

Abstract
Phosphoinositides (PIs) are concentrated in specific subcellular membranes in order to recruit and regulate cytosolic proteins responsible for vesicular trafficking, cytoskeletal rearrangement, and eukaryotic cell growth, differentiation, and survival. Phox homology (PX) domains are found in proteins that are integral players in endocytic pathways. For example, Vam7p is targeted by its PX domain to phosphatidylinositol 3-phosphate [PtdIns(3)P] in the yeast vacuole, where it interacts with other SNARE proteins and GTPases of the vesicular membrane fusion machinery. Although several PX structures have been solved, the role of dynamics in their interactions with membrane lipids is unclear. Here, we present the first detailed characterization of the backbone dynamics of a PX domain, that of Vam7p, in the presence and absence of its ligand. The structure appears to tumble more rapidly in solution upon binding PtdIns(3)P, revealing a conformational change that includes adjustments in the flexible membrane insertion loop (MIL). The flexibilities of the MIL and domain termini are pronounced in both states, while the α1 and α2 helices are rigid. Dynamic effects are spread across the binding pocket, with PtdIns(3)P inducing altered mobility of different residues on multiple timescales, including a shift in the MIL to slower timescale motions. The bound state is more dynamic overall, particularly in the β-sheet lobe, which packs against the ligand's 3-phosphate. Thus, the induced dynamic and structural effects are transduced from the buried heart of the binding pocket in the helical lobe through the β-sheet lobe to the exposed surface of the bilayer-inserted protein.
Keywords: PX domain, phosphoinositide, backbone dynamics, NMR
Thousands of peripheral membrane proteins contain domains that bind to, insert into, turn over, and are regulated by phospholipids. However, not one of their mechanisms is understood in terms of the detailed structural dynamics that are responsible for phospholipid–ligand recognition. The PX domain-containing protein family is one of the most extensive, with 45 human members including PI and Ser/Thr kinases, phospholipases, and sorting nexins (Cheever and Overduin 2005). The most common PX function is recognition of PtdIns(3)P-containing membranes within the context of the endocytic pathway, as exemplified by the Vam7p t-SNARE (Cheever et al. 2001).
Vam7p plays a central role in the regulated fusion of vesicles with the yeast vacuole. As this process progresses, Vam7p cycles through a priming step involving Sec17p and Sec18p, to trans-SNARE partnering with another membrane, and then through dissociation from and reassociation with the membrane, a process which is regulated by the Rab GTPase Ypt7p and the HOPS complex (reviewed in Collins et al. 2005). Vam7p and possible homologs in Schizosaccharomyces pombe and Candida albicans are currently the only known SNARE proteins to contain PX domains (Dietrich et al. 2003), although sorting nexins share a similar modular architecture. Most SNAREs associate with membranes through transmembrane domains or post-translationally added lipid anchors. Vam7p instead binds reversibly to the membrane, affording greater scope for dynamically regulated membrane anchoring and fusion events.
Structures of several PX domains have been solved, revealing a conserved α/β fold. There are no major rearrangements of the structured elements upon ligand binding based on the PX domain structures from Grd19p and p40phox complexed with PtdIns(3)P (Bravo et al. 2001; Zhou et al. 2003), from p47phox with bound sulphate (Karathanassis et al. 2002), and from Grd19p, CISK, p47phox, sorting nexin 1, and Vam7p, all of which are uncomplexed (Cheever et al. 2001; Hiroaki et al. 2001; Lu et al. 2002; Xing et al. 2004; Zhong et al. 2005). The conserved fold consists of a three-stranded antiparallel β-sheet draped across three α-helices. The two longest and roughly parallel helices, α1 and α2, are connected by a long variable loop that contains a type II polyproline helix and, in the case of p47phox, a short 310 helix. Variations occur in the α3–α3′ helix and termini, which contain additional helical extensions in p40phox (Bravo et al. 2001). In contrast to the conserved core, the loops have relatively variable sequences and adopt a wide variety of conformations.
In an effort to define the dynamic changes occurring in the Vam7p PX domain as a result of ligand binding, 15N spin relaxation data were collected in the absence and the presence of dibutanoyl (C4-) PtdIns(3)P. The relaxation data revealed a network of conformational changes in residues involved in both PtdIns(3)P binding and membrane insertion, as well as the exposed β1–β2 hairpin in the opposite extremity of the bilayer-associated protein.
Materials and methods
Protein purification
A DNA fragment encoding amino acids 2–122 of wild-type Vam7p from Saccharomyces cerevisiae was amplified by polymerase chain reaction using primers that added 5′EcoRI and 3′BamHI sites, and then ligated into the pGEX-2T vector (Amersham). The Vam7p sequence was preceded by the glutathione S-transferase sequence, a thrombin cleavage site and the vector-derived sequence GlySer.
The PX domain was expressed by the Escherichia coli strain BL21-CodonPlus-RP (Stratagene) in LB or 15N or 13C/15N Bio-Express-1000 media (Cambridge Isotope Laboratories). The protein was purified with Glutathione Sepharose 4B (Amersham) and cleaved with thrombin (Sigma). The domain was denatured with 6 M guanidinium hydrochloride and refolded by dialysis into the NMR sample buffer. Purification by FPLC over a 16/60 Superdex 75 column (Amersham) revealed that the protein migrated as a monomer.
NMR spectroscopy
All of the NMR samples contained between 50 and 1600 μM unlabeled or uniformly 15N- or 15N,13C-labeled protein in 50 mM potassium phosphate buffer (pH 7.0), 100 mM KCl, 1 mM DTT, 1 mM NaN3, and 10% 2H2O. All NMR spectra were recorded at 25°C on Varian INOVA 500 and 600 MHz spectrometers. Resonance and NOE assignments were made from CBCA(CO)NH, HNCACB, HSQC-NOESY-HSQC, and 15N-edited NOESY-HSQC spectra as described previously (Kay et al. 1995). The data were processed and analyzed using nmrPipe (Delaglio et al. 1995), NMRView (Johnson and Blevins 1994), and in-house software programs on Sun Microsystems Ultra, Silicon Graphics O2, and Mandrake-Linux workstations.
The interaction sites were mapped by comparing 1H,15N HSQC spectra of 15N-labeled PX domain (50 μM) while titrating in soluble C4-PtdIns(3)P (Echelon, Inc.) as previously described (Cheever 2001). The normalized chemical shift changes (NΔδ) were calculated according to Equation 1:
where δH and δN are the 1H and 15N chemical shift changes in parts per million (ppm), respectively, and γN and γH are the gyromagnetic ratios for 15N and 1H, respectively (Amezcua et al. 2002).
The dissociation constant (Kd) was determined by analysis of the chemical shift changes using an NMRView script provided by K.H. Gardner (available at http://freedom7.swmed.edu/NMRview/titration.html). This script was used to perform nonlinear regression fitting of NΔδ values with Equation 2:
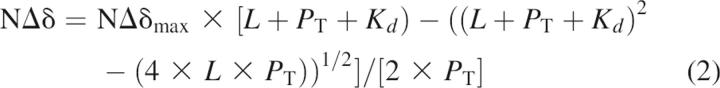 |
where NΔδmax is the change in chemical shift at saturation, L is the ligand concentration, and PT is the total protein concentration.
The 15N R1, R2, and 1H-15N steady-state NOE experiments were recorded according to established methods (Farrow et al. 1994) and analyzed as described previously (de Beer et al. 2002). The 15N R1 values were determined from the spectra recorded with T1 delay times of 11, 67, 145, 245, 368, 535, 769, 1115, and 1505 msec. 15N R2 values were determined using spectra collected with T2 delay times of 17, 35, 51, 68, 85, 102, 119, 136, 153, 170, and 187 msec. Recovery delays of 1.2 sec were used in the measurement of both R1 and R2 values. NOE values were determined from spectra collected either with a 5-sec relaxation delay alone or with a proton presaturation period of 3 sec preceded by a 2-sec relaxation delay. The R1, R2, and NOE values were determined as described previously (Mandel et al. 1995). Briefly, peak heights from R1 and R2 experiments were fit using the program Curvefit (A.G. Palmer III, Columbia University) and Equation 3:
where I(t) is the peak intensity after a delay of time t, and I0 is the intensity at time t = 0. NOE values were determined as the ratios of the peak heights from spectra recorded with and without presaturation. In the absence of PtdIns(3)P, the R1 and R2 relaxation decay data were fit adequately for 91 and 93 residues, respectively, as evidenced by χ2 values below the 0.05 critical values. In the PtdIns(3)P-bound state, relaxation decay data were fit within the 0.05 critical values for 85 R1 and 83 R2 decays.
The optimal spectral density model for fitting the relaxation data of each amino acid was chosen using the statistical approach of Mandel et al. (1995) where the five available models were described by the dynamical parameters: (1) S2 alone; (2) S2 and τe; (3) S2 and Rex; (4) S2, τe, and Rex; or (5) S2, τe, and S2f. The dynamical parameters were calculated using Modelfree 4.15 software (Palmer et al. 1991; Mandel et al. 1995). Initial estimates of the overall correlation times (τm) were calculated using trimmed and averaged R2/R1 ratios (Farrow et al. 1994). Edited lists of R2/R1 ratios were used to calculate diffusion tensors and an axially symmetric diffusion model was chosen by comparison with both isotropic and fully anisotropic diffusion models (Bruschweiler et al. 1995; Tjandra et al. 1995; Mandel et al. 1996; Lee et al. 1997). Since experimentally derived coordinates have not yet been determined for the PtdIns(3)P-complexed Vam7p PX domain, and also because of the high structural similarity of the PtdInd(3)P-free and bound Grd19p crystal structures, the structural coordinates from the PtdIns(3)P-free Vam7p PX domain were used to represent both forms of the protein in our diffusion model calculations. The τm value calculations and diffusion model evaluations were performed with the programs R2R1_tm, R2R1_diffusion and Quadric_diffusion, which, along with Modelfree 4.15 software, are available from the Web site http://cpmcnet.columbia.edu/dept/gsas/biochem/labs/palmer/.
Modeling
The position of PtdIns(3)P was modeled into the Vam7p PX domain using rigid body docking. The Vam7p PX domain's Cα atoms were aligned with those of the homologous p40phox structure (Bravo et al. 2001) using SPOCK (J.A. Christopher), and the PtdIns(3)P molecule was placed in the corresponding pocket in agreement with the chemical shift perturbations and the structural interactions observed in the Grd19p and p40phox structures (Bravo et al. 2001; Zhou et al. 2003). Superimposed structural coordinates were from PDB files 1H6H and 1KMD.
Results
Structure of the binding site
The three-dimensional structure of the unbound Vam7p PX domain has been solved (Lu et al. 2002), and chemical shift perturbations induced by PtdIns(3)P in Tris buffer are known (Cheever et al. 2001). However, significant additional perturbations were revealed through extending the backbone assignments to 98% of all residues by performing the experiments in a more physiologically relevant, phosphate-based buffer. The PtdIns(3)P site is evident from mapping the chemical shift perturbations onto the PX domain surface (Fig. 1). Major perturbations occur in the β1 and β2 strands, the proline-rich loop between α1 and α2, and the α1 and α2 helices. The residues showing the largest changes—Tyr42, Ser43, Trp46, and Glu66—are clustered together in a deep pocket that is able to accommodate the PtdIns(3)P headgroup.
Figure 1.
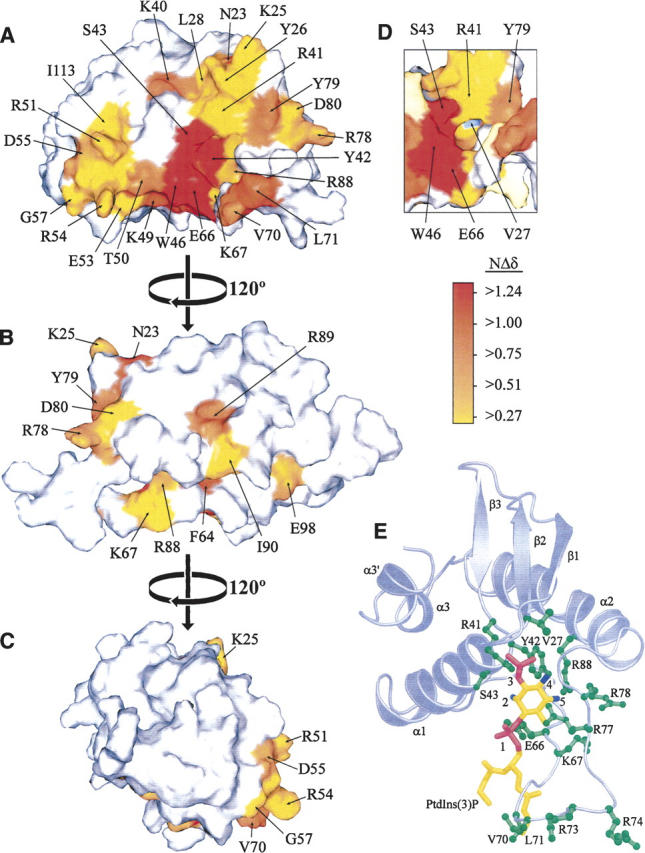
The PtdIns(3)P binding surface of the Vam7p PX domain is identified by chemical shift changes. (A–C) Three views of the Vam7p PX domain rotated by 120° about the vertical axis. Residues are colored according to the magnitude of normalized chemical shift changes (NΔδ) that were registered upon binding C4-PtdIns(3)P, as indicated by the scale. (D) A view of the cavity underneath Arg41, colored as above with the exception of Val27, which is colored blue for increased visibility. The structural images were prepared using SPOCK (Christopher et al. 1998), and the structural coordinates are from PDB file 1KMD (Lu et al. 2002). (E) The Vam7p PX domain model is oriented to show a bound C4-PtdIns(3)P molecule with phosphate groups and hydroxyl groups colored in magenta and blue, respectively, and binding residues, in green. The secondary structure elements, PtdIns(3)P binding residues, and PtdIns(3)P groups are labeled.
Based on rigid body docking, a deep invagination lined by Val27, Arg41, and Tyr42 binds the 3-phosphate (Fig. 1E). This pocket is occupied by a free phosphate even in the absence of a ligand, as evidenced by chemical shift changes induced by inorganic phosphate (Supplemental Fig. 2), which, in lieu of high salt, was required for sample stability. The highly conserved basic residue Arg88 lies deep in a cavity where it is likely to contact the inositol ring 4- and 5-hydroxyl groups. The proximal Glu66 and Lys67 residues are positioned to interact with the lipid's 1-phosphate and 2-hydroxyl group. Residues Arg41, Lys67, and Arg77 are positioned nearby to help coordinate the phosphate groups of the ligand in a canonical fashion. The acyl chains of the C4-PtdIns(3)P molecule are positioned near MIL residues Val70 and Leu71.
Figure 2.
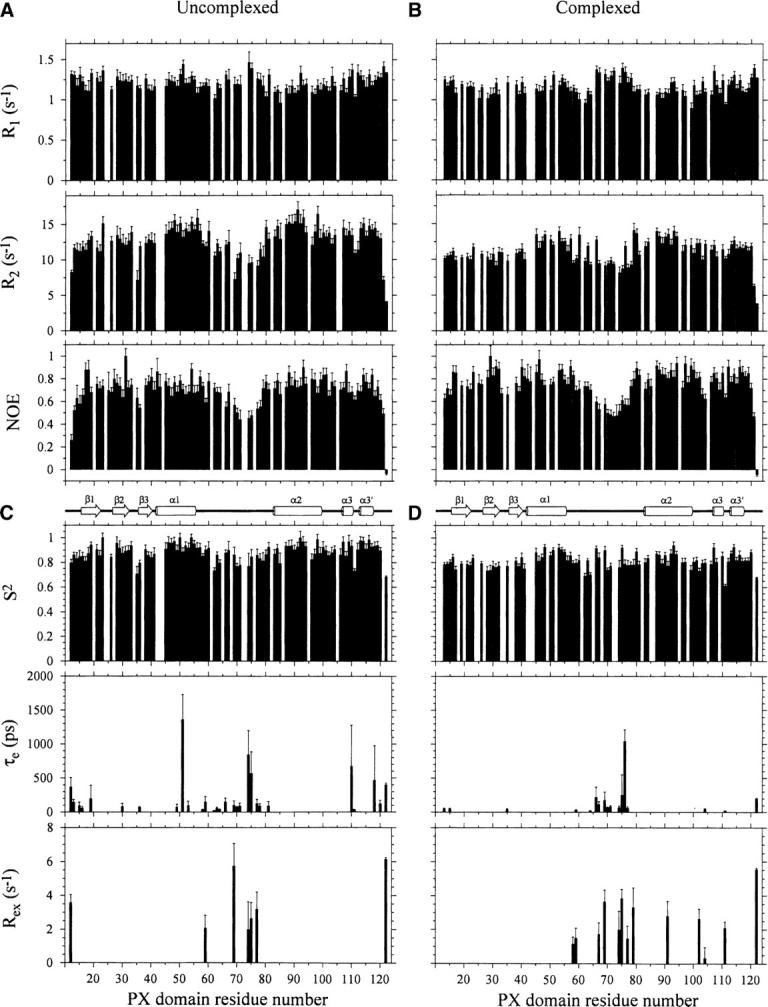
Relaxation parameters for the Vam7p PX domain (500 μM) in the absence (A) and the presence (B) of a 15-fold excess of C4-PtdIns(3)P. R1, R2, and NOE values were determined for backbone amide groups and are plotted for each residue of the protein sequence. The Vam7p PX domain secondary structure is shown below. Dynamical parameters for the Vam7p PX domain in the absence (C) and the presence (D) of a saturating concentration of C4-PtdIns(3)P. S2, τe, and Rex values for backbone amide groups are plotted.
Ligand affinity
The PtdIns(3)P-bound and -free states of the Vam7p PX domain exchange rapidly on the NMR timescale, indicating a relatively fast off-rate. Consequently, the ligand affinity could be estimated from the normalized 1H and 15N resonance perturbations induced by increasing ligand concentrations. Binding constants calculated from spectra collected at 10 C4-PtdIns(3)P concentrations yielded a Kd of 299 ± 26 μM for the interaction (Supplemental Fig. 3). This monovalent affinity would be expected to be amplified in vivo by presentation of PtdIns(3)P in a bilayer for concomitant hydrophobic insertion, as well as avidity effects afforded by protein oligomerization through Vam7p's C-terminal coiled-coil region.
Figure 3.
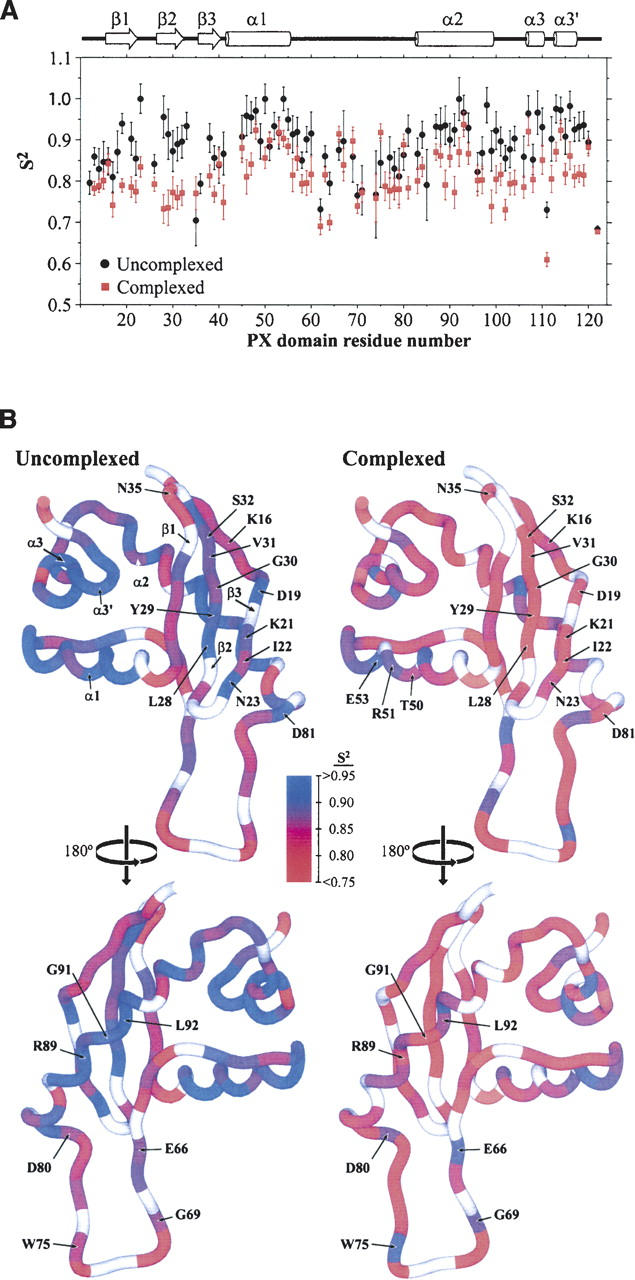
(A) Comparison of the generalized order parameter (S2) values for uncomplexed and PtdIns(3)P-complexed PX domain indicates alterations in dynamics. The S2 values for the PX domain in the absence (black) and the presence (red) of C4-PtdIns(3)P are plotted as a function of protein sequence. The Vam7p PX domain secondary structure is shown above. (B) Disorder increases in the Vam7p PX domain upon binding PtdIns(3)P. The generalized order parameter (S2) for each residue of the free (A) and PtdIns(3)P-bound (B) PX domain is color-coded on the structure according to the legend in the center. Residues having S2 values ≥0.95 are colored blue, while those with S2 values ≤0.75 are colored red. Values of S2 between 0.95 and 0.75 are indicated with a continuous color spectrum from blue to red, as indicated by the scale. Residues for which S2 could not be determined are in white. Two different 180° rotations (top and bottom) are presented for the complexed and free states, and residues in regions undergoing altered dynamics are labeled.
Relaxation rates
The effect of ligand binding on the dynamics of each residue in the Vam7p PX domain was studied using 15N NMR relaxation data collected in the absence and the presence of saturating concentrations of C4-PtdIns(3)P. For the uncomplexed protein, quantifiable peak intensity measurements from the R1, R2, and NOE spectra were obtained for 94, 93, and 95 residues, respectively. The complexed state yielded reliable R1, R2, and NOE intensity data for 89, 87, and 93 residues, respectively. The remaining 15N,1H peaks were overlapped or—in the case of Lys8, Met9, Ser10, Lys25, Tyr42, and Arg73—too weak for accurate measurement of their respective intensities.
Comparison of the average relaxation rates of the PtdIns(3)P-free and -bound Vam7p PX domain suggested a global decrease for the complex. The average R1 values of the free and bound states were 1.21 ± 0.07 and 1.16 ± 0.06 sec−1, respectively, while their R2 values were 12.73 ± 0.95 and 11.15 ± 0.57 sec−1. The R1 and R2 data together suggest that the complexed PX domain tumbles more rapidly in solution than the free state. In contrast, the heteronuclear NOE values increased from an average of 0.70 ± 0.06–0.76 ± 0.05 (Fig. 2). These values are sensitive to backbone mobility on a sub-nanosecond timescale with higher sensitivity for large amplitude motions, and indicate that generally slower internal mobility is present in the complex.
The residues in the MIL and termini exhibited anomalous relaxation rates and NOE values. In particular, there was a marked decrease in R2 values for MIL residues between Ser58 and Tyr79, as well as for terminal residues Lys12, Ser121, and Lys122, either in the absence or the presence of PtdIns(3)P. This was mirrored by a small increase in R1 values in these residues in both states. Reduced NOE values were observed for not only the expected termini but also MIL residues including Val70, Leu71, Asp72, Arg73, and Arg74, in sharp contrast with the bordering α-helices on either side. Thus this element, which is positioned to insert deep into the membrane, is also the most dynamic part of the structural domain in either the PI-complexed or -free state.
Molecular tumbling and shape
Protein tumbling can be gauged from the overall correlation time (τm) and revealed that the ligand-bound PX domain tumbles faster than the ligand-free state. The τm values were estimated to be 10.0 ± 0.5 nsec and 9.5 ± 0.4 nsec for the free and C4-PtdIns(3)P-bound PX domain, respectively, based on an axially symmetric diffusion model using all R2/R1 ratios within 1 standard deviation of the average. The coordinates of the Vam7p PX domain (Lu et al. 2002) were used to calculate an inertial tensor with principal components of 1.00:0.864:0.470. This shape was confirmed by analysis of the R2/R1 values, which indicate that the Vam7p PX domain can best be represented by the axially symmetric model, with rotational diffusion tensors D‖/D⊥ of 1.28 ± 0.05 and 1.38 ± 0.04 for the ligand-free and -bound forms, respectively. This infers that the complex not only tumbles more rapidly in solution, but its shape also deviates farther from spherical symmetry.
Model-free analysis
The best dynamical models for fitting the relaxation data of each residue were determined using a statistical approach with distinct parameter sets (Mandel et al. 1995). The simplest model using only the square of the generalized order parameter (S2) could adequately fit relaxation data for most residues in both states. However, more residues in the uncomplexed PX domain required an effective internal correlation time (τe) for fitting, particularly those in the α1 helix, the MIL, and the termini, indicating significant internal motions. Similarly, several MIL residues also required a nonzero conformational exchange parameter (Rex) to accurately describe their dynamics, both in the uncomplexed and the complexed states, reflecting motions on the microsecond-to-millisecond (μsec–msec) timescale. The microdynamic parameters S2, τe, and Rex describe the mobility of the backbone NH vectors, the correlation time of local motion, and the conformational exchange contribution to the transverse relaxation, respectively, and were calculated for each residue using the appropriate spectral density model (see Supplemental Tables).
Generalized order parameters
The PX domain appears to exhibit higher backbone mobility upon PtdIns(3)P binding. The average S2 value, which represents the backbone mobility within the picosecond-to-nanosecond (psec–nsec) timescale, was 0.89 ± 0.07 in the unbound PX domain, while the average S2 value was 0.81 ± 0.06 for the complex. All residues had lower S2 values in the PtdIns(3)P-bound state except for Asn35, Arg51, Glu53, Glu66, Gly69, Trp75, and Asp80 (Fig. 3), indicating that mobility is induced upon ligand binding and that this occurs primarily in secondary structure elements. Of the few residues that showed larger S2 values in the bound state, Asn35 is in a loop far from the binding site, and the rest are scattered across the MIL.
As expected, the largest S2 values were found in the secondary structure elements. In the ligand-free state, order parameters which significantly over the average were observed in Asn23, α1 (Leu48, Thr50, and Arg54), α2 (Leu92, Glu93, Glu94, and Glu98), α3 (Arg107 and Arg109), and α3′ (Ile113, Ala114, and Asp116). In the complex, significantly elevated order was found in α1 (Leu48, Arg51, Glu53, Arg54, and Asp55), MIL (Glu66, Gly69, Trp75, and Asp80), α2 (Glu87, Leu92, and Glu93), α3 (Arg107), α3′ (Ile113 and Ala114), and Leu120. In contrast, residues with lower than average S2 values include Asn35, Tyr62, Val70, Thr111, and Lys122 in the ligand-free form and Tyr62, Phe64, Thr111, and Lys122 in the bound state. Thus, the relative pattern of rigid helices and flexible loop elements generally correlate between the ligand-free and -bound states.
The PX domain elements that exhibit distinct degrees of backbone mobility depending on ligand occupancy are shown in Figure 3B. The α1 and α2 helices display elevated S2 values in both the PtdIns(3)P-free and -bound states, while PtdIns(3)P binding renders the more remote α3 helix slightly less mobile upon PtdIns(3)P binding. However, the most striking difference between the free and bound states is seen in the β1–β2 hairpin. In particular, these β-strands have higher S2 values than the loops in the free state, but this relative pattern is reversed in the bound form. The relative increase in the S2 values is particularly pronounced in several of the central MIL residues nearest the ligand. In summary, PtdIns(3)P binding induces a net increase in mobility across the PX domain, particularly in the β1–β2 hairpin and α3 helix, and is balanced by slight increases in the order parameters of several MIL residues.
Discussion
Structural mode of PtdIns(3)P binding by PX domains
Three-dimensional structures have been solved of several PX domains, including two complexed with C4-PtdIns(3)P (Bravo et al. 2001; Zhou et al. 2003) and six without (Cheever et al. 2001; Hiroaki et al. 2001; Karathanassis et al. 2002; Zhou et al. 2003; Xing et al. 2004; Zhong et al. 2005). All PX structures have similar folds yet possess significant differences in their ligand specificities and binding pockets. The congruence of the structural interactions in PX domains with the PI headgroup suggests a consensus binding mode among those with canonical specificity for PtdIns(3)P, including in the case of Vam7p (Fig. 1E). Membrane interactions are particularly complex, with the long disordered loop between α1 and α2 that interacts with membranes (Cheever et al. 2001; Stahelin et al. 2003) being variable in sequence and structure, and needing to reorient to bind the PI headgroup, as described below.
Global conformation changes induced by PtdIns(3)P
The ligand-bound PX domain appears to tumble faster in solution than the uncomplexed form based on its lower R1 and R2 rates, and the corresponding decrease in the τm value. Three factors that influence a protein's diffusion rate are mass, shape, and solution viscosity. Binding of a PtdIns(3)P molecule would slightly increase the overall mass, yet τm was decreased rather than increased. Similarly, PtdIns(3)P addition would be expected to increase viscosity and, hence, also τm. Thus the observed decrease in τm is instead most likely due to a different shape induced in the PX domain. The most obvious structural element that could cause a shape change is the large and dynamic MIL, which protrudes away from the globular PX domain in the uncomplexed state.
Insights can be drawn by comparison of the Vam7p PX domain structure with the free and PtdIns(3)P-bound Grd19p PX structures (Zhou et al. 2003), despite the greater length of the MIL in Vam7p. Superposition of the structures reveals that Vam7p MIL residues Glu66, Lys67, and Arg77 would have to reposition to engage PtdIns(3)P (Fig. 1E). In Grd19p, this loop undergoes large conformational changes upon PtdIns(3)P binding and moves 3.5 Å to interact with the 1-phosphate group. In the case of sorting nexin 1’s PX domain, the increased mobility of its MIL is removed upon PtdIns(3)P interaction (Zhong et al. 2005). Together, this suggests a mechanism whereby the MIL is drawn into position for PtdIns(3)P headgroup binding, inducing a more rapidly tumbling structure in PX domains.
β-Sheet disorder induced by PtdIns(3)P
Ligand binding reduces the average order parameter of the PX domain backbone, indicating a general increase in the amplitude of psec–nsec timescale motions. Interestingly, the largest decreases in S2 values are found in the β1 and β2 strands and the connecting loop. Residues here, including Asp19, Asn23, and Leu28, have some of the highest S2 values in the uncomplexed protein but exhibit relatively low values when PtdIns(3)P is bound. It is noted that interpretation of these calculated S2 values may be complicated by possible chemical exchange effects of weak phosphate interactions by the PtdIns(3)P-free protein. Residues Arg89, Gly91, and Leu92 in α2 contact the β1 and β2 strands and also have substantially lower S2 values in the PtdIns(3)P-bound complex. Moreover, the dynamics of Gly91 in the bound state also required a new Rex term, suggesting it has increased mobility in both the psec–nsec and μsec–msec timescales. Residues Tyr79, Asp80, and Asp81 also pack against β1 and undergo altered dynamics upon PtdIns(3)P binding. For example, to fit the Tyr79 dynamics, a τe term was required. Asp80 is one of the few residues with an increase in S2 value upon PtdIns(3)P binding, while the opposite effect is seen with Asp81. Thus the increase in the backbone mobility of the β1–β2 hairpin coincides with complex dynamical changes in structurally proximal residues.
Many of the β1–β2 residues also experience substantial chemical shift perturbations during titrations with PtdIns(3)P (Supplemental Fig. 1). The β2 residue Val27 is crucial as it lines the predicted 3-phosphate binding pocket (Fig. 1). The positioning of this hydrophobic residue adjacent to the charged phosphate group of a bound PtdIns(3)P molecule would be energetically unfavorable. Thus, 3-phosphate insertion could disrupt hydrophobic packing in the β1 and β2 strands, contributing to the observed increase in the amplitude of the psec–nsec backbone mobility of Vam7p. Indeed, occupancy of this pocket by inorganic phosphate alone is sufficient to perturb β1 residues Lys16, β2 residues Val27 and Leu28, and proximal Tyr42 (Supplemental Fig. 2).
Ligand-induced change in the dynamics of Vam7p's β1–β2 hairpin are consistent with structural studies of other PX domains. The crystal structure of the CISK PX domain shows a reorientation of this β1–β2 loop when a sulfate ion is bound in place of the 3-phosphate site (Xing et al. 2004). The homologous loop in Grd19p shows significant conformational change between the unbound and C4-PtdIns(3)P-bound crystal structures, although this element also forms one of the crystal packing interfaces (Zhou et al. 2003). The loop between β1 and β2 of the uncomplexed PX domain of sorting nexin 1 exhibits high frequency mobility (Zhong et al. 2005). Interestingly, all of the PX domain structures indicate that there is a β-bulge involving the residues that correspond to Vam7p's β1 residues Asp18 and Asp19. This bulge twists the β-sheet so that β2 is positioned toward the binding pocket, and these residues experience chemical shift changes that suggest an involvement in the induced dynamics of β1 and β2 residues upon PtdIns(3)P binding.
Binding pocket dynamics affected by PtdIns(3)P occupancy
Diverse effects of ligand binding were observed for residues that directly contact PtdIns(3)P in the model of the complex (Fig. 1E). Residues Arg41 and Arg88 near the 3-phosphate and 4/5-OH groups, respectively, were fit using the simplest spectral density model, suggesting that their internal motions are restricted to timescales <20 psec. Only a small S2 decrease was seen for Arg88, while Arg41’s value dropped precipitously, indicating a larger amplitude motion for this residue upon PtdIns(3)P binding. For residues Tyr42 and Ser43, NH signal intensity was too weak for reliable quantitation, suggesting conformational exchange on the intermediate NMR timescale.
Direct contacts with the PtdIns(3)P headgroup by MIL residues Glu66, Lys67, Arg77, and Arg78 are anticipated. Of these residues, ligand binding increased the S2 value of Glu66, which is near the 1-phosphate, while order in the other residues decreased. Consistently significant internal motions in Glu66 and Arg77 are suggested by their requirement for τe terms in either state, while τe was only necessary for Lys67 fitting in the bound state and for Arg78 when unbound. The Arg77 dynamics required Rex terms in both states, indicating motions on the chemical exchange timescale, but Rex was only required by Lys67 fitting in the bound state, suggesting an induced increase in its μsec–msec timescale motions. Thus, ligand binding has complex and specific effects on the motional properties of individual residues in the binding site, rather than inducing a single concerted dynamic effect.
Ligand binding increases the overall disorder as well as mobility of binding site residues in proteins (Farrow et al. 1994; Zidek et al. 1999). This increase in entropy is thought to help offset the entropic cost associated with ligand binding (Zidek et al. 1999; Stone 2001), suggesting an explanation for the diverse dynamic effects induced by PtdIns(3)P in the PX domain's residues and justifying future energetic studies. PtdIns(3)P is distinguished from small molecule ligands studied by its amphiphilic structure, bilayer setting, and the extensive interactions that link transient lipid headgroup recognition with stable membrane insertion. We suggest that lipid-induced mobility in peripheral membrane domains could play a generally important role in phospholipid recognition and the concomitant protein insertion into the membrane.
Slowing of MIL dynamics by PtdIns(3)P
The most unusual dynamic element of the PX domain is the MIL. This long and variable loop exhibits substantially increased R1 and decreased R2 and NOE values, indicating substantially increased dynamics both in the absence and the presence of PtdIns(3)P. In the uncomplexed state MIL residues Glu59, Gly69, Arg74, Trp75, and Arg77 undergo motions on the μsec–msec chemical exchange timescale, as evidenced by the requirement of their relaxation data for Rex terms. These residues—as well as Ser58, Thr62, Glu63, Phe64, Glu66, Val70, Leu71, Arg78, and Tyr79—all required a τe term, indicating significant internal mobility. Enhanced mobility on the psec–nsec timescale is also evident from the lower S2 values of Tyr62, Phe64, Val70, Leu71, and Arg74. The increased mobility of the unbound Vam7p MIL backbone is supported by its poor structural convergence in the solution structure (Lu et al. 2002) and may also be influenced by the presence of proline residues at positions 61, 65, 68, and 82. Together, this indicates that the dynamics of the MIL include motions with multiple timescales and amplitudes, and these motions differ substantially from those seen throughout the rest of the unbound Vam7p PX domain.
The dynamics of the MIL in the PtdIns(3)P-bound state have several significant differences from those of the uncomplexed protein. For example, residues Asn35, Glu66, Gly69, Trp75, and Glu80 were unique in exhibiting slight increases in S2 values on binding. These changes become more noteworthy when viewed in the context of the overall decrease of the order parameters of the complex (Fig. 3), and suggest a significant relative decrease in their psec–nsec timescale mobility. In contrast, MIL residues such as Tyr62 and Phe64 had lower S2 values on binding, indicating increased mobility. These induced dynamic changes are reflected by induced chemical shift changes, which are particularly pronounced for Phe64, Glu66, Val70, and Leu71, indicating a significant conformational change here. By analogy, MIL residues of sorting nexin 1 exhibit a high-frequency mobility that is reduced by the inherently weak PtdIns(3)P interaction of its PX domain (Zhong et al. 2005). Changes in MIL conformation are also evident by X-ray crystallography. The crystal structure of the Grd19p PX domain reveals that the electron density of two MIL leucines is unclear in the C4-PtdIns(3)P-bound state, suggesting local disorder (Zhou et al. 2003). Their corresponding electron density is well defined in the crystal structure of the unbound state, although comparison is obscured by crystal contacts involving its MIL.
A key MIL residue in Vam7p is Lys67, which is predicted to bind the 1-phosphate group by its structural analogy to Lys92 in p40phox (Bravo et al. 2001) and Lys112 in Grd19p (Zhou et al. 2003). However, Lys67’s side chain is turned away from the docked PtdIns(3)P molecule in the ligand-free Vam7p PX domain (Lu et al. 2002) and would need to be reoriented to participate directly (Fig. 1E). The Lys67 residue required a τe term in fitting its relaxation data only in the complexed state, consistent with such a reorientation. Similarly, not only Lys67 but also MIL residues Ser58 and Tyr79 required an Rex term to accurately fit their relaxation data only in the complexed state. The decrease in S2 values for Lys67 and Tyr79 may result from inclusion of the anti-correlated Rex term in the model-free analysis for these residues. Alternatively, these data suggest that the mobility of residues Ser58, Lys67, and Tyr79 increases on the slower timescale when the ligand is bound.
In summary, PtdIns(3)P binding has complex dynamic and structural effects on the PX domain. Accommodation of the 3-phosphate in a hydrophobic pocket triggers an increase in mobility in the β1–β2 hairpin, which forms a variable surface loop on the protein that is positioned away from the membrane surface. Greater mobility was also found in the nearby α3 helix, which leads to Vam7p's helical bundle oligomerization domain. Ligand engagement by the MIL induced a more rapidly tumbling shape in the structure, with reduced fast motions and increased slow motions in residues positioned to insert deep into the bilayer. We propose that these induced dynamic changes compensate for specific stabilizing PtdIns(3)P contacts in the binding pocket and provide an optimal interface for stable and regulated insertion into the surrounding membrane by the PX domain.
Acknowledgments
We thank F. Dancea, T. de Beer, D. Jones, K. Kami, and C. Ludwig for discussions, and R. Muhandiram and L.E. Kay for NMR pulse sequences. This work was supported by grants from the National Institutes of Health (M.O. and T.G.K.), BBSRC and Wellcome Trust (M.O.), and by the University of Colorado Health Sciences Center's NMR Facility.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Michael Overduin, CR-UK Institute for Cancer Studies, School of Medicine, University of Birmingham, Birmingham B15 2TT, UK; e-mail: M.Overduin@bham.ac.uk; fax: +44 (0) 121 414 4486.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062194906.
Abbreviations: HSQC, heteronuclear single quantum coherence; MIL, membrane insertion loop; NOE, nuclear Overhauser effect; NOESY, nuclear Overhauser effect spectroscopy; NMR, nuclear magnetic resonance spectroscopy; PI, phosphoinositide; PtdIns(3)P, phosphatidylinositol 3-phosphate; SNARE, soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor; TOCSY, total correlation spectroscopy.
References
- Amezcua C.A., Harper S.M., Rutter J., Gardner K.H. 2002. Structure and interactions of PAS kinase N-terminal PAS domain: Model for intramolecular kinase regulation. Structure 10: 1349–1361. [DOI] [PubMed] [Google Scholar]
- Bravo J., Karathanassis D., Pacold C.M., Pacold M.E., Ellson C.D., Anderson K.E., Butler P.J.G., Lavenir I., Perisic O., Hawkins P.T. et al. 2001. The crystal structure of the PX domain from p40(phox) bound to phosphatidylinositol 3-phosphate. Mol. Cell 8: 829–839. [DOI] [PubMed] [Google Scholar]
- Bruschweiler R., Liao X., Wright P.E. 1995. Long-range motional restrictions in a multidomain zinc-finger protein from anisotropic tumbling. Science 268: 886–889. [DOI] [PubMed] [Google Scholar]
- Cheever M.L. and Overduin M. 2005. PX domain. In Modular signaling domains (eds. Cesareni G. et al.) . . Wiley, New York.
- Cheever M.L., Sato T.K., de Beer T., Kutateladze T.G., Emr S.D., Overduin M. 2001. Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat. Cell Biol. 3: 613–618. [DOI] [PubMed] [Google Scholar]
- Collins K.M., Thorngren N.L., Fratti R.A., Wickner W.T. 2005. Sec17p and HOPS, in distinct SNARE complexes, mediate SNARE complex disruption or assembly for fusion. EMBO J. 24: 1775–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beer T., Fang J., Ortega M., Yang Q., Maes L., Duffy C., Berton N., Sippy J., Overduin M., Feiss M. et al. 2002. Insights into specific DNA recognition during the assembly of a viral genome packaging machine. Mol. Cell 9: 981–991. [DOI] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Dietrich L.E., Boeddinghaus C., LaGrassa T.J., Ungermann C. 2003. Control of eukaryotic membrane fusion by N-terminal domains of SNARE proteins. Biochim. Biophys. Acta 1641: 111–119. [DOI] [PubMed] [Google Scholar]
- Farrow N.A., Muhandiram R., Singer A.U., Pascal S.M., Kay C.M., Gish G., Shoelson S.E., Pawson T., Forman-Kay J.D., Kay L.E. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33: 5984–6003. [DOI] [PubMed] [Google Scholar]
- Hiroaki H., Ago T., Ito T., Sumimoto H., Kohda D. 2001. Solution structure of the PX domain, a target of the SH3 domain. Nat. Struct. Biol. 8: 526–530. [DOI] [PubMed] [Google Scholar]
- Johnson B.A. and Blevins R.A. 1994. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4: 603–614. [DOI] [PubMed] [Google Scholar]
- Karathanassis D., Stahelin R.V., Bravo J., Perisic O., Pacold C.M., Cho W., Williams R.L. 2002. Binding of the PX domain of p47phox to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J. 21: 5057–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L.K., Rance M., Chazin W.J., Palmer III A.G. 1997. Rotational diffusion anisotropy of proteins from simultaneous analysis of 15N and 13Cα nuclear spin relaxation. J. Biomol. NMR 9: 287–298. [DOI] [PubMed] [Google Scholar]
- Lu J., Garcia J., Dulubova I., Sudhof T.C., Rizo J. 2002. Solution structure of the Vam7p PX domain. Biochemistry 41: 5956–5962. [DOI] [PubMed] [Google Scholar]
- Mandel A.M., Akke M., Palmer III A.G. 1995. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 246: 144–163. [DOI] [PubMed] [Google Scholar]
- Mandel A.M., Akke M., Palmer III A.G. 1996. Dynamics of ribonuclease H: Temperature dependence of motions on multiple time scales. Biochemistry 35: 16009–16023. [DOI] [PubMed] [Google Scholar]
- Palmer III A.G., Rance M., Wright P.E. 1991. Intramolecular motions of a zinc finger DNA-binding domain from Xfin characterized by proton-detected natural abundance 13C heteronuclear NMR spectroscopy. J. Am. Chem. Soc. 113: 4371–4380. [Google Scholar]
- Stahelin R.V., Burian A., Bruzik K.S., Murray D., Cho W. 2003. Membrane binding mechanisms of the PX domains of NADPH oxidase p40phox and p47phox. J. Biol. Chem. 278: 14469–14479. [DOI] [PubMed] [Google Scholar]
- Stone M.J. 2001. NMR relaxation studies of the role of conformational entropy in protein stability and ligand binding. Acc. Chem. Res. 34: 379–388. [DOI] [PubMed] [Google Scholar]
- Tjandra N., Feller S.E., Pastor R.W., Bax A. 1995. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J. Am. Chem. Soc. 117: 12562–12566. [Google Scholar]
- Xing Y., Liu D., Zhang R.G., Joachimiak A., Songyang Z., Xu W.Q. 2004. Structural basis of membrane targeting by the phox homology domain of cytokine-independent survival kinase (CISK-PX). J. Biol. Chem. 279: 30662–30669. [DOI] [PubMed] [Google Scholar]
- Zhong Q., Watson M.J., Lazar C.S., Hounslow A.M., Waltho J.P., Gill G.N. 2005. Determinants of the endosomal localization of sorting nexin 1. Mol. Biol. Cell 16: 2049–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.Z., La Sierra-Gallay I.L., Quevillon-Cheruel S., Collinet B., Minard P., Blondeau K., Henckes G., Aufrere R., Leulliot N., Graille M. et al. 2003. Crystal structure of the yeast Phox homology (PX) domain protein Grd19p complexed to phosphatidylinositol-3-phosphate. J. Biol. Chem. 278: 50371–50376. [DOI] [PubMed] [Google Scholar]
- Zidek L., Novotny M.V., Stone M.J. 1999. Increased protein backbone conformational entropy upon hydrophobic ligand binding. Nat. Struct. Biol. 6: 1118–1121. [DOI] [PubMed] [Google Scholar]