

Abstract
Background & Aims:
The complement pathways are important components of the innate and adaptive immune response. Here we tested the hypothesis that activation of complement is required for development of ethanol-induced fatty liver.
Methods:
Wild type and mice lacking the 3rd (C3) and 5th (C5) components of the complement activation pathway, as well as mice lacking decay accelerating factor (CD55/DAF), a complement regulatory protein, were fed Lieber-DeCarli ethanol-containing diets for 6 weeks or pair-fed control diets.
Results:
Ethanol feeding to wild type mice increased C3a in plasma. Wild type and C5-/- mice fed the ethanol diet developed hepatic steatosis characterized by micro- and macro-vesicular lipid accumulation and increased triglyceride content. C3-/- did not develop steatosis, while CD55/DAF-/- mice accumulated even more hepatic triglyceride after ethanol feeding than wild type mice. Serum ALT and hepatic TNFα, indicators of hepatocyte injury and inflammation, respectively, were increased in wild type and CD55/DAF-/- mice, but not in C5-/- mice after ethanol feeding. In contrast to the protective effect of C3-/- against ethanol-induced steatosis, both ALT and TNFα were increased in C3-/- mice after ethanol feeding.
Conclusions:
Here we have identified several elements of the complement system as important contributors to ethanol-induced fatty liver. C3 contributed primarily to the accumulation of triglyceride in the liver, while C5 was involved in inflammation and injury to hepatocytes. Further, the absence of CD55/DAF exacerbated these responses, suggesting that CD55/DAF serves as a barrier to ethanol-induced fatty liver.
Introduction
The complement system, comprised of a network of more than 30 proteins, belongs to both the innate and adaptive arms of the immune system. Activation of the complement pathway can occur via the classical, lectin or alternative pathways; all three pathways culminate in the activation of C3. Upon activation, C3 is cleaved into C3a and C3b, C3b then acts in the activation/cleavage of C5 to C5a and C5b; these processes ultimately result in the formation of the pore-forming C5b-9 membrane attack complex, which mediates lysis of target and infected cells. In addition to their role in the formation of the membrane attack complex, the complement cleavage products C3a, C3b and C5a, resulting from the activation of C3, together contribute to a number of pro-inflammatory and protective responses, including phagocyte recruitment, opsonization of pathogens and lysis of certain pathogens and cells. The C3a- and C5a-mediated responses are characterized by the stimulation of cytokine production (35; 39), either alone or in the presence of other inflammatory mediators, such as lipopolysaccharide (LPS) (39), as well as increased expression of chemokines and adhesion molecules (11; 23).
The development of ethanol-induced liver injury is mediated, at least in part, by an inflammatory response involving the innate immune system. Ethanol exposure, both in humans and animal models, is associated with increased production of pro-inflammatory cytokines and chemokines (43; 45), as well as increased production of reactive oxygen species (18; 20). Kupffer cells, the resident macrophages in the liver, are critical to the onset of ethanol-induced liver injury. Ablation of Kupffer cells prevents the development of fatty liver and inflammation, early events in the progression of ethanol-induced liver damage, in rats exposed to ethanol via intragastric feeding (1). Endotoxin (LPS), a component of gram-negative bacterial cell walls, is an important activator of Kupffer cells, stimulating the production of inflammatory and fibrogenic cytokines, as well as reactive oxygen species. LPS concentration is increased in the blood of alcoholics (4; 14) and rats exposed to ethanol via gastric infusion (37), probably due to impaired barrier function of the intestinal mucosa (4).
Our interest in a possible role for the complement system in the development of ethanol-induced liver injury was inspired by the large body of data implicating the complement system in the development of acute and chronic inflammatory responses to bacteria/bacterial products, as well as in response to cell injury, both hallmarks of ethanol-induced liver injury. At the time we initiated our experiments, there was a report by Jarvelainen, et al., (22) that ethanol feeding to rats increased deposition of complement proteins C3 and C8, but not C1, in the liver. Lack of C1 deposition suggests that ethanol feeding activates the complement cascade via the lectin and/or alternative pathways (22). As LPS is a potent activator of the alternative pathway, ethanol-induced activation of the alternative pathway would be consistent with the increased LPS exposure observed in humans after alcohol consumption, as well as in animal models of ethanol exposure (4; 14; 37).
If the complement system contributes to ethanol-induced liver injury, then mice lacking key components of the complement activation system should be protected from ethanol-induced liver injury. In contrast, mice lacking decay accelerating factor (CD55/DAF), a complement regulatory protein that inhibits the cleavage-induced activation of C3 and C5, thus intering with the activation of the rest of the complement cascade, including the formation of the terminal C5b-9 complex, might be more susceptible to ethanol-induced liver injury. A recent report found that mice lacking the 3rd component of the complement pathway (C3-/-) did not develop hepatic steatosis or increased ALT concentrations in response to acute or chronic ethanol exposure (5). Here we have further examined the role of complement in ethanol-induced liver injury, making use of mouse lines that lack the 3rd (C3) and 5th (C5) components of the complement activation pathway, as well as mice lacking CD55/DAF, to investigate the role of complement activation in the progression of ethanol-induced liver injury.
Methods and Materials
Materials
Female C57BL/6 mice, C5-/- mice (B10.D2-Hc0H2dH2-T18c/0SnJ) and their haplotype controls (B10.D2-Hc1H2dH2-T18c/nSnJ) (nSnJ)(8-12 weeks old) were purchased from Jackson Labs (Bar Harbor, Maine). CD55/DAF-/- mice have been previously described and were back-crossed 4 generations with the C57BL/6 strain (28). The C57/129 wild type mice used in the current study were back-crossed in parallel with CD55/DAF-/-. C3-/- have been previously described (49). Lieber-DeCarli high-fat ethanol and control diets were purchased from Dyets (Bethlehem, PA). Antibodies were from the following sources: CYP2E1 (Research Diagnostics, Inc., Flanders, NJ), anti-rat Fc and horseradish peroxidase conjugated goat-anti mouse C3 (MP Biomedicals, Irvine, CA), rat anti-mouse C3a (mAb 3/11) (Cell Sciences, Canton, MA) and HSC70 (Santa Cruz Biotechnology, Santa Cruz, CA). Antibody pairs used for ELISAs for TNFα, IL-6 and IFNγ were from Biolegend (San Diego, CA). Anti-rabbit and mouse IgG-peroxidase were purchased from Boehringer Mannheim (Indianapolis, IN).
Ethanol feeding
All procedures using animals were approved by the Case Western Reserve University Institutional Animal Care and Use Committee. 8-12 week old female wild type mice (C57BL/6, C57/129, nSnJ) and mice lacking specific complement pathway components (abbreviated as “complement knock-out mice”) were housed in shoe-box cages (2 animals/cage) with microisolator lids. Standard microisolator handling procedures were used throughout the study. Mice were randomized into ethanol-fed and pair-fed groups and then adapted to control liquid diet for 2 days. The ethanol-fed group was allowed free access to ethanol containing diet with increasing concentrations of ethanol: 1% (vol/vol) and 2% each for 2 days, then 4% ethanol for 7 d, and finally 5% ethanol for a further 4 weeks. The macronutrient composition Lieber-DeCarli high fat control and ethanol-containg diets are 18% of calories as protein and 35% as fat. Carbohydrates comprise 47% of calories in the control diet. In the ethanol-containing diets, ethanol and carbohydrate content vary with ethanol dose to a final composition of 28% of calories as ethanol and 19% as carbohydrate, at the highest concentration of ethanol fed (5% vol/vol). Control mice were pair-fed diets which iso-calorically substituted maltose dextrins for ethanol over the entire feeding period. Mice were then anesthetized, blood samples taken into non-heparinized syringes from the posterior vena cava, livers blanched with saline via the portal vein and then excised. Portions of each liver were then either fixed in formalin or frozen in optimal cutting temperature (OCT) compound (Sakura Finetek U.S.A., Inc., Torrance CA) for histology or flash frozen in liquid nitrogen and stored at -80°C until further analysis. Blood was either allowed to clot on ice for 60 min and then centrifuged to isolate serum or transferred to heparin-containing tubes for the isolation of plasma. Plasma was then stored with (for measurement of C3a (see below)) or without 20 mM EDTA at -80°C.
Short term ethanol exposure
Mice were exposed to 6 g/kg ethanol via an intragastric gavage. After 90 min, mice were anesthetized and blood samples taken into non-heparinized syringes from the posterior vena cava. Serum was isolated and then stored at -80 °C.
ALT activity
Serum samples were assayed for ethanol and alanine aminotransferase (ALT) using commercially available enzymatic assay kits (Diagnostic Chemicals, LTD, Oxford, CT).
C3a ELISA
The concentration of C3a in the plasma was assessed using a specific rat anti-C3a specific monoclonal antibody that recognizes neo-activation epitopes on the C3a peptide (29; 31). An ELISA assay was carried out exactly as described by Mastellos, et al. (31). Briefly, anti-rat Fc antibody was used to coat microtiter plates, then rat anti-C3a monoclonal antibody (mAb 2/11) was added to the plates as the capture antibody. Plasma samples were treated with 20 mM EDTA to prevent further activation of C3 during the ELISA assay and serial dilutions of plasma added to the ELISA plate (10-25 ul). A horseradish peroxidase conjugated goat anti-mouse C3 antibody was used as the detection antibody. Plates were washed four times in 1% bovine serum albumin in phosphate buffered saline containing 0.05% Tween 20 after each step. Serial dilutions of plasma treated with zymosan (2mg/ml) for 30 min at 36°C was used to generate a C3a standard curve. C3a concentration in plasma samples is expressed as percent of total C3a in zymosan-activated plasma. Non-specific binding in the ELISA assay, assessed with plasma from C3-/- mice, was included as a negative control. C3-/- mice did not have detectable C3a in their plasma under the conditions of this ELISA (data not shown).
Liver histology and triglycerides
Formalin-fixed tissues were paraffin embedded, sectioned and stained with hemotoxylin and eosin. Sections were coded prior to analysis and examined by two independent individuals. For Oil Red O staining, 10 micron sections were cut from frozen OCT samples and affixed to a microscope slide. Slides were then stored at 4°C until staining. Liver sections were then air dried for 5-10 min at room temperature and stained in fresh Oil Red O (Sigma, St. Louis, MO) for 12 minutes, rinsed in water and then counterstained with hematoxylin. Total liver triglycerides were measured using the Triglyceride Reagent Kit from Pointe Scientific Inc. (Lincoln Park, Michigan).
Liver homogenization and Western blotting
0.5-1.0 g of frozen liver tissue was homogenized in 10ml/g tissue in lysis buffer (50 mM Tris-HCl, pH7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA with added protease inhibitors (Complete™ (Roche Diagnostics, Mannheim, Germany), 17.5 μg/ml aprotinin, 5μg/ml bestatin, 10 μg/ml leupeptin, 1 mg/ml bacitracin, and 20 μg/ml E64) and phosphatase inhibitors (1 mM vanadate and 10 mM Na pyrophosphate)) using 15 strokes in a glass on glass homogenizer (loose pestle). After 15 min on ice, samples were centrifuged at 16,000 x g for 15 min to remove insoluble material. Protein concentrations were measured. Samples were then used to measure hepatic cytokine concentrations (see below) or normalized and samples prepared in Laemmli buffer and boiled for 5 min. Samples were then separated by SDS-polyacrylamide gel electrophoresis and transferred to membranes for Western blotting. Membranes were probed with specific antibodies against CYP2E1 or HSC70, used as a control for equal protein loading, overnight at 4°C, then washed and incubated for 1 h in secondary antibodies coupled to horseradish peroxidase. Bound antibody was detected by chemiluminescence. Immunoreactive protein quantity was assessed by scanning densitometry.
Hepatic cytokine concentration
Liver homogenates were used to measure the concentration of hepatic cytokines by ELISA. 20-30 μg of hepatic protein were used in ELISA assays to measure TNFα, interleukin 6 (IL-6) and interferon-γ (IFN-γ) peptides accumulated in the liver.
Statistical analysis
Values reported are means + SEM. Because of the limitations in the number of mice available, several feeding trials were carried out over the course of 18 months and combined for the final data analysis. Pair-fed controls within each genotype were always carried out in parallel with ethanol feeding. Data from C5-/- mice and their haplotype controls (nSnJ) were analyzed separately from the other genotypes because of differences in the response of the nSnJ mice compared to C57BL/6 and C57/129 wild type strains to ethanol feeding. Data were analyzed by general linear models procedure (SAS, Carey, IN), blocking for trial effects when data from more than one feeding trial was used in a data set. Data were log transformed if needed to obtain a normal distribution. Follow-up comparisons were made by least square means testing.
Results
Activation of the complement pathway via the classical, lectin and alternative pathways culminates in the generation of a C3 convertase that enzymatically cleaves C3 to form C3a and C3b (48). C3b then initiates the second part of the complement cascade resulting in the production of the pathophysiologically key C5a and C5b-C9 components. Here we first asked whether ethanol feeding activated the complement pathway by measuring C3a concentration in plasma of wild type mice. Plasma concentrations of C3a were measured by ELISA in wild type mice. C3a concentrations were undetectable in pair-fed mice, but increased after ethanol feeding to 7.6% of the total C3a in mouse plasma after activation with zymosan, an agent that stimulates the proteolytic cleavage of C3 (Figure 1).
Figure 1.
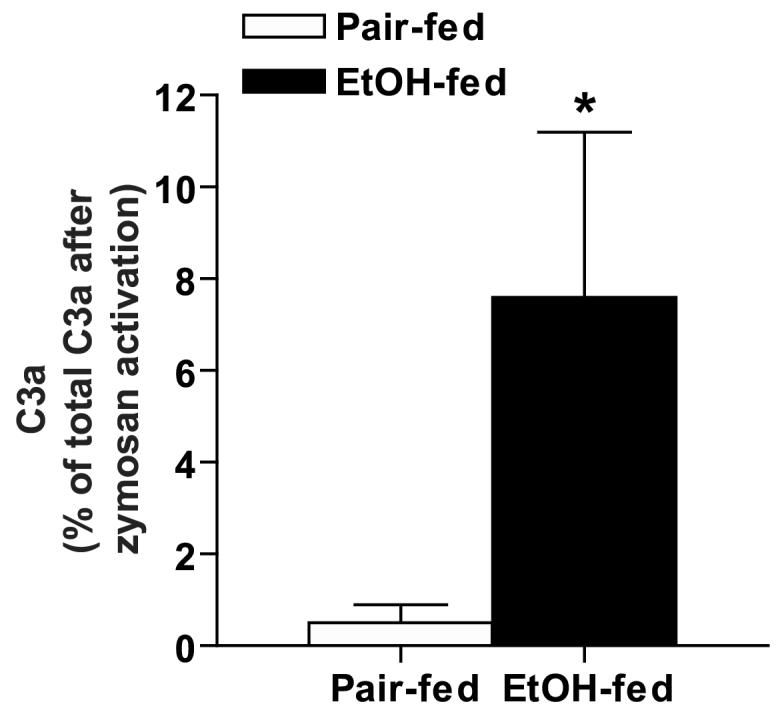
Plasma C3a in C57BL/6 wild type mice after ethanol feeding. C3a, an activation product of C3, was measured in plasma of C57BL/6 mice either allowed free access to ethanol-containing diets or pair-fed control diets as described in Methods. C3a concentration was measured by ELISA and is expressed as % of total C3a in zymosan-activated plasma of control mice. Plasma from C3-/- mice was used as a negative control in the ELISA assay; C3a was not detectable in the plasma of C3-/- mice under the conditions of this assay. Values represent means ± SEM, n=5 for pair-fed and 10 for ethanol-fed, *p<0.05 compared to pair-fed.
If complement activation contributes to ethanol-induced fatty liver, then mice lacking C3 or C5 should be protected from ethanol-induced steatosis. Further, mice lacking CD55/DAF, an intrinsic complement regulatory protein that terminates the complement cascade at the level of C3 and C5 cleavage, should be more susceptible to ethanol-induced liver injury. To test this hypothesis, wild type mice and mice lacking specific complement pathway components were adapted to an ethanol-containing liquid diet for 2 weeks and then maintained on a 5% ethanol liquid diet for 4 weeks or pair-fed control diets. Ethanol- and pair-fed mice gained equivalent body mass during the ethanol feeding protocol in each of the mouse strains studied (Table 1). After ethanol feeding, all three strains of wild type mice developed steatosis characterized by micro- and macro-vesicular structures observed on H&E stained sections of liver (Figure 2), although the nSnJ mice (C5 haplotype controls) accumulated less lipid compared to C57BL/6 and C57/129 mice (Figure 2 and 3). Characteristic of the ad libitum Lieber DeCarli feeding model, little evidence of inflammation was observed in wild type mice after ethanol feeding. While steatosis developed in C5-/- mice, micro- and macro-vesicular steatosis was not observed in liver sections from C3-/- mice (Figure 2). In contrast, CD55/DAF-/- mice developed more severe liver injury, accumulating more macro-vesicular structures compared to wild type mice after ethanol feeding (Figure 2).
Table 1.
Growth of wild type and complement knock-out mice during ethanol feeding and serum ethanol concentrations in response to an acute ethanol challenge by gavage.
| Body weight (g) | Serum EtOH (mM) | ||||
|---|---|---|---|---|---|
| Genotype | Pair-fed | EtOH-fed | |||
| Initial | Final | Initial | Final | ||
| C57BL/6 | 20.1±0.6 | 25.2±0.8(n=9) | 19.9±0.3 | 23.3±0.7(n=9) | 82.0 ± 4.5(n=8) |
| C57/129 | 20.0±1.0 | 24.0±1.0(n=5) | 19.7±0.7 | 23.7±1.2(n=5) | 103.6 ± 7.3*(n=4) |
| C3-/- | 19.8±0.8 | 25.7±1.1(n=6) | 19.2±0.3 | 25.0±1.5(n=6) | 83.3 ± 3.2(n=4) |
| CD55/DAF-/- | 21.3±1.0 | 26.3±1.9(n=5) | 21.3±0.5 | 25.0±1.1(n=5) | 71.3 ± 12.1(n=4) |
| nSnJ | 19.3±0.6 | 22.5±0.7(n=5) | 19.3±0.6 | 24.6±0.6(n=5) | 56.1 ± 5.8(n=3) |
| C5-/- | 18.1±0.2 | 24.5±0.9(n=10) | 18.7±0.2 | 26.2±0.8(n=10) | 65.6 ±7.8(n=4) |
p<0.05 compared to all C5-/- and CD55/DAF-/-
Figure 2.
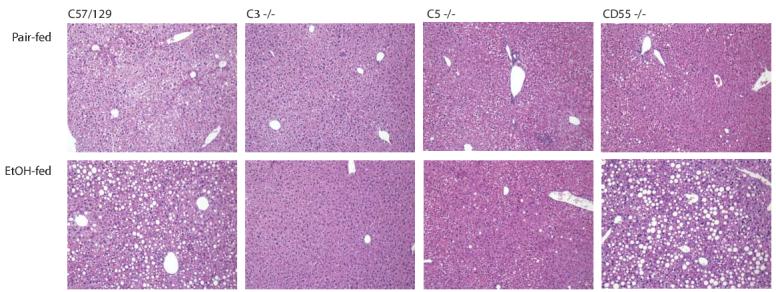
Liver histology in wild type and complement knock-out mice after ethanol feeding. Livers from mice allowed free access to ethanol-containing diets or pair-fed control diets were perfused with saline, then fixed in formalin. Liver sections were prepared and stained with hemotoxylin and eosin. Images were taken at a 100 x magnification. Figures are representative of at least 5 mice in each experimental group.
Figure 3.

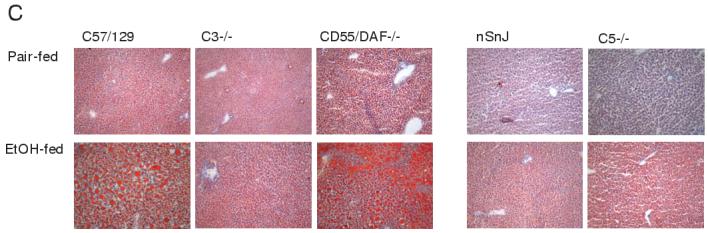
Liver triglycerides (A/B) and Oil Red O staining (C) in wild type and complement knock-out mice after ethanol feeding. A portion of the livers from wild type and complement knock-out mice (A: C57BL/6, C57/129, C3-/-, CD55/DAF-/- and B: nSnJ and C5-/- ), allowed free access to ethanol-containing diets or pair-fed control diets, were perfused with saline, weighed and then frozen in liquid nitrogen or frozen in OCT reagent and then stored at -80°C until assay. A/B: Total triglycerides were measured using the Trinder reagent. Values represent means ± SEM, A: n=7-9 and B: n=5-13. *p<0.05 compared to pair-fed mice of the same strain. C: Frozen liver sections were prepared and stained in Oil Red O. Images were taken at a 100 x magnification. Figures are representative of 2-3 mice in each experimental group.
Oil red O staining of frozen liver sections and quantitative assessment of total hepatic triglyceride paralleled the histological observations in H&E stained samples (Figure 3). Total hepatic triglyceride concentration (Figure 3A and B) and Oil Red O staining of neutral lipids (Figure 3C) increased in all three wild type strains in response to ethanol feeding, compared to pair-fed controls, although triglyceride concentrations in nSnJ mice were lower than that in C57BL/6 and C57/129 strains (Figure 3). C3-/- mice did not accumulate excess triglyceride compared to pair-fed controls, while hepatic triglycerides were even further increased in CD55/DAF-/- compared to wild type mice (Figure 3). In contrast to C3-/- mice, C5-/- mice were not protected from ethanol-induced triglyceride accumulation (Figure 3). Liver to body weight ratios, a general measure of liver injury, were increased in C57BL/6 and C57/129 wild type mice after ethanol feeding, with no change in C3-/- mice (Figure 4A). In contrast, neither the C5-/- mice or their haplotype controls exhibited increased liver to body weight ratios (Figure 4B). Serum ALT activities were increased after ethanol feeding in C57/BL6 and C57/129 wild type mice (Figure 5A), as well as in the nSnJ mice (Figure 5B). In C3-/- mice, ALT activity was increased with ethanol feeding compared to their pair-fed controls, but to a lesser extent then in the wild type controls. ALT activity was also increased in response to ethanol feeding in CD55/DAF-/- mice; however, this increase in ALT activity in response to ethanol exposure was even greater than that observed in wild type mice. Finally, despite the development of steatosis, C5-/- mice did not exhibit increased ALT activity after ethanol feeding (Figure 5B).
Figure 4.

Liver to body weight ratios in wild type and complement knock-out mice after ethanol feeding. Livers from wild type and complement knock-out mice (A: C57BL/6, C57/129, C3-/-, CD55/DAF-/- and B: nSnJ and C5-/- ), allowed free access to ethanol-containing diets or pair-fed control diets, were perfused with saline, excised, blotted on tissue paper and weighed. Values represent means ± SEM, A: n=4-6 (except CD55/DAF-/- where n=2) and B: n=5-13. *p<0.05 compared to pair-fed mice of the same strain.#n=2 for ethanol-fed CD55/DAF-/- mice; this treatment was not included in the statistical analysis.
Figure 5.

Serum ALT activity in wild type and complement knock-out mice after ethanol feeding. Wild-type and complement knock-out mice (A: C57BL/6, C57/129, C3-/-, CD55/DAF-/- and B: nSnJ and C5-/- ) were allowed free access to ethanol-containing diets or pair-fed control diets. ALT activity in serum was measured enzymatically. Values represent means ± SEM, A: n=6-9 and B:n=5-13. *p<0.05 compared to pair-fed mice of the same strain, #p<0.05 compared to EtOH-fed C57Bl/6 and C57/129 wild-type mice.
These differential effects of ethanol feeding in C3-/- and C5-/- mice suggested that the C3- and C5-activation pathways may contribute to distinct aspects of ethanol-induced liver injury. Since increased expression of pro-inflammatory cytokines, such as TNFα, contributes to hepatocellular injury during ethanol exposure (45), we assessed the effects of ethanol feeding on hepatic TNFα, IL-6 and IFN-γ expression in C3-/-, C5-/- and CD55/DAF-/- mice compared to wild type controls. As expected, ethanol feeding increased hepatic TNFα, IL-6 and IFN-γ peptide concentrations in all wild type strains, as well as in CD55/DAF-/- mice (Figure 6). In the C3-/- mice, ethanol feeding also increased hepatic cytokines (Figure 6A), while C5-/- mice were protected and did not exhibit increased TNFα, IL-6 or IFN-γ (Figure 6B).
Figure 6.

Hepatic TNFα, IL-6 and IFN-γ concentrations in wild type and complement knock-out mice after ethanol feeding. Wild-type and complement knock-out mice (A: C57/129, C3-/-, CD55/DAF-/- and B: nSnJ and C5-/- ) were allowed free access to ethanol-containing diets or pair-fed control diets. Livers were perfused with saline and frozen in liquid nitrogen. Lysates were prepared from frozen livers and TNFα, IL-6 and IFN-γ peptides measured by ELISA. The concentrations of each cytokine in the liver lysates (pg/mg protein) were used in the statistical analysis. The concentrations of cytokines in pair-fed controls did not differ between wild type and complement knock-out mice. Values represent means ± SEM of the percent increase in cytokine concentration in ethanol-fed compared to pair fed mice within each genotype, A: n=6-9 and B: n=3-7 for nSnJ and 9-12 for C5-/-. *p<0.05 compared to pair-fed mice of the same strain.
In order to ensure that differences in susceptibility to ethanol-induced liver injury between wild type and complement knock-out mice were not due to differences in ethanol metabolism, serum ethanol concentrations in response to a single exposure to ethanol were assessed. Chow-fed, ethanol-naïve mice were treated with 6 g/kg ethanol by gavage and serum concentrations of ethanol measured after 90 min. No ethanol was detected in the serum of C57BL/6 wild type mice treated with saline (data not shown). Ethanol concentrations ranged from 56-104 mM (Table 1). The concentration of ethanol at 90 min did not differ between the C57BL/6 wild type, C3-/-, C5-/- and CD55/DAF-/- transgenic lines, but was slightly (p<0.05) elevated in the mixed background C57/129 wild type compared to C5-/- and CD55/DAF-/- all other strains (Table 1). However, this small difference in ethanol concentration did not appear to be biologically important, as the C57/129 mice were not more susceptible to liver injury after long-term ethanol feeding compared to the other wild type mice used in these studies. Ethanol feeding increased the expression of CYP2E1, measured by Western blotting, in both wild type and complement knock-out mice (Figure 7), suggesting that the complement pathway is not required for CYP2E1 induction in response to ethanol exposure. Taken together, the similar serum ethanol concentrations observed in response to short term ethanol exposure and equivalent induction of CYP2E1 expression suggest that the metabolic handling of ethanol is similar between wild type and the complement knock-out mice used in this study.
Figure 7.
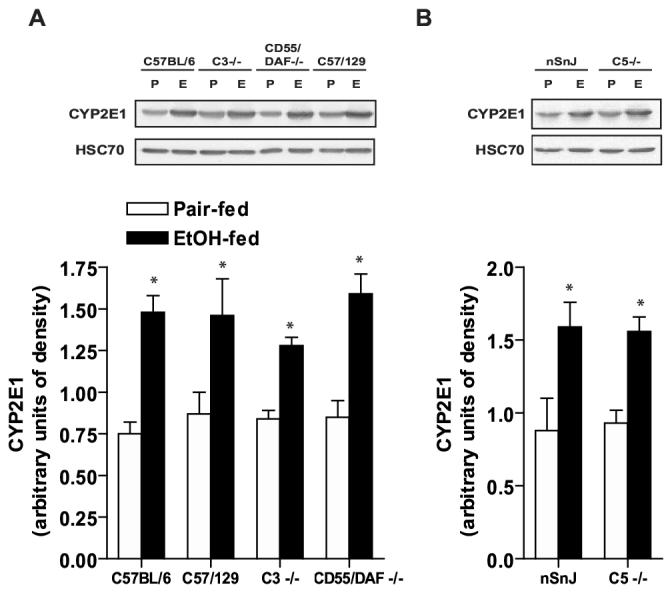
CYP2E1 expression is induced in livers of wild type and complement knock-out mice after ethanol feeding. Wild-type and complement knock-out mice (A: C57BL/6, C57/129, C3-/-, CD55/DAF-/- and B: nSnJ and C5-/- ) were allowed free access to ethanol-containing diets or pair-fed control diets. Livers were perfused with saline and frozen in liquid nitrogen. Homogenates were prepared from livers of ethanol- and pair-fed mice and the relative expression of CYP2E1 assessed by Western blot analysis. Western blots were also probed with antibody against HSC70 as a loading control. Values represent means ± SEM, n=4. *p<0.05 compared to pair-fed controls of the same strain.
Discussion
Components of the innate immune response, including NK and NKT cells (34), Kupffer cells (resident hepatic macrophages) (36) and the complement system (29; 40), as well as T-cells and antibody-dependent adaptive immune responses (46), are involved in the hepatic response to various types of injury, including bacterial and viral infections, exposure to toxins, partial hepatectomy and ischemia-reperfusion. Previous studies have demonstrated a primary role for several components of the innate immune response, including Kupffer cells (36) and NKT cells (34) in the progression of ethanol-induced liver injury. Here we demonstrate that the complement pathway is intimately involved in the progression of ethanol-induced fatty liver . First, we show that C3a concentration is increased in the serum after ethanol feeding, providing evidence for C3 activation. Second, making use of mice lacking the 3rd and 5th components of the complement system, we report that C3 and C5 have differential contributions to the pathophysiological progression of fatty liver. C3 contributes to the development of hepatic steatosis, while C5 acts primarily to mediate increased pro-inflammatory cytokine production and injury to hepatocytes. Finally, we show that CD55/DAF, a complement regulatory protein, serves as a brake to inhibit the progression of steatosis and hepatocyte injury in response to ethanol exposure in mice.
Activation of the complement pathway can occur via the classical, lectin or alternative pathways, with all three pathways culminating in the activation of C3. These pathways leading to C3 cleavage are “triggered enzyme cascades”, analogous to the regulated activation of the coagulation pathway (48). Making use of monoclonal antibodies developed to neo-epitopes revealed on C3a and C3b/iC3b (31), here we report the presence of C3a in the serum of mice after ethanol exposure (Figure 1). These data are consistent with a previous report using an in vitro complement activation assay demonstrating a deposition of C3 and C8, but not C1, were observed in periportal regions of the liver after ethanol feeding (22). Jarvelainen, et al (22) proposed that this lack of C1 activation suggests that ethanol activates complement via the alternative pathway. However, the sensitivity and/or the timing of these assays for C1 deposition may have precluded the identification of a role for the classical pathway in mediating the effects of ethanol in liver injury. Future studies will be required to delineate the precise mechanisms and time course for complement activation in response to ethanol exposure.
The progression of alcohol-induced liver injury follows a characteristic pattern from fatty liver/steatosis, inflammation, hepatocyte necrosis and apoptosis, regenerating nodules, fibrosis and eventually cirrhosis in some individuals (30). Current models for the progression of injury suggest that accumulation of triglyceride in the hepatocyte is “lipotoxic” and acts as an initiating event in the disease process (27). However, several lines of evidence suggest that there is a critical relationship between the increased production of inflammatory cytokines and activation of components of the innate immune response with the development of steatosis. For example, ablation of Kupffer cells by treatment with gadolinium chloride, or treatment of mice with antibody to TNF-α prevents the development of both steatosis and hepatic inflammation in response to ethanol exposure (17). Similarly, mice lacking TLR4, CD14 or the TNF receptor I are protected from ethanol-induced steatosis, as well as inflammation and early stages of fibrosis (43; 45). Here we find differential roles for the 3rd and 5th component of the complement pathway in the development of ethanol-induced steatosis and increased inflammatory cytokine production. Mice lacking C3 did not develop steatosis in response to ethanol feeding, but still exhibited a modestly increased ALT in the circulation, a marker of hepatocyte injury, as well as increased expression of inflammatory cytokines in the liver. In contrast, mice lacking C5 had increased hepatic triglycerides, but were completely protected from ethanol-induced increases in ALT and inflammatory cytokines. These data suggest that the development of steatosis requires C3. Further, the data from C3-/- mice are consistent with the hypothesis that neither ethanol-induced increases in hepatic inflammatory cytokines or hepatocellular damage are dependent on the development of steatosis.
The mechanisms by which C3 contributes to ethanol-induced steatosis are not known. One of the breakdown products of C3, termed acylation stimulating protein (ASP or C3adesArg) is involved in the regulation of glucose and lipid metabolism (reviewed in (6). While adipose tissue is the primary target of ASP, in vivo studies suggest that it also regulates fatty acid uptake and utilization in the liver (6), although this role is controversial (25; 25). In one study, “ASP deficient” mice (a C3-/- strain on a 129 background) exhibited a decrease in the uptake of fatty acids into adipose tissue in response to an oral fat load. In contrast, the uptake of fatty acids, as well as the accumulation of fatty acid oxidation products, in liver was increased in ASP/C3-/- mice (38). These data suggest that fatty acid oxidation was upregulated in the ASP/C3-/- mice; however, the mechanisms for this increase were not investigated (38).
While the effects of C3 deficiency on lipid homeostasis are still controversial, it is possible that the absence of ASP/C3adesArg contributes to the protection of the C3-/- mice from ethanol-induced steatosis. It has been hypothesized that one of the mechanisms for ethanol-induced steatosis is failure to up-regulate genes involved in fatty acid oxidation in the liver (8). Indeed, the therapeutic efficacy of PPARα agonists (12) and adiponectin (51) in preventing ethanol-induced steatosis is thought to be due to the ability of these agents to up-regulate genes required for fatty acid oxidation in the liver. Therefore, the absence of C3-/- could lead to an increase in fatty acid oxidation in ethanol-fed mice and thus decrease the fatty acids available for synthesis of triglyceride and reduce the development of steatosis.
C3a and C5a, termed the anaphylatoxins, are important regulators of the inflammatory response, exhibiting both pro- and anti-inflammatory properties. C3a and C5a stimulate the production of cytokines in a number of cell types (35; 39), either alone or in the presence of other inflammatory mediators, such as LPS (39). C5a is generally much more pro-inflammatory than C3a with respect to effects on blood flow, lymphatic drainage and vascular permeability and neutrophil activation, and consequently, with respect to local concentrations of agonists and complement components (21). C5a has also been correlated with increased expression of cytokines and chemokines (3; 7) and reactive oxygen species (9). C5 has been associated with injury to lung (15) and kidney (10)(11).
While C3a and C5a have numerous pro-inflammatory properties, they also have anti-inflammatory activity. C3a suppresses LPS-induced TNFα, IL-1 and IL-6 secretion from isolated peripheral blood mononuclear cells and lymphocytes (13; 41; 42). Further, C3a receptor knock-out mice show increased LPS-induced pro-inflammatory responses, suggesting important anti-inflammatory function for C3a-mediated activation of the C3a receptor (24). Similarly, C5-/- mice exhibit an enhanced inflammatory response to bleomycin-induced lung injury (2) and C5a suppresses LPS-stimulated synthesis of IL-12 family cytokines by macrophages (16).
Expression of inflammatory cytokines in the livers of mice during ethanol exposure was different in the C3-/- and C5-/- mice. C3-/- mice, while protected from ethanol-induced steatosis, had increased expression of inflammatory cytokines and moderately increased ALT. In contrast, C5-/- mice did not increase liver cytokines or ALT, even though they exhibited steatosis. These data suggest that C5, but not C3, activation contributes to increased production of inflammatory cytokines during ethanol exposure.
While reduced liver cytokines in the C5-/- after ethanol exposure clearly point to a pro-inflammatory function of C5a during ethanol exposure, the maintenance of increased inflammatory cytokines in the livers of C3-/- mice may be due to several possible mechanisms. First, we would expect that C3-/- mice would be functionally deficient in both C3a and C5a (See Table 2), since C3b-dependent C5 convertase activity would be lacking in C3-/- mice. However, , alternative non-complement mechanisms for activation of C5 have been described, including a role for plasmin-(50) and thrombin-mediated C5 proteolytic cleavage (19). The presence of C5a, with potent pro-inflammatory functions, in the C3-/- mice would be consistent with the increased liver cytokines after ethanol feeding. In addition, it is also possible that C3a has an anti-inflammatory function during ethanol exposure. Given the importance of LPS-stimulated cytokine production in the progression of ethanol-induced liver injury (44), the absence of an anti-inflammatory function for C3a in the C3-/- mice could allow for the maintenance of LPS-stimulated cytokine production during ethanol exposure. This hypothesis is consistent with the particularly effective ability of C3a to inhibit LPS-stimulated cytokine production (24).
Table 2.
Summary of the role of complement and chronic ethanol-induced liver injury
| Genotype | Complement phenotype | Hepatic triglyceride | Hepatic cytokines | ALT |
|---|---|---|---|---|
| Wild type | C3+/C5+ | ⇈ | ⇈ | ⇈ |
| C3-/- | C3-/C5-/+? | ↔ | ⇈ | ↑ |
| C5-/- | C3+/C5- | ⇈ | ↔ | ↔ |
| CD55/DAF-/- | C3++/C5++ | ↑↑↑ | ⇈ | ↑↑↑ |
Induction of CYP2E1 expression and associated production of reactive oxygen species is another important mechanism thought to contribute to ethanol-induced liver injury (18). However, several lines of evidence suggest that CYP2E1-induced oxidant stress is not sufficient to generate ethanol-induced liver injury, suggesting that additional pathways are required to develop the pathological responses of the liver to ethanol. For example, CYP2E1 deficient mice still develop ethanol-induced liver injury (26). Further, while mice deficient in LPS-binding protein, iNOS, or Egr-1 are protected from ethanol-induced liver injury, CYP2E1 is still induced after ethanol exposure in each of these knock-out strains (32; 33; 47). The response of each of the complement deficient strains to an acute ethanol exposure (Table 1), as well as the equivalent induction of CYP2E1 (Figure 7) suggests that the complement pathway does not regulate ethanol metabolism. Further, these data show that the induction of CYP2E1 in the different strains does not correlate well with the development of ethanol-induced fatty liver.
Previous studies have demonstrated that the progression of ethanol-induced liver injury involves several components of the innate immune response including Kupffer cells (36) and NKT cells (34). Recent studies have also suggested that activation of C3 in the complement pathway, an integral element of the innate immune response, may also contribute to ethanol-induced liver injury (5; 22). Here we demonstrate that ethanol exposure to mice results in the activation of the complement pathway and that C3 and C5 appear to differentially contribute to the development of steatosis, increased production of pro-inflammatory cytokines and injury to hepatocytes. CD55/DAF, a complement regulatory protein, serves as a brake on ethanol-induced steatosis and injury to hepatocytes . Taken together, these data demonstrate an important role for complement activation in ethanol-induced fatty liver and suggest that understanding the role of specific components in the complement pathways may facilitate the development of novel therapeutic strategies in the treatment of alcoholic liver disease.
Footnotes
Grant support:This work was supported by NIH grants AA013868 and AA11975 to LEN and AI23598 MEM.
- ALT:
- alanine aminotransferase
- C3:
- third component of the complement system
- C5:
- fifth component of the complement system
- CYP2E1:
- cytochrome P450 2E1
- DAF:
- decay accelerating factor
- FBS:
- fetal bovine serum
- IL-6:
- interleukin 6
- IFNγ:
- interferon γ
- LPS:
- lipopolysaccharide
- OCT:
- optimal cutting temperature
- PBS:
- phosphate buffered saline
- SEM:
- standard error of the mean
- TNFα:
- tumor necrosis factor α
Reference List
- 1.Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology. 1994;20:453–460. [PubMed] [Google Scholar]
- 2.Addis-Lieser E, Kohl J, Chiaramonte MG. Opposing regulatory roles of complement factor 5 in the development of bleomycin-induced pulmonary fibrosis. J Immunol. 2005;175:1894–902. doi: 10.4049/jimmunol.175.3.1894. [DOI] [PubMed] [Google Scholar]
- 3.Albrecht EA, Chinnaiyan AM, Varambally S, Kumar-Sinha C, Barrette TR, Sarma JV, Ward PA. C5a-induced gene expression in human umbilical vein endothelial cells. Am J Pathol. 2004;164:849–59. doi: 10.1016/S0002-9440(10)63173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and nonalcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J. Hepatol. 1987;4:8–14. doi: 10.1016/s0168-8278(87)80003-x. [DOI] [PubMed] [Google Scholar]
- 5.Bykov I, Junnikkala S, Pekna M, Lindros KO, Meri S. Complement C3 contributes to ethanol-induced liver steatosis in mice. Ann Med. 2006;38:280–6. doi: 10.1080/07853890600664608. [DOI] [PubMed] [Google Scholar]
- 6.Cianflone K, Xia Z, Chen LY. Critical review of acylation-stimulating protein physiology in humans and rodents. Biochim Biophys Acta. 2003;1609:127–43. doi: 10.1016/s0005-2736(02)00686-7. [DOI] [PubMed] [Google Scholar]
- 7.Clark JD, Qiao Y, Li X, Shi X, Angst MS, Yeomans DC. Blockade of the complement C5a receptor reduces incisional allodynia, edema, and cytokine expression. Anesthesiology. 2006;104:1274–82. doi: 10.1097/00000542-200606000-00024. [DOI] [PubMed] [Google Scholar]
- 8.Crabb DW, Galli A, Fischer M, You M. Molecular mechanisms of alcoholic fatty liver: role of peroxisome proliferator-activated receptor alpha. Alcohol. 2004;34:35–8. doi: 10.1016/j.alcohol.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Daniel DS, Dai G, Singh CR, Lindsey DR, Smith AK, Dhandayuthapani S, Hunter RL, Jr, Jagannath C. The reduced bactericidal function of complement C5-deficient murine macrophages is associated with defects in the synthesis and delivery of reactive oxygen radicals to mycobacterial phagosomes. J Immunol. 2006;177:4688–98. doi: 10.4049/jimmunol.177.7.4688. [DOI] [PubMed] [Google Scholar]
- 10.de Vries B, Kohl J, Leclercq WK, Wolfs TG, van Bijnen AA, Heeringa P, Buurman WA. Complement factor C5a mediates renal ischemia-reperfusion injury independent from neutrophils. J Immunol. 2003;170:3883–9. doi: 10.4049/jimmunol.170.7.3883. [DOI] [PubMed] [Google Scholar]
- 11.DiScipio RG, Daffern PJ, Jagels MA, Broide DH, Sriramarao P. A comparison of C3a and C5a-mediated stable adhesion of rolling eosinophils in postcapillary venules and transendothelial migration in vitro and in vivo. J Immunol. 1999;162:1127–36. [PubMed] [Google Scholar]
- 12.Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem. 2003;278:27997–8004. doi: 10.1074/jbc.M302140200. [DOI] [PubMed] [Google Scholar]
- 13.Fischer WH, Hugli TE. Regulation of B cell functions by C3a and C3a(desArg): suppression of TNF-alpha, IL-6, and the polyclonal immune response. J Immunol. 1997;159:4279–86. [PubMed] [Google Scholar]
- 14.Fukui H, Brauner B, Bode J, Bode C. Plasma endotoxin concentrations in patients with alcoholic and nonalcoholic liver disease: reevaluation with an improved chromogenic assay. J. Hepatol. 1991;12:162–169. doi: 10.1016/0168-8278(91)90933-3. [DOI] [PubMed] [Google Scholar]
- 15.Harkin DW, Marron CD, Rother RP, Romaschin A, Rubin BB, Lindsay TF. C5 complement inhibition attenuates shock and acute lung injury in an experimental model of ruptured abdominal aortic aneurysm. Br J Surg. 2005;92:1227–34. doi: 10.1002/bjs.4938. [DOI] [PubMed] [Google Scholar]
- 16.Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Kohl J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity. 2005;22:415–26. doi: 10.1016/j.immuni.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Hines IN, Wheeler MD. Recent advances in alcoholic liver disease III. Role of the innate immune response in alcoholic hepatitis. Am J Physiol Gastrointest Liver Physiol. 2004;287:G310–4. doi: 10.1152/ajpgi.00094.2004. [DOI] [PubMed] [Google Scholar]
- 18.Hoek JB, Pastorino JG. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol. 2002;27:63–68. doi: 10.1016/s0741-8329(02)00215-x. [DOI] [PubMed] [Google Scholar]
- 19.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–7. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 20.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 21.Janeway CA, Travers P, Walport M, Shlomchik M. Garland Publishing; New York: 2001. Innate Immunity in Immunobiology; pp. 57–58. [Google Scholar]
- 22.Jarvelainen HA, Vakeva A, Lindros KO, Meri S. Activation of complement components and reduced regulator expression in alcohol-induced liver injury in the rat. Clin Immunol. 2002;105:57–63. doi: 10.1006/clim.2002.5267. [DOI] [PubMed] [Google Scholar]
- 23.Jauneau AC, Ischenko A, Chan P, Fontaine M. Complement component anaphylatoxins upregulate chemokine expression by human astrocytes. FEBS Lett. 2003;537:17–22. doi: 10.1016/s0014-5793(03)00060-7. [DOI] [PubMed] [Google Scholar]
- 24.Kildsgaard J, Hollmann TJ, Matthews KW, Bian K, Murad F, Wetsel RA. Cutting edge: targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J Immunol. 2000;165:5406–9. doi: 10.4049/jimmunol.165.10.5406. [DOI] [PubMed] [Google Scholar]
- 25.Kildsgaard J, Zsigmond E, Chan L, Wetsel RA. A critical evaluation of the putative role of C3adesArg (ASP) in lipid metabolism and hyperapobetalipoproteinemia. Mol Immunol. 1999;36:869–76. doi: 10.1016/s0161-5890(99)00108-x. [DOI] [PubMed] [Google Scholar]
- 26.Kono H, Bradford BU, Yin M, Sulik KK, Koop DR, Peters JM, Gonzalez FJ, McDonald T, Dikalova A, Kadiiska MB, Mason RP, Thurman RG. CYP2E1 is not involved in early alcohol-induced liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 1999;277:G1259–G1267. doi: 10.1152/ajpgi.1999.277.6.G1259. [DOI] [PubMed] [Google Scholar]
- 27.Koteish A, Diehl AM. Animal models of steatosis. Semin Liver Dis. 2001;21:89–104. doi: 10.1055/s-2001-12932. [DOI] [PubMed] [Google Scholar]
- 28.Lin F, Fukuoka Y, Spicer A, Ohta R, Okada N, Harris CL, Emancipator SN, Medof ME. Tissue distribution of products of the mouse decay-accelerating factor (DAF) genes. Exploitation of a Daf1 knock-out mouse and site-specific monoclonal antibodies. Immunology. 2001;104:215–25. doi: 10.1046/j.0019-2805.2001.01287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markiewski MM, Mastellos D, Tudoran R, DeAngelis RA, Strey CW, Franchini S, Wetsel RA, Erdei A, Lambris JD. C3a and C3b activation products of the third component of complement (C3) are critical for normal liver recovery after toxic injury. J Immunol. 2004;173:747–54. doi: 10.4049/jimmunol.173.2.747. [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Hernandez A, Amenta PS. The hepatic extracellular matrix II. Ontogenesis, regeneration and cirrhosis. Virchows Archiv A Pathol Anat. 1993;423:77–84. doi: 10.1007/BF01606580. [DOI] [PubMed] [Google Scholar]
- 31.Mastellos D, Prechl J, Laszlo G, Papp K, Olah E, Argyropoulos E, Franchini S, Tudoran R, Markiewski M, Lambris JD, Erdei A. Novel monoclonal antibodies against mouse C3 interfering with complement activation: description of fine specificity and applications to various immunoassays. Mol Immunol. 2004;40:1213–21. doi: 10.1016/j.molimm.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 32.McKim SE, Gabele E, Isayama F, Lambert JC, Tucker LM, Wheeler MD, Connor HD, Mason RP, Doll MA, Hein DW, Arteel GE. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125:1834–44. doi: 10.1053/j.gastro.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 33.McMullen MR, Pritchard MT, Wang Q, Millward CA, Croniger CM, Nagy LE. Early growth response-1 transcription factor is essential for ethanol-induced Fatty liver injury in mice. Gastroenterology. 2005;128:2066–76. doi: 10.1053/j.gastro.2005.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minagawa M, Deng Q, Liu ZX, Tsukamoto H, Dennert G. Activated natural killer T cells induce liver injury by Fas and tumor necrosis factor-alpha during alcohol consumption. Gastroenterology. 2004;126:1387–99. doi: 10.1053/j.gastro.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 35.Monsinjon T, Gasque P, Chan P, Ischenko A, Brady JJ, Fontaine MC. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003;17:1003–14. doi: 10.1096/fj.02-0737com. [DOI] [PubMed] [Google Scholar]
- 36.Nagy LE. New insights into the role of the innate immune response in the development of alcoholic liver disease. Expt Biol Med. 2003;228:882–890. doi: 10.1177/153537020322800803. [DOI] [PubMed] [Google Scholar]
- 37.Nanji AA, Khettry U, Sadrzadeh SMH, Yamanaka T. Severity of liver injury in experimental alcoholic liver disease: Correlation with plasma endotoxin, prostaglandin E2, leukotriene B4 and thromboxane B2. Am. J. Path. 1993;142:367–373. [PMC free article] [PubMed] [Google Scholar]
- 38.Pajvani UB, Hawkins M, Combs TP, Rajala MW, Doebber T, Berger JP, Wagner JA, Wu M, Knopps A, Xiang AH, Utzschneider KM, Kahn SE, Olefsky JM, Buchanan TA, Scherer PE. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J Biol Chem. 2004;279:12152–62. doi: 10.1074/jbc.M311113200. [DOI] [PubMed] [Google Scholar]
- 39.Schieferdecker HL, Schlaf G, Jungermann K, Gotze O. Functions of anaphylatoxin C5a in rat liver: direct and indirect actions on nonparenchymal and parenchymal cells. Int Immunopharmacol. 2001;1:469–81. doi: 10.1016/s1567-5769(00)00038-2. [DOI] [PubMed] [Google Scholar]
- 40.Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, Lambris JD. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198:913–23. doi: 10.1084/jem.20030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takabayashi T, Vannier E, Burke JF, Tompkins RG, Gelfand JA, Clark BD. Both C3a and C3a(desArg) regulate interleukin-6 synthesis in human peripheral blood mononuclear cells. J Infect Dis. 1998;177:1622–8. doi: 10.1086/515316. [DOI] [PubMed] [Google Scholar]
- 42.Takabayashi T, Vannier E, Clark BD, Margolis NH, Dinarello CA, Burke JF, Gelfand JA. A new biologic role for C3a and C3a desArg: regulation of TNF-alpha and IL-1 beta synthesis. J Immunol. 1996;156:3455–60. [PubMed] [Google Scholar]
- 43.Thurman RG. Mechanisms of Hepatic Toxicity II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am. J. Physiol. 1998;275:G605–G611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 44.Thurman RG. Mechanisms of Hepatic Toxicity II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am. J. Physiol. 1998;275:G605–G611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 45.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. New Engl. J. Med. 2000;343:1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 46.Tuma DJ, Casey CA. Dangerous byproducts of alcohol breakdown--focus on adducts. Alcohol Res Health. 2003;27:285–90. [PMC free article] [PubMed] [Google Scholar]
- 47.Uesugi T, Froh M, Arteel GE, Bradford BU, Wheeler MD, Gabele E, Isayama F, Thurman RG. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol. 2002;168:2963–2969. doi: 10.4049/jimmunol.168.6.2963. [DOI] [PubMed] [Google Scholar]
- 48.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 49.Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A. 1995;92:11490–4. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wetsel RA, Kolb WP. Expression of C5a-like biological activities by the fifth component of human complement (C5) upon limited digestion with noncomplement enzymes without release of polypeptide fragments. J Exp Med. 1983;157:2029–48. doi: 10.1084/jem.157.6.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.You M, Considine RV, Leone TC, Kelly DP, Crabb DW. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology. 2005;42:568–77. doi: 10.1002/hep.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]