

Abstract
There is strong evidence for a genetic predisposition to autism and an intense interest in discovering heritable risk factors that disrupt gene function. Based on neurobiological findings and location within a chromosome 7q31 autism candidate gene region, we analyzed the gene encoding the pleiotropic MET receptor tyrosine kinase in a family based study of autism including 1,231 cases. MET signaling participates in neocortical and cerebellar growth and maturation, immune function, and gastrointestinal repair, consistent with reported medical complications in some children with autism. Here, we show genetic association (P = 0.0005) of a common C allele in the promoter region of the MET gene in 204 autism families. The allelic association at this MET variant was confirmed in a replication sample of 539 autism families (P = 0.001) and in the combined sample (P = 0.000005). Multiplex families, in which more than one child has autism, exhibited the strongest allelic association (P = 0.000007). In case-control analyses, the autism diagnosis relative risk was 2.27 (95% confidence interval: 1.41–3.65; P = 0.0006) for the CC genotype and 1.67 (95% confidence interval: 1.11–2.49; P = 0.012) for the CG genotype compared with the GG genotype. Functional assays showed that the C allele results in a 2-fold decrease in MET promoter activity and altered binding of specific transcription factor complexes. These data implicate reduced MET gene expression in autism susceptibility, providing evidence of a previously undescribed pathophysiological basis for this behaviorally and medically complex disorder.
Keywords: autism spectrum disorder, association, candidate gene, hepatocyte growth factor, hepatocyte growth factor receptor
Autism is a complex, behaviorally defined neurodevelopmental disorder characterized by social deficits, language impairments, and repetitive behaviors with restricted interests. There has been a dramatic increase in the diagnosis of autism (1). The population prevalence of autism is debated; recent reports indicate that ≈1 in 500 individuals have autism and as many as 1 in 166 individuals have an autism spectrum disorder (ASD) (1, 2). Here, we broadly use the term “autism” to refer to any individual with ASD; for the purposes of our genetic analyses, we use a binary code to designate as “affected” any individual diagnosed with autism or ASD and “unaffected” any individual lacking such a diagnosis. The etiology of this complex disease likely involves environmental factors, but autism is highly heritable. Twin studies demonstrate concordance rates of 82–92% in monozygotic twins and 1–10% concordance rate in dizygotic twins (3). Sibling recurrence risk (6–8%) is 35 times the population prevalence (1, 4), making autism among the most heritable of all neuropsychiatric disorders.
The most widely accepted hypotheses regarding autism etiology include oligogenic inheritance and epistatic interactions among common vulnerability-conferring genetic variants and, possibly, gene–environment interactions. Genomewide linkage scans, an unbiased approach to localize genetic factors, have identified several chromosomal regions as promising locations for autism vulnerability genes, including peaks on chromosomes 2q, 7q, 15q, and 17q (5–8). This genetic approach to identify susceptibility genes is very powerful, but the heterogeneity present within autism families has led thus far to mixed success in identifying candidate genes (9, 10). In pursuing specific candidates, most studies have focused on genes expressed predominantly in the brain (11, 12). Our own evaluation of linkage peaks extended beyond genes with selective brain expression to consider the complex medical conditions seen in autism patients. In addition to the well known behavioral core features, some individuals with autism exhibit gastrointestinal, immunological, or nonspecific neurological symptoms (13–15). Although the degree to which individuals with autism exhibit more medical complications compared with typical individuals is debated, it is possible that autism vulnerability could include genes involved more broadly in multiple biological processes that impact the development and function of the brain and other organ systems in parallel.
We applied the convergence of developmental biological and genetic information to analyze the gene encoding the MET receptor tyrosine kinase (OMIM 164860; GenBank accession NM_000245; chromosome 7q31) as an autism candidate gene. MET is best understood for its role in the metastasis of a variety of cancers (16), due to somatic gain-of-function mutations, and in mediating hepatocyte growth factor (HGF)/scatter factor signaling in peripheral organ development and repair (17–19). MET signaling contributes to immune function (20–22) and gastrointestinal repair (18, 23, 24). Recent studies by our group and others revealed that MET also contributes to development of the cerebral cortex (25, 26) and cerebellum (27), both of which exhibit developmental disruptions in autism (28, 29). Hypomorphic MET/HGF signaling in the cerebral cortex results in abnormal interneuron migration from the ganglionic eminence and reduced interneurons in the frontal and parietal regions of cortex (25, 26). Hypomorphic MET/HGF signaling in the cerebellum causes decreased proliferation of granule cells and a concomitant reduction in the size of the cerebellum, particularly in the vermis (27). Both of these neuropathologic abnormalities are consistent with those observed in brains of individuals with autism (28, 29). We therefore pursued MET as an autism candidate gene.
Results
Screen of the MET Gene for Variants in Autism.
To identify variants in the MET gene, we screened the 21 exons and key regulatory regions of the gene in 86 individuals with autism by using temperature gradient capillary electrophoresis and direct resequencing. Primers used to amplify the exonic regions of the MET gene are listed in Table 2, which is published as supporting information on the PNAS web site. Two rare nonsynonymous variants were identified in exon 14: C3095T, a nonconserved arginine-to-cysteine substitution at amino acid 988 (R988C), and C3162T, a threonine-to-isoleucine substitution at amino acid 1010 (T1010I). These same variants were reported in small cell lung cancer cell lines as functional somatic mutations (30). We directly resequenced 277 cases and 319 unrelated controls (Table 3, which is published as supporting information on the PNAS web site) to determine the frequencies of the two nonsynonymous exon 14 variants. For each of the variants, R988C and T1010I, the rare allele was present in five cases (1.8%) and two controls (0.6%). These differences are not significant either for genotypic (χ2 = 1.773; df = 2; P = 0.412) or for allelic frequencies (χ2 = 1.762; df = 1; P = 0.184). Thus, there is no genetic evidence to support autism association for these rare alleles. Synonymous SNPs were identified in exon 2 (rs11762213), exon 7 (rs13223756), exon 20 (rs41736), and exon 21 (rs2023748 and rs41737). The initial screen also identified variants in the promoter region (rs184953 and rs1858830) and a variant (rs41739) in the 3′ untranslated region of the MET gene.
Family Based Association Analyses.
To determine a possible association between MET and autism, we tested for transmission disequilibrium in a two-stage study design by using nine markers that span the entire MET locus (Fig. 1). SNPs were genotyped in an original sample consisting of 204 families (178 simplex and 26 multiplex), followed by a replication sample of 539 families (87 simplex and 452 multiplex) (Table 3). Analysis of intermarker linkage disequilibrium (LD) revealed that MET contains two distinct LD blocks (Fig. 1): a 17-kb block at the 5′ end of the gene and an expansive 110-kb block that includes exons 2–21, the entire coding region of the MET gene. Transmissions of haplotypes within each LD block were examined with the haplotype-based association test (HBAT) (31). HBAT analysis revealed significant transmission distortion in LD block 1 (χ2 = 17.521; df = 6; P = 0.008), indicating the presence of an autism-associated variant in the MET promoter region.
Fig. 1.

MET locus genomic structure, genotyping markers, and definition of haplotype blocks. The MET locus consists of 21 exons spanning 125-kb on chromosome 7q31. Nine SNPs spanning the MET locus were chosen to perform association studies and Taqman Assays-on-Demand were used to determine genotype. The nine genotyping markers defined two distinct linkage disequilibrium blocks. Pairwise linkage disequilibrium (D′) values are indicated. Pairwise r2 values are provided in Table 1, which is published as supporting information on the PNAS web site.
The LD block 1 haplotype association supported the possibility of identifying a variant that disrupts MET gene regulation. We thus examined transmissions of single markers by using the family based association test (FBAT) (32). Parent-to-affected offspring transmissions observed (TOBS) were compared with transmissions expected (TEXP), generating a P value representing the probability of observing the transmission disequilibrium by chance. We observed a significant overtransmission of the rs1858830 C allele to affected individuals (Fig. 2; see Tables 4–6, which are published as supporting information on the PNAS web site). Transmission disequilibrium for rs1858830 under a dominant model was significant in both the original 204-family sample (TOBS = 81, TEXP = 65, P = 0.0005) and in the replication sample of 539 families (TOBS = 225, TEXP = 198, P = 0.001); combined analysis of these samples was highly significant (TOBS = 306, TEXP = 263, P = 0.000005) (Fig. 2a). The rs1858830 variant is a common G/C SNP, situated just 20 bp 5′ to the MET transcriptional start site, and promoter variants often function as dominant mutations (33). However, FBAT analyses using an additive model yielded similar results: the original sample showed significant association (TOBS = 139, TEXP = 119, P = 0.006), a trend in the replication sample (TOBS = 490, TEXP = 464, P = 0.072) and again a significant association in the combined samples (TOBS = 629, TEXP = 583, P = 0.005) (Fig. 2b). Among the markers in LD with rs1858830, only the most informative based on allele frequency (rs437; minor allele frequency 0.295; pairwise r2 = 0.270) was significantly associated with autism; less informative markers (rs184953 and rs40238; minor allele frequency <0.178; pairwise r2 to rs1858830 < 0.164; Table 1) failed to show an association. No other marker consistently reached significant transmission disequilibrium after corrections for multiple comparisons (Fig. 2; Tables 4–6). Examination of transmissions of all nine markers to unaffected siblings showed no significant transmission distortion, indicating that the overtransmission of the rs1858830 C-allele is specific to autism and not due to altered viability.
Fig. 2.

Plots of FBAT and HBAT P values. Plotted are log10 P values for overtransmitted alleles (points) and global haplotype analyses (lines). Significance thresholds for Bonferroni corrected P values (P = 0.025) are indicated. (a) FBAT dominant model: MET promoter variant rs1858830 (marker 3) allele C was overtransmitted to individuals with autism in the original sample (P = 0.00005), replication sample (P = 0.001), and combined sample (P = 0.000005). (b) FBAT and HBAT additive model: MET promoter variant rs1858830 (marker 3) allele C was overtransmitted to individuals with autism in the original sample (P = 0.006) and combined sample (P = 0.005). Global haplotype analyses indicated significant transmission disequilibrium (P = 0.008) in LD block 1, which includes rs1858830. (c) FBAT and HBAT additive model: MET promoter variant rs1858830 (marker 3) allele C was overtransmitted to individuals with autism in multiplex families (P = 0.001) but not simplex families (P = 0.886). A marker in linkage disequilbrium with rs1858830 (rs437; marker 1) exhibited significant transmission disequilibrium in multiplex families (P = 0.009) but not in simplex families (P = 0.377). Global haplotype analyses indicated transmission disequilibrium in LD block 1 in multiplex families (P = 0.007) and in simplex families (P = 0.022).
To further understand the heritability of the MET promoter allele in our large sample (1,231 individuals with autism from 743 families), we examined the 265 simplex (one affected child, irrespective of number of siblings) and 478 multiplex (more than one affected child) families independently (Fig. 2c; Tables 7 and 8, which are published as supporting information on the PNAS web site). Within the framework of a genetically complex, heterogeneous, polygenic disorder like autism, common genetic predisposing factors are likely to be enriched in multiplex families, whereas a fraction of the simplex family cases are more likely to be caused by rare chromosomal rearrangements or other sporadic events (34). Association of the rs1858830 C allele was restricted to multiplex families, with simplex families displaying no association under either the dominant model (multiplex: TOBS = 236, TEXP = 198, P = 0.000007; simplex: TOBS = 70, TEXP = 65, P = 0.202) or the additive model (multiplex: TOBS = 494, TEXP = 447, P = 0.001; simplex: TOBS = 135, TEXP = 136, P = 0.886). In addition, we used the Autism Genetic Resource Exchange database to identify 299 individuals with autism from 182 families who are positive for a narrow diagnosis, based on Autism Diagnostic Interview-Revised, Autism Diagnostic Observational Schedule, and a clinical diagnosis. The rs1858830 C allele was significantly overtransmitted to individuals with narrowly defined autism (TOBS = 94, TEXP = 75, P = 0.0002) (Table 9, which is published as supporting information on the PNAS web site). For comparison, exclusion of the 182 families with narrow diagnosis from the entire 743-family sample resulted in a significant but somewhat weaker association (TOBS = 212, TEXP = 188, P = 0.003). Thus, both subpopulations contribute to the association of the rs1858830 C allele in the combined sample (TOBS = 306, TEXP = 263, P = 0.000005).
Case-Control Association of MET Promoter Variant rs1858830.
Genotype at the rs1858830 locus was determined in a group of 189 unrelated Italian and American control individuals. A single individual with autism was randomly selected from each of the pedigrees genotyped in the combined family based association sample. Significant differences in genotypic and allelic frequencies were detected between the individuals with autism and controls (genotypic: χ2 = 12.150; df = 2; P = 0.002; allelic: χ2 = 10.440; df = 1; P = 0.001; Table 10, which is published as supporting information on the PNAS web site). Compared with the GG genotype, the autism diagnosis relative risk was 2.27 [95% confidence interval (CI): 1.41, 3.65] for the CC genotype and 1.67 (95% CI: 1.11, 2.49) for the CG genotype when analyzing cases and unrelated controls (Table 10). This relative risk may be biologically relevant in a polygenic disease such as autism. Thus, the rs1858830 C allele is common and overrepresented in individuals with autism.
Transcription Assays.
Given the location of the rs1858830 G/C variant (20-bp 5′ to the transcription start site), we hypothesized that the associated allele would affect transcription of the MET gene. To test this hypothesis, we generated two reporter constructs containing 726 bp of the human MET promoter, differing only at the rs1858830 nucleotide, and transfected them into mouse neural cell lines N2A and SN56 and the HEK cell line. The reporter construct containing the C allele produced less than half the luciferase activity than the construct containing the G allele (P < 0.05 for each of the three cell lines; Fig. 3). The 2-fold reduction in promoter activity indicates that the autism-associated rs1858830 C allele is less efficient in driving transcription than the G allele, demonstrating that the rs1858830 variant is a functional regulatory element of MET transcription.
Fig. 3.

The autism-associated MET promoter variant rs1858830 allele C produced a 2-fold decrease in transcript. Two independent mouse neural cell lines, SN56 and N2A, and the human embryonic kidney (HEK) cell line were transfected with firefly luciferase reporter constructs carrying 762-bp of the MET promoter with either the G allele or the C allele at rs1858830. Data are presented as fold-induction compared with promoterless vector. Error bars represent SEM (n = 4). ∗, P < 0.05 compared with G allele construct by two-tailed unpaired t test.
Identification of Transcription Factors That Differentially Bind the rs1858830 Variant.
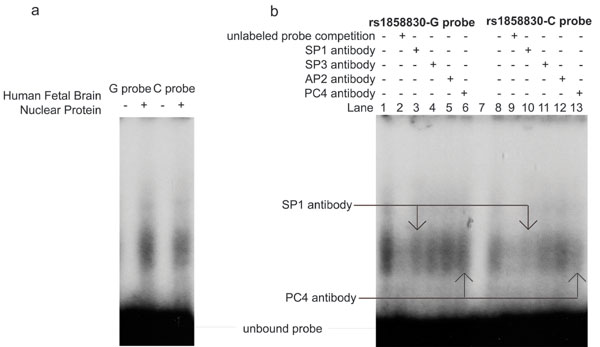
We next attempted to identify the mechanisms through which the rs1858830 MET variant might influence transcription by examining this region for transcription factor consensus sequences. The transcription factor database TRANSFAC (35) predicted that the G and C alleles would differentially bind the transcription factors SP1 and AP2 (Fig. 4a). Indeed, in EMSA, a G allele-containing oligonucleotide probe robustly bound a single protein complex in HeLa nuclear extracts, whereas an oligonucleotide probe containing the C allele weakly bound at least two protein complexes, one similar in size to that bound by the G allele oligonucleotide probe and another that migrated more slowly (Fig. 4b). EMSA with human fetal brain nuclear protein similarly showed that the G allele oligonucleotide probe bound a single transcription factor complex more robustly than the C allele probe (Fig. 5a, which is published as supporting information on the PNAS web site). To determine the specific transcription factors involved in the DNA–protein complexes, we performed supershift assays with antibodies directed to the predicted transcription factors, SP1 and AP2, as well as the SP1-family member SP3 and a transcription factor identified in a preliminary screen, PC4. Incubation of the DNA–protein complexes with specific antibodies revealed that the predominant protein in the complex is the SP1 transcription factor: Addition of SP1 antibody created a visibly supershifted band, representing a DNA–protein–antibody complex, upon incubation with the G allele oligonucleotide probe in HeLa nuclear extracts (Fig. 4c). Addition of SP1 antibody in supershift assays with the C allele oligonucleotide probe caused markedly decreased DNA–protein complex formation, indicating a specific interaction of the antibody with the DNA–protein complex. Similar results were observed in SP1-antibody supershift assays with human fetal brain nuclear protein (Fig. 5b). The antibody directed to PC4 consistently competed more effectively with the C allele probe–protein complex than with the G allele probe–protein complex (Figs. 4c and 5b), indicating a differential interaction of the PC4 transcription factor with the rs1858830 alleles.
Fig. 4.

The MET promoter variant rs1858830 alleles G and C differentially bind transcription factor complexes. (a) The double-stranded rs1858830 oligonucleotide probes used in EMSAs and supershift assays. The probes correspond to MET promoter nucleotides −35 to −6 (with zero defined as the transcription start site) and differ only at the rs1858830 locus. Predicted transcription factor binding sites are indicated (35). The rs1858830-G oligonucleotide probe is predicted to contain a single SP1-binding site, whereas the rs1858830-C probe is predicted to have two different SP1-binding sites. (b) HeLa nuclear extract EMSA revealed that rs1858830 G allele probe binds robustly a single transcription factor complex, whereas the rs1858830 C allele probe binds two transcription factor complexes. (c) HeLa cell nuclear extract supershift assays by using antibodies directed to specific transcription factors. To test the hypothesis that a DNA–nuclear protein complex contains a specific transcription factor, an antibody to the transcription factor is incubated with the complex. Observation of a slower migrating (supershifted) band representing a DNA–protein–antibody complex confirms the presence of the transcription factor. Alternatively, a reduction in the amount of the DNA–protein complex indicates that the specific transcription factor-directed antibody decreases stability of the DNA–protein complex. A supershifted band was observed upon incubation of the G allele probe–protein complex with antibody directed to the SP1 transcription factor (compare lane 1 to lane 3). Reduced DNA–protein complex was observed upon incubation of the C allele probe–protein complex with antibodies directed to transcription factors SP1 and PC4 (compare lane 8 to lanes 10 and 13). Antibodies directed to transcription factors SP3 and AP2 had moderate effects on DNA–nuclear protein complex stability (lanes 4, 5, 11, and 12). For comparison, the reduction in probe-complex formation caused by competition with 100× molar excess unlabeled probes is provided (lanes 2 and 9). Thus transcription factors SP1 and PC4 are likely regulators of MET transcription with differential binding of the rs1858830 variant alleles.
Discussion
The genetic and molecular data reported here indicate genetic association of a common, functional variant of MET with autism with a calculated relative risk of 2.27. There are several unusually attractive aspects of these findings. First, neuropathological findings in autism indicate altered organization of both the cerebral cortex and cerebellum, both of which are disrupted in mice with decreased MET signaling activity. There is co-occurrence of autism with a number of neurological and cognitive disorders, including epilepsy, atypical sleep patterns, and mental retardation (36). Together with well known dysfunction of cortical information processing, the role of MET signaling in interneuron development is relevant as a central component of the hypothesized GABAergic pathophysiological changes in autism (37). Second, the rise in autism diagnosis likely represents changing diagnostic criteria, increased awareness and an increased incidence (1, 4). Although yet to be identified environmental factors likely contribute to the development of autism, heritability studies suggest that the impact of those factors must be imposed upon individuals genetically predisposed to the disorder. Only a limited number of disease-related functional alleles have been identified to date in autism cases, and they only account for a small fraction of cases (38). We hypothesize that the common, functionally disruptive rs1858830 C allele can, together with other vulnerability genes and epigenetic and environmental factors, precipitate the onset of autism. The existence of epistatic interactions among common genetic variants at several different loci is further supported by the association between the rs1858830 C allele and autism in multiplex families and not in simplex families. Third, although admittedly still debated in terms of prevalence, individuals with autism can present complex medical profiles, such as gastrointestinal, immune, and nonspecific neurological dysfunctions (14, 15). In addition to brain development, the pleiotropic MET receptor tyrosine kinase has specific roles in digestive system development and repair (18, 23, 24) and modulation of T cell-activated peripheral monocytes and dendritic antigen-presenting cells (20, 22). We raise the possibility, still to be tested, that increased risk for autism, due to a functional polymorphism in the MET gene, may impart in certain individuals shared etiology of a parallel, although independent, disruption of brain and peripheral organ development and function. Further investigations in clinical populations will be needed to determine the contribution of the functional promoter variant of MET reported here to specific characteristics of the complex phenotype in autism.
Methods
Subjects.
Families recruited by the centers listed in Table 2 were used for this study. Clinical characterization has been described in detail in refs. 11 and 12. All research was approved by the Vanderbilt University Institutional Review Board.
Screening for Variants in the MET Gene.
Genomic DNA samples from 40 individuals with autism from the Italian sample and 46 individuals with autism from the Autism Genetic Resource Exchange Consortium were screened for exonic variants. Primers and amplification conditions used to amplify the 21 exons of the MET gene are listed in Table 2. Reveal temperature gradient capillary electrophoresis (SpectruMedix, State College, PA) was used to screen for variants in the exons of the MET gene. Amplicons identified as variant-positive then were directly resequenced to identify the variant.
SNP Genotyping.
Genotyping was performed by using TaqMan SNP Genotyping Assays on the ABI Prism 7900HT and analyzed with SDS software. Assays-On-Demand SNP Genotyping were obtained from Applied Biosystems (Foster City, CA). Eight of the nine assays provided reproducible results; the Assay-on-Demand for rs1858830 consistently failed to give reliable genotypes from genomic DNA template. Neither a TaqMan Assay-by-Design nor an Epoch Eclipse Quencher assay (Nanogen, San Diego, CA) was able to reliably provide rs1858830 genotype from genomic DNA, probably because of an inability to generate a specific amplicon within this ≈85% GC region. We therefore generated a 652-bp amplicon, including rs1858830, from genomic DNA for each sample and used separately generated 652-bp amplicons as templates for Taqman Assay-on-Demand and Epoch Eclipse Quencher genotyping assays. To ensure proper genotype calls, we also genotyped rs1858830 in each sample by using Reveal temperature gradient capillary electrophoresis (SpectruMedix). If inconsistency in any of the three indirect genotyping assays was detected, then the genotype at rs1858830 was determined by direct resequencing.
Association Analyses.
All single and haplotype association analyses were performed by using the FBAT (32) and HBAT (31) (FBAT version 1.5.5). HBAT and FBAT analyses were performed by using the empirical variance (“-e” option; Fig. 2 and Tables 4–8) because linkage has been established in the chromosomal region of the MET gene and because the empirical variance provides a more conservative estimate of association. However, little evidence for linkage at the MET locus has been reported in the samples tested here for association (Supporting Text, which is published as supporting information on the PNAS web site). Therefore, HBAT and FBAT analyses were repeated without the -e option of FBAT (Tables 11–16, which are published as supporting information on the PNAS web site). The conclusions with and without the assumption of the presence of linkage remain the same.
Corrections for Multiple Comparisons.
Appropriate corrections for multiple comparisons are an ongoing debate in human genetics. The presence of two distinct LD blocks indicates that a Bonferroni correction for multiple comparisons of two is appropriate; we consider significant only those associations with P < 0.025 (= 0.05/2). More stringent corrections for multiple comparisons are possible, but do not change the conclusions. An a priori design to independently test simplex and multiplex families as well as a post hoc decision to analyze the data by using two models of association could be argued to bring the appropriate Bonferroni correction factor to 8 (23). Thus, a very stringent correction for multiple comparisons would lead to a significance threshold of P < 0.006 (0.05/8). All associations at rs1858830 exceed this more stringent significance threshold in the original sample, combined sample, and multiplex families.
Transcription Assays.
A 762-bp fragment of the MET promoter was cloned into the pGL4.10[luc2] luciferase reporter vector (Promega, Madison, WI). Luciferase assays were conducted by using the Dual-Glo Luciferase Assay kit (Promega) according to the manufacturer's protocol.
Electrophoretic Mobility Shift and Antibody Supershift Assays.
All reactions included double-stranded, 32P-labeled oligonucleotide probe at 50,000 cpm. EMSAs were performed by using the Promega Gel Shift Assay System, according to the manufacturer's protocol. HeLa nuclear extract was purchased from Promega. Human fetal brain nuclear protein, obtained from a spontaneously aborted 22-week female fetus, was purchased from BioChain Institute, Inc. (Hayward, CA; catalog no. P2244035; lot no. A304059). Nuclear protein (5 μg) was incubated at room temperature either alone or with 100× molar excess unlabeled competitor probe for 20 min before addition of 32P-labeled probe, then incubated an additional 20 min at room temperature before loading on a 4% nondenaturing acrylamide gel. Supershift assays were performed identically except for the addition of a 60-min incubation at 4°C with 2 μg of antibody before loading on the gel.
Supporting Information.
See Supporting Text for detailed methods and Figs. 6 and 7 and Table 17, which are published as supporting information on the PNAS web site, for additional data.
Supplementary Material
Acknowledgments
We thank the patients and families participating in this study for their valuable and generous contributions. Drs. Randy Blakely, Kathleen Dennis, Bernie Devlin, Kathie Eagleson, Chun Li, Laura Lillien, Wendy Stone, and Barbara Thompson provided comments, and Shaine Jones, Cara Ballard-Sutcliffe, Denise Malone, Stefania Salamena, and Ping Mayo provided technical assistance. The Autism Genetic Resource Exchange is a program of Cure Autism Now and is supported in part by National Institute of Mental Health (NIMH) Grant MH64547 (to Daniel H. Geschwind). This work was supported in part by NIMH Grant MH65299 (to P.L.), National Institute of Child Health and Human Development Core Grant HD15052 (to P.L.), the Marino Autism Research Institute (P.L.), Telethon-Italy Grant GGP02019 (to A.M.P.), Cure Autism Now (A.M.P.), the National Alliance for Autism Research (A.M.P.), the Fondation Jerome Lejeune (A.M.P.), a National Alliance for Research on Schizophrenia and Depression Young Investigator fellowship (P.J.E.), and NIMH Grant MH61009 (to J.S.S.).
Abbreviations
- FBAT
family based association test
- HBAT
haplotype-based association test
- LD
linkage disequilibrium
- TEXP
transmissions expected
- TOBS
transmissions observed.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
See Commentary on page 16621.
References
- 1.Muhle R, Trentacoste SV, Rapin I. Pediatrics. 2004;113:e472–e486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- 2.Yeargin-Allsopp M, Rice C, Karapurkar T, Doernberg N, Boyle C, Murphy C. J Am Med Assoc. 2003;289:49–55. doi: 10.1001/jama.289.1.49. [DOI] [PubMed] [Google Scholar]
- 3.Le Couteur A, Bailey A, Goode S, Pickles A, Robertson S, Gottesman I, Rutter M. J Child Psychol Psychiatry. 1996;37:785–801. doi: 10.1111/j.1469-7610.1996.tb01475.x. [DOI] [PubMed] [Google Scholar]
- 4.Fombonne E. J Autism Dev Disord. 2003;33:365–382. doi: 10.1023/a:1025054610557. [DOI] [PubMed] [Google Scholar]
- 5.Barrett S, Beck JC, Bernier R, Bisson E, Braun TA, Casavant TL, Childress D, Folstein SE, Garcia M, Gardiner MB, et al. Am J Med Genet. 1999;88:609–615. doi: 10.1002/(sici)1096-8628(19991215)88:6<609::aid-ajmg7>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- 6.International Molecular Genetics Study of Autism Consortium. Hum Mol Genet. 2001;10:973–982. [Google Scholar]
- 7.Yonan AL, Alarcon M, Cheng R, Magnusson PK, Spence SJ, Palmer AA, Grunn A, Juo SH, Terwilliger JD, Liu J, et al. Am J Hum Genet. 2003;73:886–897. doi: 10.1086/378778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hutcheson HB, Olson LM, Bradford Y, Folstein SE, Santangelo SL, Sutcliffe JS, Haines JL. BMC Med Genet. 2004;5:12. doi: 10.1186/1471-2350-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benayed R, Gharani N, Rossman I, Mancuso V, Lazar G, Kamdar S, Bruse SE, Tischfield S, Smith BJ, Zimmerman RA, et al. Am J Hum Genet. 2005;77:851–868. doi: 10.1086/497705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma DQ, Whitehead PL, Menold MM, Martin ER, Ashley-Koch AE, Mei H, Ritchie MD, Delong GR, Abramson RK, Wright HH, et al. Am J Hum Genet. 2005;77:377–388. doi: 10.1086/433195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutcliffe JS, Delahanty RJ, Prasad HC, McCauley JL, Han Q, Jiang L, Li C, Folstein SE, Blakely RD. Am J Hum Genet. 2005;77:265–279. doi: 10.1086/432648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Persico AM, D'Agruma L, Maiorano N, Totaro A, Militerni R, Bravaccio C, Wassink TH, Schneider C, Melmed R, Trillo S, et al. Mol Psychiatry. 2001;6:150–159. doi: 10.1038/sj.mp.4000850. [DOI] [PubMed] [Google Scholar]
- 13.Valicenti-McDermott M, McVicar K, Rapin I, Wershil BK, Cohen H, Shinnar S. J Dev Behav Pediatr. 2006;27:S128–S136. doi: 10.1097/00004703-200604002-00011. [DOI] [PubMed] [Google Scholar]
- 14.Jyonouchi H, Geng L, Ruby A, Zimmerman-Bier B. Neuropsychobiology. 2005;51:77–85. doi: 10.1159/000084164. [DOI] [PubMed] [Google Scholar]
- 15.White JF. Exp Biol Med (Maywood) 2003;228:639–649. doi: 10.1177/153537020322800601. [DOI] [PubMed] [Google Scholar]
- 16.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 17.Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Proc Natl Acad Sci USA. 2004;101:4477–4482. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tahara Y, Ido A, Yamamoto S, Miyata Y, Uto H, Hori T, Hayashi K, Tsubouchi H. J Pharmacol Exp Ther. 2003;307:146–151. doi: 10.1124/jpet.103.054106. [DOI] [PubMed] [Google Scholar]
- 19.Zhang YW, Vande Woude GF. J Cell Biochem. 2003;88:408–417. doi: 10.1002/jcb.10358. [DOI] [PubMed] [Google Scholar]
- 20.Okunishi K, Dohi M, Nakagome K, Tanaka R, Mizuno S, Matsumoto K, Miyazaki J, Nakamura T, Yamamoto K. J Immunol. 2005;175:4745–4753. doi: 10.4049/jimmunol.175.7.4745. [DOI] [PubMed] [Google Scholar]
- 21.Beilmann M, Odenthal M, Jung W, Vande Woude GF, Dienes HP, Schirmacher P. Blood. 1997;90:4450–4458. [PubMed] [Google Scholar]
- 22.Beilmann M, Vande Woude GF, Dienes HP, Schirmacher P. Blood. 2000;95:3964–3969. [PubMed] [Google Scholar]
- 23.Arthur LG, Schwartz MZ, Kuenzler KA, Birbe R. J Pediatr Surg. 2004;39:139–143. doi: 10.1016/j.jpedsurg.2003.10.001. discussion 139–143. [DOI] [PubMed] [Google Scholar]
- 24.Ido A, Numata M, Kodama M, Tsubouchi H. J Gastroenterol. 2005;40:925–931. doi: 10.1007/s00535-005-1705-x. [DOI] [PubMed] [Google Scholar]
- 25.Powell EM, Mars WM, Levitt P. Neuron. 2001;30:79–89. doi: 10.1016/s0896-6273(01)00264-1. [DOI] [PubMed] [Google Scholar]
- 26.Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P. J Neurosci. 2003;23:622–631. doi: 10.1523/JNEUROSCI.23-02-00622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ieraci A, Forni PE, Ponzetto C. Proc Natl Acad Sci USA. 2002;99:15200–15205. doi: 10.1073/pnas.222362099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmen SJ, van Engeland H, Hof PR, Schmitz C. Brain. 2004;127:2572–2583. doi: 10.1093/brain/awh287. [DOI] [PubMed] [Google Scholar]
- 29.Courchesne E, Redcay E, Kennedy DP. Curr Opin Neurol. 2004;17:489–496. doi: 10.1097/01.wco.0000137542.14610.b4. [DOI] [PubMed] [Google Scholar]
- 30.Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, Johnson BE, Salgia R. Cancer Res. 2003;63:6272–6281. [PubMed] [Google Scholar]
- 31.Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM. Genet Epidemiol. 2004;26:61–69. doi: 10.1002/gepi.10295. [DOI] [PubMed] [Google Scholar]
- 32.Horvath S, Xu X, Laird NM. Eur J Hum Genet. 2001;9:301–306. doi: 10.1038/sj.ejhg.5200625. [DOI] [PubMed] [Google Scholar]
- 33.Masotti C, Armelin-Correa LM, Splendore A, Lin CJ, Barbosa A, Sogayar MC, Passos-Bueno MR. Gene. 2005;359:44–52. doi: 10.1016/j.gene.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 34.Risch N. Theor Popul Biol. 2001;60:215–220. doi: 10.1006/tpbi.2001.1538. [DOI] [PubMed] [Google Scholar]
- 35.Grabe N. In Silico Biol. 2002;2:S1–S15. [PubMed] [Google Scholar]
- 36.Tuchman R, Rapin I. Lancet Neurol. 2002;1:352–358. doi: 10.1016/s1474-4422(02)00160-6. [DOI] [PubMed] [Google Scholar]
- 37.Levitt P, Eagleson KL, Powell EM. Trends Neurosci. 2004;27:400–406. doi: 10.1016/j.tins.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 38.Persico AM, Bourgeron T. Trends Neurosci. 2006;29:349–358. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}