal4/CyO flies were crossed to w; UAS-dFOXO/UAS-dFOXO flies and the progeny were heat shocked at 37°C for 4 hours during the early third instar. (B) Flies of the genotype w; hsGal4/+; UAS-dFOXO/+ (left) were smaller than w; hsGal4/+ (right) flies when raised at 29°C. (C) The wings of w; hsGal4/+; UAS-dFOXO/+ flies raised at 29°C were smaller than control wings (scale bar = 1 mm). (D) Flies expressing dFOXO (red bars) also showed a significant reduction in body weight, wing area, cell number, and cell size when compared to control flies (grey bars) (p = 0.005). (E) Flies expressing dFOXO had smaller eyes than control flies (scale bar = 150 μm), and (F) their eyes were reduced in both the number of ommatidia and the area of the ommatidia (red bars) when compared to controls (grey bars). Genotypes are; (A-left, B-left, C-top, D-red bars, E-left, F-red bars) w; hsGal4/+; UAS-dFOXO/+, (A-right, B-right, C-bottom, D-grey bars, E-right, F-grey bars). w; hs-Gal4/+.
Regulation of FOXO by the insulin signaling pathway is conserved between mammals and flies
When dFOXO is expressed in the developing eye under the control of the GMR-Gal4 driver[50], the eye is smaller, lacking many ommatidia and nearly all of the mechanosensory bristles (Figure 4E). The remaining ommatidia are arranged in the typical hexahedral array and cross sectional analysis revealed that all of the normal photoreceptor cells are present (Figure 4E, data not shown). Thus, it appears that dFOXO expression causes a reduction in the number of cells but does not interfere with cellular differentiation and the organization of the ommatidia themselves. We have used this eye phenotype to test for interactions between dFOXO and other components of the insulin signaling pathway.
Figure 4.
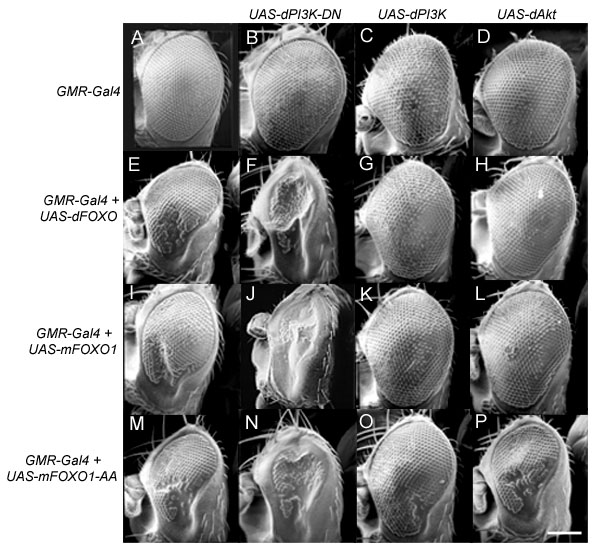
Regulation of dFOXO through insulin signaling is conserved between mammals and flies. The GMR-Gal4 driver was used to drive the expression of (B) dPI3K-DN, (C) wild type dPI3K, (D) dAkt, (E) dFOXO, (I) mFoxo1, and (M) mFoxo1-AA, both alone and in various combinations (F-H, J-L, N-P) as indicated through the rows and columns in the figure (scale bar = 150 μm). Genotypes are: (A) w; GMR-Gal4/+, (B) w; UAS-dPI3K-DN/GMR-Gal4, (C) w; UAS-dPI3K/GMR-Gal4, (D) w; UAS-dAkt/GMR-Gal4, (E) w; GMR-Gal4/+; UAS-dFOXO/+, (F) w; UAS-dPI3K-DN/GMR-Gal4; UAS-dFOXO/+, (G) w; UAS-dPI3K/GMR-Gal4; UAS-dFOXO/+, (H) w; UAS-dAkt/GMR-Gal4; UAS-dFOXO/+ (I) w; GMR-Gal4, UAS-mFoxo1/+, (J) w; GMR-Gal4, UAS-mFoxo1/UAS-dPI3K-DN, (K) w; GMR-Gal4, UAS-mFoxo1/UAS-dPI3K, (L) w; GMR-Gal4, UAS-mFoxo1/UAS-dAkt, (M) w, UAS-mFoxo1-AA/w; GMR-Gal4/+, (N) w, UAS-mFoxo1-AA/w; GMR-Gal4/UAS-dPI3K-DN, (O) w, UAS-mFoxo1-AA/w; GMR-Gal4/UAS-dPI3K, (P) w, UAS-mFoxo1-AA/w; GMR-Gal4/UAS-dAkt.
Expression of dPI3K-DN under the control of GMR-Gal4 leads to the formation of relatively normal eyes with fewer and smaller cells[46] (Figure 4B). When dFOXO is co-expressed in the developing eye with dPI3K-DN the eye is nearly obliterated (Figure 4F). In contrast, co-expression of dAkt, and wild type dPI3K with dFOXO causes nearly complete rescue of the phenotype, restoring the ommatidia and nearly all of the mechanosensory bristles (Figures 4G and 4H). Thus, diminishing insulin signaling (through overexpression of dPI3K-DN) allows for greater activity of dFOXO, and enhancing insulin signaling (through overexpression of dAkt or dPI3K) leads to inhibition of dFOXO activity. Similar results were obtained using a Murine Foxo1 (mFoxo1) construct (Figure 4 I-L), indicating that the regulatory mechanisms between these two proteins is conserved and that they are functionally interchangeable.
Growth effects of dPI3K and dAkt are masked by expression of mFoxo1-AA
The constitutively active mFoxo1-AA construct [45] was also expressed in the developing eye. Expression of this construct causes a phenotype similar to that of dFOXO and mFoxo1, with characteristic lack of ommatidia and mechanosensory bristles (Figure 4M). When mFoxo1-AA is co-expressed with dPI3K-DN the eye is nearly obliterated (Figure 4N), as seen with dFOXO and mFoxo1 (Figures 4F and 4J). Co-expression of mFoxo1-AA with dPI3K leads to a partial rescue of the phenotype, with still an obvious lack of ommatidia and mechanosensory bristles (Figure 4O). In contrast, co-expression of mFoxo1-AA with dAkt does not cause rescue of the ommatidia or mechanosensory bristles (Figure 4P), indicating that this construct is not responsive to dAkt signaling. The partial rescue of the dFOXO phenotype by dPI3K appears to be mediated through alterations in cell size (Figure 5) rather than cell number, as there is still an obvious lack of ommatidia and mechanosensory bristles (Figure 4O). This data indicates that inactivation of dFOXO is required for the full effects of growth mediated by dPI3K and dAkt.
Figure 5.

dFOXO inactivation is essential for dAkt, but not dPI3K, mediated increases in cell size. Ommatidia area was measured as a means to determine the effect of FOXO overexpression on cell size. Expression of dFOXO (bar 2), mFoxo1 (bar 3), and mFoxo1-AA (bar 4) under the control of GMR-Gal4 causes a significant decrease in ommatidia area when compared to the expression of Gal4 alone (bar 1). In addition, GMR-Gal4 was used to drive the expression of dPI3K (bars 5–8), and UAS-dAkt (bars 9–12), either alone (grey bars), or in the presence of UAS-dFOXO (red bars), UAS-mFoxo1 (light green bars), or UAS-mFoxo1-AA (dark green bars). Two sided t-tests were preformed to determine statistical significance (p = 0.001). Genotypes are: (1) w; GMR-Gal4/+, (2) w; GMR-Gal4/+; UAS-dFOXO/+, (3) w; GMR-Gal4, UAS-mFoxo1/+, (4) w, UAS-mFoxo1-AA/w; GMR-Gal4/+, (5) w; UAS-dPI3K/GMR-Gal4, (6) w; UAS-dPI3K/ GMR-Gal4; UAS-dFOXO/+, (7) w; GMR-Gal4, UAS-mFoxo1/UAS-dPI3K, (8) w, UAS-mFoxo1-AA/w; GMR-Gal4/UAS-dPI3K, (9) w; UAS-dAkt/GMR-Gal4, (10) w; UAS-dAkt/GMR-Gal4; UAS-dFOXO/+ (11) w; GMR-Gal4, UAS-mFoxo1/UAS-dAkt (12) w, UAS-mFoxo1-AA/w; GMR-Gal4/UAS-dAkt.
dPI3K can increase cell size in the presence of constitutively active Foxo
To examine the effect of dFOXO overexpression on cell size we measured the area of the ommatidia. Expression of dFOXO, mFoxo1, and mFoxo1-AA caused a significant reduction in the area of the ommatidia (p = 0.001) (Figure 5). Expression of dPI3K caused a significant increase in ommatidia size over wild type (p = 0.001) (Figure 5). This result is consistent with previous studies showing that dPI3K affects cell size in a cell autonomous manner[46]. Co-expression of dFOXO, mFoxo1, and mFoxo1-AA with dPI3K had no significant effect on the enlarged ommatidia (p = 0.001) (Figure 5). Thus, it appears that FOXO proteins have a very minimal effect on cell size in the presence of high levels of dPI3K. Surprisingly, this is the case even with the mFoxo1-AA construct, which is only partially responsive to PI3K signaling [45]. This indicates that the dPI3K mediated increase in cell size can occur through dAkt independent mechanisms.
Expression of dAkt in the developing eye caused a significant increase in ommatidia size, similar to that seen with dPI3K (p = 0.001) (Figure 5). Co-expression of dAkt with either dFOXO or mFoxo1, cause a slight, but insignificant decrease in the size of the enlarged ommatidia (Figure 5). However, co-expression of dAkt with mFoxo1-AA resulted in ommatidia that were approximately the same size as the ommatidia in eyes expressing Gal4 alone (Figure 5), and significantly smaller than the ommatidia in eyes expressing dAkt alone (p = 0.001) (Figure 5). This indicates that the deactivation of FOXO by dAkt is essential for dAkt to induce an increase in cell size.
dFOXO may reduce cell number through inhibition of the cell cycle and not apoptosis
The lack of ommatidia and mechanosensory bristles caused by dFOXO expression suggest a reduction in cell number during eye development (Figure 6A). Reduction of cell number can occur through either increased cell death, or decreased of cell proliferation. The Drosophila inhibitors of apoptosis, Diap1 and Diap2 (data not shown), and the baculovirus inhibitor of apoptosis, p35 (Figure 6B), were unable to rescue the phenotype caused by dFOXO expression. In addition, acridine orange staining of eye imaginal discs expressing dFOXO showed no increase in apoptosis when compared to controls (data not shown). Drosophila Epidermal Growth Factor Receptor (dEGFR) signaling acts to protect differentiated cells from death during eye development [51]. We thought that the pro-survival effects of dEGFR may be sufficient to suppress the phenotype caused by dFOXO overexpression. Co-expression of dEGFR with dFOXO, however, does not rescue the dFOXO phenotype as ommatidia and bristles are clearly still missing (Figure 6D). Conversely, dFOXO does not appear to affect the phenotype of dEGFR overexpression as the general disorganization of the ommatidia appears to be the same (Figure 6C). Thus, it appears that these two mechanisms are acting independently. Taken together, these results suggest that dFOXO overexpression does not cause cell death during eye development as direct inhibitors of the apoptotic machinery (p35 and Diap1/2) and a known cell survival factor (dEGFR) were unable to rescue the dFOXO phenotype.
Figure 6.
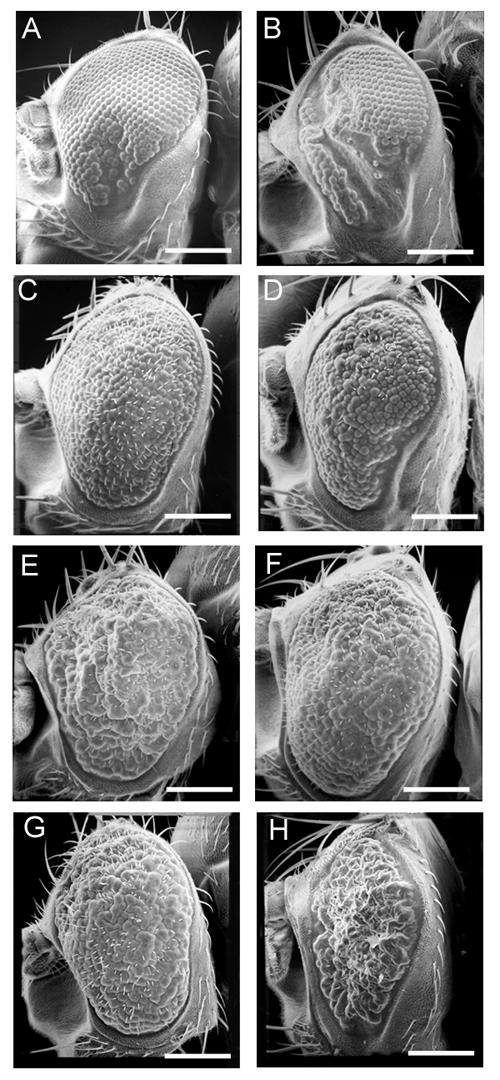
dFOXO responds to dRas2 signaling, but not to inhibitors of apoptosis. GMR-Gal4 was used to drive the expression of UAS-dFOXO (A) alone, and in the presence of (B) UAS-p35, (D) UAS-dEGFR, (F) UAS-Ras2V14. UAS-Ras2V14 was also expressed in combination with UAS-mFoxo1 (G) and UAS-mFoxo1-AA (H). Scale bars equal 150 μm. Genotypes are: (A) w; GMR-Gal4/+; UAS-dFOXO/+, (B) w; GMR-Gal4/UAS-p35; UAS-dFOXO/+, (C) w; GMR-Gal4/UAS-dEGFR, (D) w; GMR-Gal4/UAS-dEGFR; UAS-dFOXO/+, (E) w; GMR-Gal4/UAS-RasV14, (F) w; GMR-Gal4/UAS-Ras2V14; UAS-dFOXO/+, (G) w; GMR-Gal4, UAS-mFoxo1/UAS-Ras2V14, and (H) w, UAS-mFoxo1-AA/w; GMR-Gal4/ UAS-Ras2V14.
Since inhibition of apoptosis could not rescue the phenotype caused by dFOXO overexpression in the eye, we examined if activating the cell cycle could inhibit the phenotype. Expression of the E2F and Dp transcription factors has been shown to promote cell proliferation in the wing imaginal disc[4]. Co-expression of E2F and Dp with dFOXO was not sufficient to rescue the dFOXO phenotype (data not shown). Overexpression of constitutively active dRas1 (dRas1V12) has been shown to induce ectopic cell proliferation[52] and G1/S progression in the Drosophila wing disc[53,54]. Co-expression of dRas1V12 with dFOXO was lethal, so we used a constitutively active version of dRas2 (dRas2V14). Although dRas2 has not been characterized for its role in cell cycle control, it is possible that it has a similar function to dRas1. Expression of UAS-dRas2V14 under the control of GMR-Gal4 led to extreme overgrowth of the eye, lack of ommatidial organization, and the formation of huge ommatidia (Figure 6E). Co-expression of dRas2V14 with dFOXO was sufficient to restore many of the ommatidia and mechanosensory bristles lost through overexpression of dFOXO alone (Figure 6A and 6F). A similar effect was observed upon co-expression of dRas2V14 with mFoxo1 (Figure 6G). In contrast, the loss of ommatidia and bristles seen upon over expression of mFoxo1-AA was not rescued by dRas2V14 (Figure 6H). This suggests that dRas2V14 inhibits dFOXO via a dAkt phosphorylation dependent mechanism.
Discussion
For the most part, the genetic mechanisms that control size in multicellular organisms are not well understood [2]. Recently, components of the insulin signaling pathway have been shown to regulate body size in Drosophila melanogaster through alterations in cell size and cell number [1,6]. We have identified dFOXO as a negative controller of growth and organism size, which is regulated by components of the Drosophila insulin signaling pathway, dPI3K and dAkt. Through overexpression studies in the developing eye, we have shown that dFOXO is regulated by dPI3K and dAkt in a manner that is consistent with the regulatory mechanisms deduced through studies in C. elegans and mammalian cell culture. In addition, overexpression of dFOXO in the larvae reduces larval growth, phenocopies the effects of nutritional stress, and causes alterations in feeding behavior. With this in mind, we propose that dFOXO is involved in the response of Drosophila larvae to nutritional stress.
Conservation of FOXO in Drosophila
The FOXO homologues appear to play an evolutionarily conserved role in the control of cellular processes under conditions of low levels of insulin signaling [30,31]. Our experiments provide three lines of evidence supporting the conservation of this mechanism in Drosophila. First, dFOXO shows strong sequence homology to Daf-16 and the human FOXO homologues (Figure 1B). One significant characteristic is the high conservation of the three consensus Akt phosphorylation sites, suggesting that dAkt is most likely able to phosphorylate dFOXO in vivo, as shown biochemically with the mammalian FOXO homologues[33-35]. Second, our experiments show that dFOXO and mFoxo1 cause nearly identical phenotypic responses when overexpressed in the developing Drosophila eye (Figure 4, 5 and 6). This suggests that the activity of these proteins is highly conserved as is observed when the C. elegans FOXO homologue, Daf-16, is expressed in mammalian cell culture[32]. Third, the phenotypic effects of FOXO overexpression can be modulated by alterations in the insulin signaling pathway. Reduced insulin signaling leads to a drastic enhancement of the phenotype that results from expression of FOXO factors (Figure 4). In contrast, increased insulin signaling tends to mask these phenotypes, in a manner that is dependent on the integrity of the Akt phosphorylation sites (Figures 4 and 5). As a result, we believe that regulation of FOXO is conserved in Drosophila, and that this will be a very useful system in elucidating the function of FOXO transcription factors in a model organism.
Regulation of size by dFOXO
Our results show that ectopic dFOXO expression can mediate reduction in cell size and cell number (Figures 3, 4, and 5). However, the mechanisms by which these reductions occur are still unclear. Net reduction in cell number may occur through decreased cell proliferation or increased apoptosis. Insulin and other growth factors that activate PI3K and Akt have been implicated as potent survival factors in mammalian cell culture [10,11]. They prevent cell death, in part, by inhibition of FOXO factors and it has been shown that FOXO3a can upregulate expression of the pro-apoptotic protein Bim[55]. In Drosophila, reduction of insulin signaling can lead to apoptosis in the developing embryo [13,14,56,57]. It is possible that this increase in apoptosis is a result of dFOXO activation, however, when dFOXO is expressed in the developing eye there is no apparent increase in apoptosis, nor is the phenotype suppressed by inhibition of caspases, or by co-expression of a known cell survival factor, dEGFR (unpublished observations, Figure 6). These apparent discrepancies may be the result of tissue specific differences. In mammalian cell culture, induction of cell death by FOXO factors seems to be limited to non-transformed haematopoietic cell lineages [31]. In Drosophila, loss of dAkt function, inhibition of dPI3K, or overexpression of dPTEN, all induce cell death in the embryo[13,14]. However, in imaginal disc cells lacking PI3K function, there is no increase in apoptosis[58]. Thus, the cells in the embryo and imaginal discs may react differently to reduced levels of insulin signaling. Although we do not observe induction of apoptosis upon dFOXO expression, it is possible that increased levels of dFOXO activity (eg. through dominant negative inhibition of PI3K) do cause apoptosis.
Studies in mammalian cell culture have implicated FOXO factors in control of the cell cycle through increased expression of the cyclin dependent kinase inhibitor p27Kip1 [59,60]. It is possible that the reduction of cell number seen upon dFOXO expression is a result of cell cycle inhibition. Co-expression of an activated version of Drosophila Ras2 (dRas2V14) was sufficient to increase cell number in the presence of dFOXO (Figure 6). dRas1 has been shown to induce growth in Drosophila imaginal discs [52-54] through activation of dPI3K and the transcription factor dMyc [53]. Although there is very little information available about dRas2, it is possible that the function of dRas2 overlaps with that of dRas1. Expression of dRas2V14 in the developing eye does cause a phenotype that suggests overgrowth of cells (Figure 6E), and the dRas2V14 interaction with dFOXO appears to be dependent on dAkt signaling (Figure 6H). This is not surprising considering that dRas1 [53] and mammalian Ras [61] have been shown to activate PI3K signaling. Interestingly, increasing the cell cycle through overexpression of the transcription factors E2F and Dp did not rescue the cell number deficit seen upon overexpression of dFOXO (unpublished observations). This suggests the possibility that activation of dFOXO may override the function of other growth promoting factors, such as dMyc, which mediates dRas1 induced G1/S progression [53]. Supporting this, we have observed that increased growth as mediated by dAkt is entirely dependent on its ability to inactivate dFOXO (Figures 4P and 5). Furthermore, increased growth mediated by dPI3K appears to be dependent on dFOXO inactivation with respect to increased cell number, but not cell size (Figures 4O and 5). In humans, inactivation of FOXO factors may play an important role in tumor suppression by down regulating expression of D-type cyclins, thus inhibiting cell cycle progression and transformation[62]. It will be interesting to test the interactions between dFOXO and other cell cycle promoters to determine the extent of dFOXO dominance over cell proliferation.
In addition to its effect on cell number, dFOXO is able to control cell size (Figures 3 and 5). The ability of dAkt to increase cell size is dependent on dFOXO inactivation, however, dPI3K does not need to inactivate dFOXO to increase cell size (Figure 5). The difference between dPI3K and dAkt might be attributed to greater activity of the UAS-dPI3K transgene. However, expression of these constructs individually yields very similar results (Figures 4 and 5) indicating that this is probably not the case. This suggests that dPI3K may control size through dAkt-independent mechanisms. One possibility is through the positive growth regulator, dS6k[63]. dAkt appears to increase growth through inhibition of a TSC1/TSC2 (tuberous sclerosis) complex[64,65]. This complex acts through inhibition of dTOR (target of rampamycin) [66], which promotes growth through activation of dS6K [67,68]. Although it appears that dAkt can upregulate growth through dS6K, dS6K activity is not reduced in larvae lacking dAkt or dPI3K [67]. These results do not necessarily suggest that dPI3K and dAkt can not activate dS6K, as dS6K levels may be maintained through amino acid signals [66,68]. dS6K activity was shown to be dependent on phosphoinositide dependent kinase (dPDK1) [67], which interacts genetically with dAkt, dPI3K, dPTEN, and dInr [56,69] Thus, it is possible that dPI3K can modulate dS6K activity through dPDK1, independently of dAkt.
Insulin signaling and stress response
Studies in C. elegans indicate that insulin signaling is a critical mediator of longevity and stress resistance[70,71]. One of the most well-studied stress responses is the Daf-16 mediated formation of the dauer larvae under conditions of starvation and/or crowding. Several lines of evidence indicate that dFOXO may play a similar role in Drosophila larvae. When Drosophila larvae are deprived of food prior to 70 hours AEL, they live in a state of developmental arrest for several days before death. However, when starved after 70 hours AEL, the larvae are able to develop into adults that are reduced in size. This alteration in developmental response has been termed the "70 hour change" and is likely determined by the minimum size required for a Drosophila larvae to enter pupation[16]. We have mimicked the "70 hour change" through overexpression of dFOXO at different stages of larval development, in the presence of ample food (Figures 2 and 3). For example, ubiquitous high level expression of dFOXO in the early larvae (i.e. before 70 hours AEL) leads to developmental arrest, whereas heat shock induced expression of dFOXO during the third instar (i.e. after 70 hours AEL) leads to the development of small adults. Second, the normal development of starved larvae can resume upon the acquisition of food. Similarly, developmental arrest caused by expression of dFOXO prior to the "70 hour change" can be reversed if dFOXO expression is discontinued (Figure 2). Developmental arrest caused by expression of mFoxo1-AA before the "70 hour change" is not reversible suggesting a constitutive starvation type response as seen in C. elegans when Daf-16 phosphorylation sites are mutated[49]. Interestingly, the reversibility of FOXO induced arrest has also been observed in mammalian cell culture[72]. Third, under conditions of poor nutrition or crowding larval development does not cease, but the larval period is extended and small adults are produced [15]. We have replicated this effect through low level expression of dFOXO during the course of development (Figure 3). Finally, feeding behavior is drastically altered in larvae expressing dFOXO (Figure 2), causing them to wander away from their food. These larvae are often found crawling on the sides and lids of Petri dishes. This response may provide a selective advantage in the search for food as seen in C. elegans dauer larvae, which often crawl up to the highest point possible in hopes of attaching to passing organisms that could move the larvae to new locations with better food supply [19]. Taken together, these results suggest that dFOXO activity may act to promote survival during times of nutritional stress in a manner that recapitulates the formation of dauer larvae in C. elegans. It is tempting to speculate that dFOXO plays a role in response to other forms of stress, as observed with Daf-16[70,71]. Mammalian FOXO factors have been implicated in the protective response to oxidative stress [73-75] and FOXO factors are upregulated in response to caloric restriction in rat skeletal muscle [76]. Thus, it is possible that FOXO factors provide an evolutionarily conserved switch, by which an organism can alter its developmental program in order to promote survival under harsh conditions.
Insulin signaling and feeding behavior
Previously, it was observed that activation of insulin signaling caused larvae to wander away from their food [18]. We have observed a similar effect through overexpression of dFOXO, which acts in opposition to insulin signaling. As described previously, it is possible that hyperactivation of insulin signaling may lead to depletion of the haemolymph by increasing the cellular uptake of nutrients [18]. This would lead to increased hunger and cause the larvae to wander in search of food. Since PI3K activity is lost under conditions of starvation [18] it stands to reason that dFOXO would be active under these conditions. Being a transcription factor, endogenous dFOXO could activate a host of genes under conditions of starvation leading to a "genetic starvation profile". Indeed gene expression is drastically altered upon starvation[42]. Thus, dFOXO may induce larval wandering through expression of a sub-set of genes which are normally active during starvation, whereas activation of insulin signaling may induce larval wandering by causing physiological changes that lead to a false sense of starvation.
Conclusions
We have shown that dFOXO is conserved in sequence and regulatory mechanisms when compared to homologues from mammals and C. elegans. Drosophila melanogaster provides a powerful tool for the analysis of genes in a whole organism. Thus, future studies in this organism should provide new insights into the biological function of the FOXO transcription factors. This may have implications to the study of cancer and diseases related to insulin, such as diabetes and obesity. Our data, taken together with that of others, suggests that dFOXO plays a protective role in the developmental response of Drosophila larvae to nutritional stress. Thus, it is possible that dFOXO plays a functional role in response to multiple forms of stress. In a world plagued with massive pollution and hunger it is important that we understand how our bodies react to starvation and environmental stress.
Methods
Identification and sequence analysis of dFOXO
The human FOXO4 gene was used to search the NCBI (National Center for Biotechnology Information) genomic data bank for Drosophila homologues. Drosophila genomic sequences with high homology to FOXO4 were identified and used to search the Berkeley Drosophila Genome Project (BDGP) for homologous cDNAs. This procedure allowed us to identify the clone, LD05569, which was sub-cloned and sent for sequencing to Cortec DNA Laboratories, Inc., Kingston, Ontario. Restriction mapping and sequencing revealed a cDNA of approximately 3.6 kb translating into a theoretical protein sequence of 463 amino acids (Fig 1B). Note that there are two other potential start codons that may act as sites for translation initiation, and are located slightly upstream of the start site we have identified.
Creation of transgenic Drosophila lines and overexpression studies
mFoxo1, and mFoxo1-T24A/S253A (AA) clones were generously provided by Dr. William H. Biggs III [45] and the dFOXO cDNA, LD05569, was obtained from Research Genetics. The cDNAs were ligated into the p[PUAST] expression vector for use of the UAS/Gal4 ectopic expression system [43]. Transgenic flies were created by injecting p[PUAST]-FOXO constructs into w1118 Drosophila embryos. Driver lines, GMR-Gal4 [50], heat shock-Gal4 (hsGal4)[47], and Act5C-Gal4 (ActGal4)[44] were obtained from the Bloomington stock center, as were the UAS lines UAS-dEGFR, UAS-dRas2V14, UAS-E2F, UAS-Dp, UAS-p35, UAS-Diap1, and UAS-Diap2. UAS-dPI3K and UAS-dPI3K-DN (UAS-dp110D954A) were generously provided by Dr. Sally Leevers. Heat shock treatment was conducted in a 37°C water bath.
Phenotypic analysis
All experiments were performed at 25°C unless otherwise stated. For scanning electron micrographs, flies were desiccated overnight and coated in gold. Ommatidia area was measured using NIHimage 6.2 and each value shown is the mean of 9 measurements, taken from 3 individual eyes. Due to the low survival rate of males expressing dFOXO, only females were included in the analysis of wings and body weight. Flies were raised under non-crowded conditions and a minimum of 12 flies were weighed individually to determine average body weight. Wing area was measured using ImageJ 1.28u, from the National Institute of Health. Cell size and cell number were calculated as previously described[63]. A minimum of 10 wings were analyzed per genotype. Two-sided t-tests were performed to determine significant differences.
Feeding behavior and phenocopy of starvation using ActGal4
The Gal4 driver line w; ActGal4/CyO was crossed to w1118, w; UAS-dFOXO/UAS-dFOXO, w, UAS-mFoxo1-AA/w, UAS-mFoxo1-AA, and yw; UAS-dPI3K-DN/UAS-dPI3K-DN. Since the ActGal4 insertion is not homozygous, we assumed that only half of the hatched larvae contained the insertion. This assumption was supported by observation of the adults arising from each cross. For w; ActGal4/CyO X w1118 the number of adults produced was nearly equal to the number of hatched embryos, with approximately half bearing the CyO balancer chromosome. For w; ActGal4/CyO X w; UAS-dFOXO/UAS-dFOXO and w; ActGal4/CyO X UAS-mFoxo1-AA/w, UAS-mFoxo1-AA only flies bearing the CyO chromosome survived and the number of adults was approximately half the number of the total hatched larvae. Small wandering larvae were observed only for w; ActGal4/CyO X w; UAS-dFOXO/UAS-dFOXO and w; ActGal4/CyO X UAS-mFoxo1-AA/w, UAS-mFoxo1-AA, and in these crosses, only the larvae present in the food were growing. Thus, we assumed that small wandering larvae were of the genotypes w; ActGal4/+; UAS-dFOXO/+, and w, UAS-mFoxo1-AA/w; ActGal4/+.
For the feeding behavior assay, embryos were collected on apple juice agar over ~2 hour time periods, counted, and transferred to a Petri dish with filter paper that was soaked in 20% sucrose in PBS. In the center of the Petri dish was a small piece of standard Drosophila food. At 48 hours AEL the number of hatched eggs was counted to account for unfertilized embryos. At both 48 hours and 72 hours AEL the number of larvae not on the food were counted. The percent wandering larvae was calculated based on the number of larvae off the food, the number of hatched eggs, and the assumption that only half of the total larvae contained the ActGal4 transgene. The results presented are the average from three separate trials and statistical significance was determined using a two-sided t-test. Individual values were taken from analysis of approximately 50 larvae.
Authors' Contributions
JMK conducted all genetic experiments and drafted the manuscript, as well as playing a partial role in sequence analysis of dFOXO and the creation of transgenic fly lines. JTD was responsible for the cloning and sequence analysis of dFOXO. JML participated in the creation of transgenic fly lines. BES initiated investigation of the dFOXO gene, and created and initiated characterization of UAS-dFOXO transgenics, as well as acting as supervisor and primary investigator.
Acknowledgments
Acknowledgments
We thank Roy Ficken and Lisa Lee from the Department of Biology at the Memorial University of Newfoundland for their technical assistance. Thanks to Dr. Ellen Larsen for advice and support during the early stages of this project. Thanks to Drs. William Biggs III and Fredrick G. Barr for the mFoxo1 clones and the Berkeley Drosophila Genome Project and Research Genetics for the dFOXO clone. Thanks also to Drs. Sally Leevers and Michael Waterfield, and to the Bloomington Drosophila stock center at Indiana University for providing fly stocks. Thanks to Dr. Ernst Hafen, Dr. Helene Volkoff, Martin Junger, and Annika Haywood for discussion and/or comments on the manuscript. This research was funded by the Natural Sciences and Engineering Research Council of Canada, the Banting Research Foundation, the Dean of Science of Memorial University of Newfoundland (start-up funds) to BES. JMK was partially funded by the School of Graduate Studies at the Memorial University of Newfoundland.
Contributor Information
Jamie M Kramer, Email: x04jmk@mun.ca.
Jason T Davidge, Email: jason_davidge@hotmail.com.
Joseph M Lockyer, Email: f54jml@hotmail.com.
Brian E Staveley, Email: bestave@mun.ca.
References
- Oldham S, Bohni R, Stocker H, Brogiolo W, Hafen E. Genetic control of size in Drosophila. Philos Trans R Soc Lond B Biol Sci. 2000;355:945–952. doi: 10.1098/rstb.2000.0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon I, Raff M. Size control in animal development. Cell. 1999;96:235–244. doi: 10.1016/s0092-8674(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Stern DL, Emlen DJ. The developmental basis for allometry in insects. Development. 1999;126:1091–1101. doi: 10.1242/dev.126.6.1091. [DOI] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–1193. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- Weigmann K, Cohen SM, Lehner CF. Cell cycle progression, growth and patterning in imaginal discs despite inhibition of cell division after inactivation of Drosophila Cdc2 kinase. Development. 1997;124:3555–3563. doi: 10.1242/dev.124.18.3555. [DOI] [PubMed] [Google Scholar]
- Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol. 2001;11:213–221. doi: 10.1016/S0960-9822(01)00068-9. [DOI] [PubMed] [Google Scholar]
- Ikeya T, Galic M, Belawat P, Nairz K, Hafen E. Nutrient-dependent expression of insulin-like peptides from neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Curr Biol. 2002;12:1293–1300. doi: 10.1016/S0960-9822(02)01043-6. [DOI] [PubMed] [Google Scholar]
- Fernandez R, Tabarini D, Azpiazu N, Frasch M, Schlessinger J. The Drosophila insulin receptor homolog: a gene essential for embryonic development encodes two receptor isoforms with different signaling potential. Embo J. 1995;14:3373–3384. doi: 10.1002/j.1460-2075.1995.tb07343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Jack J, Garofalo RS. The Drosophila insulin receptor is required for normal growth. Endocrinology. 1996;137:846–856. doi: 10.1210/endo.137.3.8603594. [DOI] [PubMed] [Google Scholar]
- Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, Beckingham K, Hafen E. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell. 1999;97:865–875. doi: 10.1016/s0092-8674(00)80799-0. [DOI] [PubMed] [Google Scholar]
- Staveley BE, Ruel L, Jin J, Stambolic V, Mastronardi FG, Heitzler P, Woodgett JR, Manoukian AS. Genetic analysis of protein kinase B (AKT) in Drosophila. Curr Biol. 1998;8:599–602. doi: 10.1016/s0960-9822(98)70231-3. [DOI] [PubMed] [Google Scholar]
- Scanga SE, Ruel L, Binari RC, Snow B, Stambolic V, Bouchard D, Peters M, Calvieri B, Mak TW, Woodgett JR, Manoukian AS. The conserved PI3'K/PTEN/Akt signaling pathway regulates both cell size and survival in Drosophila. Oncogene. 2000;19:3971–3977. doi: 10.1038/sj.onc.1203739. [DOI] [PubMed] [Google Scholar]
- Robertson FW. The ecological genetics of growth in Drosophila. 6. The genetic correlation between the duration of the larval periodand body size in relation to larval diet. Genet Res. 1963;4:74–92. [Google Scholar]
- Beadle G, Tatum E, Clancy C. Food level in relation to rate of development and eye pigmentation in Drosophila. Biol Bull. 1938;75:447–462. [Google Scholar]
- Britton JS, Edgar BA. Environmental control of the cell cycle in Drosophila: nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development. 1998;125:2149–2158. doi: 10.1242/dev.125.11.2149. [DOI] [PubMed] [Google Scholar]
- Britton JS, Lockwood WK, Li L, Cohen SM, Edgar BA. Drosophila's insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell. 2002;2:239–249. doi: 10.1016/s1534-5807(02)00117-x. [DOI] [PubMed] [Google Scholar]
- Riddle DL. The Dauer Larvae. In: Wood WB, editor. The Nematode Caenorhabiditis elegans. New York, Cold Spring Harbour Laboratory Press; 1988. pp. 393–412. [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Gottlieb S, Ruvkun G. daf-2, daf-16 and daf-23: genetically interacting genes controlling Dauer formation in Caenorhabditis elegans. Genetics. 1994;137:107–120. doi: 10.1093/genetics/137.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JZ, Tissenbaum HA, Ruvkun G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature. 1996;382:536–539. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- Paradis S, Ruvkun G. Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 1998;12:2488–2498. doi: 10.1101/gad.12.16.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Ailion M, Toker A, Thomas JH, Ruvkun G. A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes Dev. 1999;13:1438–1452. doi: 10.1101/gad.13.11.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Sublett JE, Jeon IS, Shapiro DN. The alveolar rhabdomyosarcoma PAX3/FKHR fusion protein is a transcriptional activator. Oncogene. 1995;11:545–552. [PubMed] [Google Scholar]
- Borkhardt A, Repp R, Haas OA, Leis T, Harbott J, Kreuder J, Hammermann J, Henn T, Lampert F. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11)(q13;q23) Oncogene. 1997;14:195–202. doi: 10.1038/sj.onc.1200814. [DOI] [PubMed] [Google Scholar]
- Biggs W. H., 3rd, Cavenee WK, Arden KC. Identification and characterization of members of the FKHR (FOX O) subclass of winged-helix transcription factors in the mouse. Mamm Genome. 2001;12:416–425. doi: 10.1007/s003350020002. [DOI] [PubMed] [Google Scholar]
- Arden K.C., Biggs W.H. Regulation of the FoxO family of transcription factors by phosphatidylinositol-3 kinase-activated signaling. Arch Biochem Biophys. 2002;403:292–298. doi: 10.1016/S0003-9861(02)00207-2. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27:352–360. doi: 10.1016/S0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Tzivion G, Nasrin N, Ogg S, Dore J, Ruvkun G, Alexander-Bridges M. Phosphatidylinositol 3-kinase signaling inhibits DAF-16 DNA binding and function via 14-3-3-dependent and 14-3-3-independent pathways. J Biol Chem. 2001;276:13402–13410. doi: 10.1074/jbc.M010042200. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- Brownawell AM, Kops GJ, Macara IG, Burgering BM. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol Cell Biol. 2001;21:3534–3546. doi: 10.1128/MCB.21.10.3534-3546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Bioinformatics Institute: ClustalW submission form http://www.ebi.ac.uk/clustalw
- Woods YL, Rena G, Morrice N, Barthel A, Becker W, Guo S, Unterman TG, Cohen P. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem J. 2001;355:597–607. doi: 10.1042/bj3550597. http://www.ebi.ac.uk/clustalw [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- Rena G, Prescott AR, Guo S, Cohen P, Unterman TG. Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targetting. Biochem J. 2001;354:605–612. doi: 10.1042/0264-6021:3540605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ruiter ND, Burgering BM, Bos JL. Regulation of the Forkhead transcription factor AFX by Ral-dependent phosphorylation of threonines 447 and 451. Mol Cell Biol. 2001;21:8225–8235. doi: 10.1128/MCB.21.23.8225-8235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinke I, Schutz CS, Katzenberger JD, Bauer M, Pankratz MJ. Nutrient control of gene expression in Drosophila: microarray analysis of starvation and sugar-dependent response. Embo J. 2002;21:6162–6173. doi: 10.1093/emboj/cdf600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Ito K, Awano W, Suzuki K, Hiromi Y, Yamamoto D. The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurones and glial cells. Development. 1997;124:761–771. doi: 10.1242/dev.124.4.761. [DOI] [PubMed] [Google Scholar]
- Biggs W. H., 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD. The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. Embo J. 1996;15:6584–6594. [PMC free article] [PubMed] [Google Scholar]
- Kraus ME, Lis JT. The concentration of B52, an essential splicing factor and regulator of splice site choice in vitro, is critical for Drosophila development. Mol Cell Biol. 1994;14:5360–5370. doi: 10.1128/mcb.14.8.5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JM, Staveley BE. GAL4 causes developmental defects and apoptosis when expressed in the developing eye of Drosophila melanogaster. Genet Mol Res. 2003;2:43–47. [PubMed] [Google Scholar]
- Lee RY, Hench J, Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol. 2001;11:1950–1957. doi: 10.1016/S0960-9822(01)00595-4. [DOI] [PubMed] [Google Scholar]
- Freeman M. Reiterative use of the EGF receptor triggers differentiation of all cell types in the Drosophila eye. Cell. 1996;87:651–660. doi: 10.1016/s0092-8674(00)81385-9. [DOI] [PubMed] [Google Scholar]
- Dominguez M, Wasserman JD, Freeman M. Multiple functions of the EGF receptor in Drosophila eye development. Curr Biol. 1998;8:1039–1048. doi: 10.1016/s0960-9822(98)70441-5. [DOI] [PubMed] [Google Scholar]
- Karim FD, Rubin GM. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development. 1998;125:1–9. doi: 10.1242/dev.125.1.1. [DOI] [PubMed] [Google Scholar]
- Prober DA, Edgar BA. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 2002;16:2286–2299. doi: 10.1101/gad.991102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prober DA, Edgar BA. Ras1 promotes cellular growth in the Drosophila wing. Cell. 2000;100:435–446. doi: 10.1016/s0092-8674(00)80679-0. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/S0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- Cho KS, Lee JH, Kim S, Kim D, Koh H, Lee J, Kim C, Kim J, Chung J. Drosophila phosphoinositide-dependent kinase-1 regulates apoptosis and growth via the phosphoinositide 3-kinase-dependent signaling pathway. Proc Natl Acad Sci U S A. 2001;98:6144–6149. doi: 10.1073/pnas.101596998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Potter CJ, Tao W, Li DM, Brogiolo W, Hafen E, Sun H, Xu T. PTEN affects cell size, cell proliferation and apoptosis during Drosophila eye development. Development. 1999;126:5365–5372. doi: 10.1242/dev.126.23.5365. [DOI] [PubMed] [Google Scholar]
- Weinkove D, Neufeld TP, Twardzik T, Waterfield MD, Leevers SJ. Regulation of imaginal disc cell size, cell number and organ size by Drosophila class I(A) phosphoinositide 3-kinase and its adaptor. Curr Biol. 1999;9:1019–1029. doi: 10.1016/S0960-9822(99)80450-3. [DOI] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, Coffer PJ. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1) Mol Cell Biol. 2000;20:9138–9148. doi: 10.1128/MCB.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Marte BM, Warne PH, Downward J. Phosphatidylinositol 3' kinase: one of the effectors of Ras. Philos Trans R Soc Lond B Biol Sci. 1996;351:225–31; discussion 231-2. doi: 10.1098/rstb.1996.0020. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91. doi: 10.1016/S1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: a regulator of cell size. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- Gao X, Pan D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 2001;15:1383–1392. doi: 10.1101/gad.901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS, Ru B, Pan D. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol. 2002;4:699–704. doi: 10.1038/ncb847. [DOI] [PubMed] [Google Scholar]
- Radimerski T, Montagne J, Rintelen F, Stocker H, van der Kaay J, Downes CP, Hafen E, Thomas G. dS6K-regulated cell growth is dPKB/dPI(3)K-independent, but requires dPDK1. Nat Cell Biol. 2002;4:251–255. doi: 10.1038/ncb763. [DOI] [PubMed] [Google Scholar]
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev. 2000;14:2689–2694. doi: 10.1101/gad.845700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintelen F, Stocker H, Thomas G, Hafen E. PDK1 regulates growth through Akt and S6K in Drosophila. Proc Natl Acad Sci U S A. 2001;98:15020–15025. doi: 10.1073/pnas.011318098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TE, Henderson S, Murakami S, de Castro E, de Castro SH, Cypser J, Rikke B, Tedesco P, Link C. Longevity genes in the nematode Caenorhabditis elegans also mediate increased resistance to stress and prevent disease. J Inherit Metab Dis. 2002;25:197–206. doi: 10.1023/A:1015677828407. [DOI] [PubMed] [Google Scholar]
- Yanase S, Yasuda K, Ishii N. Adaptive responses to oxidative damage in three mutants of Caenorhabditis elegans (age-1, mev-1 and daf-16) that affect life span. Mech Ageing Dev. 2002;123:1579–1587. doi: 10.1016/S0047-6374(02)00093-3. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Medema RH, Glassford J, Essers MA, Dijkers PF, Coffer PJ, Lam EW, Burgering BM. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol Cell Biol. 2002;22:2025–2036. doi: 10.1128/MCB.22.7.2025-2036.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H, Brunet A, Grenier JM, Datta SR, Fornace A. J., Jr., DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- Furukawa-Hibi Y, Yoshida-Araki K, Ohta T, Ikeda K, Motoyama N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J Biol Chem. 2002;277:26729–26732. doi: 10.1074/jbc.C200256200. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- Furuyama T, Yamashita H, Kitayama K, Higami Y, Shimokawa I, Mori N. Effects of aging and caloric restriction on the gene expression of Foxo1, 3, and 4 (FKHR, FKHRL1, and AFX) in the rat skeletal muscles. Microsc Res Tech. 2002;59:331–334. doi: 10.1002/jemt.10213. [DOI] [PubMed] [Google Scholar]