

Abstract
Over time it has become clear, that much like other organ systems, the function and responsiveness of the immune system is impaired during the course of sepsis and that this is a precipitous event in the decline of the critically ill patient/animal. One hypothesis put forward to explain the development of septic immune dysfunction is that it is a pathological result of increased immune cell apoptosis. Alternatively, it has been proposed that the clearance of increased numbers of apoptotic cells may actively drive immune suppression through the cells that handle them. Here we will review the data from studies involving septic animals and patients, which indicate that loss of immune cells, as well as non-immune cells, in some cases, is a result of dysregulated apoptosis. Subsequently, we will consider the cell death pathways, i.e., ‘extrinsic’ and/or ‘intrinsic’, which are activated and what cell populations may orchestrate this dysfunctional apoptotic process, immune and/or non-immune. Finally, we will discuss potentially novel therapeutic targets, such as caspases, death receptor family members [e.g., TNF, Fas], pro-/anti-apoptotic Bcl-family members, etc., and approaches, such as caspase inhibitors, use of fusion proteins, peptidomimetics, siRNA, etc., which might be considered for the treatment of the septic patient.
Keywords: Sepsis, mice, human, apoptosis, death-receptor pathway, mitochondrial pathway, organ dysfunction
Introduction
The incidence of sepsis in the United States of America as of the year 2000 was ∼700,000 cases per year, with an overall mortality rate reported to be nearly 30% (Angus et al. 2001). And as the American population ages, it is anticipated that the occurrence of sepsis will only increase (Angus et al. 2001). Unfortunately, with respect to the development of new drug therapies that might substantially impact morbidity and mortality of sepsis, with the exception of recombinant human activated protein C, this has proven to be a difficult task (Rice and Bernard 2005). Even appreciating the important advancements made in the treatment of the critically ill patient through the application of low-dose corticosteroids (Annane et al. 2002) and the improved control of blood glucose (van den Berghe et al. 2001), these still provide a relatively modest survival benefit. Another important difference between these therapies, where some benefit has been seen, is that they have divergent modes of action. With the possible exception of low-dose steroids, they do not directly act on inflammation, in a fashion comparable to that of the failed anti-inflammatory drugs, such as anti-tumor necrosis factor (TNF), anti-interleukin (IL)-1 receptor antagonist, etc., trialed in septic patients over the last 10 years. Taken another way, this suggests that the pathology behind the septic condition in the patient/animal is not simply about inflammation, thus, much more needs to be understood about sepsis and processes underpinning its development if we are either to optimize these present clinical therapies or develop novel approaches.
In this respect, it is now understood that essentially all cells in the body have within them the capacity to undergo cell suicide. This process is referred to apoptosis or programmed cell death and is described as an energy dependent means (unlike necrotic cell injury) that can be initiated by intra-as well as extra-cellular events. This is also largely a non-inflammatory event in which cells are actively eliminated via this pathway during processes as diverse as morphogenesis, tissue remodeling, and the regulation/resolution of the immune response. However, it is also now evident that overt activation or suppression of the apoptotic pathway can contribute to a variety of pathological conditions, such as HIV immune depression, cancer, autoimmune disorders, neurodegenerative diseases, inflammatory bowel disease and ischemic injury (Li et al. 1995;Liles 1997;Barinaga 1998;Barr and Tomei 1994). Therefore, the objective of this review is to discuss the evidence that has been accumulated over the last 10 years that supports not only the concepts that change in the septic animal/patient’s apoptotic process and/or the interaction of the immune system/body with these apoptotic materials contributes to septic morbidity and mortality, but that components of the apoptotic process represent potential novel therapeutic targets.
Apoptotic Pathways
Apoptotic cell death occurs primarily through three general pathways: the extrinsic or death receptor pathway (type I cells), the intrinsic or mitochondrial pathway (type II cells) and the endoplasmic reticulum (ER) or stress-induced pathway (Figure 1) (Hotchkiss et al. 2003;Wesche et al. 2005). In brief, with respect to the extrinsic pathway it is thought that ligation of the death receptors, i.e. Fas, TNF receptor (TNFR), Trail, etc., leads to activation of apoptosis (type I cells) through the recruitment of Fas associated death domain protein (FADD), procaspase-8 (an initiator caspase), etc. via formation of a death inducing signaling complex (DISC) and activation of caspase-3. Several counter-regulatory proteins, such as FADD-like interleukin-1-converting enzyme (FLICE)-inhibitory protein (FLIP), inhibitor of apoptosis protein (IAP), etc., are also present at the level of the DISC and caspase-8 activation.
Figure 1.
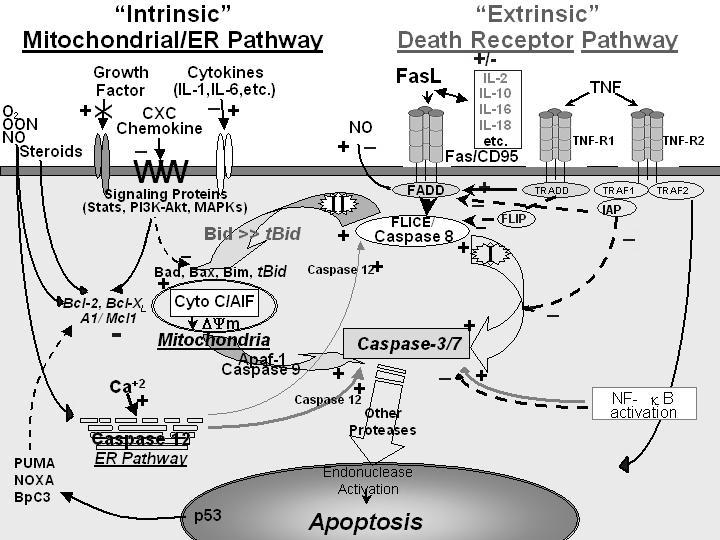
Apoptotic signaling pathways as seen through death receptor ligation of TNFR or Fas (extrinsic signaling, type I cells) or through activation of the mitochondrial pathway through Bcl-2 family members (intrinsic signaling, type II cells).
Alternatively, activation of the intrinsic pathway (type II cells) is regulated by the interaction of various anti-(B-cell CLL/lymphoma 2 [Bcl-2], myeloid cell leukemia sequence 1 [Mcl-1], etc.) and/or pro-apoptotic members (Bcl-2/Bcl-XL-associated death promoter [Bad], Bcl-2-associated X protein [Bax], Bcl-2 interacting mediator of cell death [Bim], BH3 interacting domain death agonist [Bid], etc.) of the Bcl-2 family of proteins, which control mitochondrial ion permeability/flux (ΔΨm) and function. If this process is dys-functional it culminates in the release of various proteins that contribute to apoptosome formation (cytochrome C, Smac/Diablo, apaf-1 and caspase-9) and the eventual activation of caspase-3/7 (an executioner caspase). Intrinsic pathway regulation appears to be a general product of cytokine/chemokine/growth factor regulation via signaling or the lack there of (due to growth factor removal); through phosphatidylinositol-3 kinase (PI3K)/Akt, the mitogen activated protein kinases (MAPK), steroid receptors, nuclear factor kappa-B (NFκB), signal transducers and activators of transcription (STAT), etc. These in turn act either on the various Bcl-2 family members or effect initiator/regulatory caspases, i.e., caspases 1, 8, 10 or IAP activity. The intrinsic pathway is also effected by changes in p53 status mediated through NOXA, PUMA, BbC3, etc., which also act on Bcl-2 family members’ function/expression, that link to processes effecting the cell cycle. Activation of caspase-12 in the ER potentially represents a third possible pathway that is activated by oxidant/calcium stress; however, this appears to be a species-specific apoptotic effecter. That said, these pathways are not wholly autonomous as the cleavage of BID to truncated BID (tBID) by caspase-8 exhibits a significant aspect of cell death pathway crosstalk and a number of the regulators mentioned in the intrinsic pathway can directly/indirectly affect the extrinsic pathway’s activity.
Evidence that Apoptosis Contributes to the Pathology of Sepsis
Studies in recent years have suggested that dysregulated apoptotic immune cell death may play a role in contributing to the immune dysfunction and multiple organ failure observed during sepsis and that blocking it can improve survival of experimental animals (Chung et al. 2003;Hotchkiss et al. 2003). With respect to the immune system, those cells most commonly reported to exhibit evidence of dysregulated apoptotic cell death appear to be lymphocytes (Figure 2). That the loss of lymphocytes is detrimental to the survival of septic mice is documented by the observation that RAG-/-mice are markedly more susceptible to lethal effects of polymicrobial septic challenge, cecal ligation and puncture (CLP), than their background controls (Hotchkiss et al. 2003). Typically, lymphocyte apoptosis has been seen following the onset of experimental sepsis in the thymus, spleen, and gut-associated lymphoid tissues (GALT). The most basic hypothesis arising from these observations is that the overt apoptotic loss of these lymphocytes in the septic animal/patient reduces the number of functional immune cells available to ward off the lethal effects of septic challenge (Figure 3A). Probably the most potent support for this hypothesis comes from the data indicating that if the development of lymphocyte apoptosis is block via the restricted over-expression of Bcl-2, this ameliorates much of the mortality seen in the CLP model of sepsis in mice (Hotchkiss et al. 2003).
Figure 2.
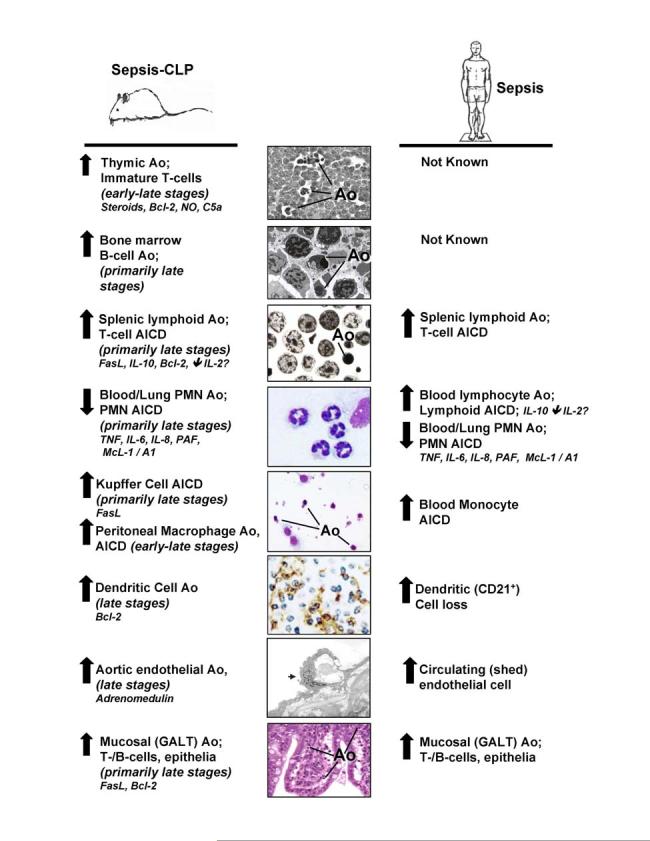
The levels of apoptosis (Ao) seen in experimental septic mice and septic patients, as well as the mediators that affect the onset and frequency of Ao in various immune cell types.
Figure 3.
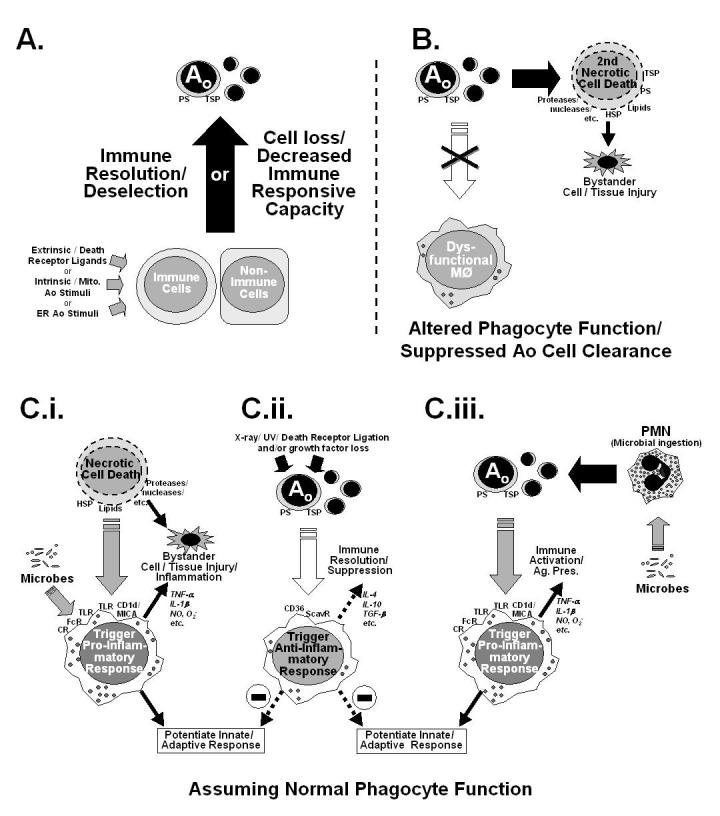
A depiction of several possible mechanisms of immune suppression. (A) Illustrates the simple hypothesis (mechanism) that the immune dysfunction observed is a result of advertant/inadvertent apoptotic (Ao) loss of immune cell potential/capacity resultant from extrinsic and/or intrinsic Ao pathway activation. Here no consideration is made for Ao cell clearance. (B) Depicts a scheme in which phagocytic function is compromised, so as to block apoptotic cell clearance, subsequently allowing apoptotic cells to move into secondary necrosis, that may in turn produce by-stander tissue injury. The scenarios illustrated in C.i.-C.iii. represent the proposed effects that clearance of necrotic (C.i.) and/or apoptotic cell materials (induced by classic Ao stimuli [C.ii.] or ingestion of microbes [C.iii.])has on the developing macrophage functional phenotype (pro-inflammatory vs. anti-inflammatory/immune suppressive) is considered when phagocytic function is normal. CD36, cell differentiation antigen 36; CR, complement receptor(s); CD1d/MICA non-variant major histocompatability class 1-like antigen family; FcR, immunoglobulin constant region receptor(s); HSP, heat shock protein(s); PS, phosphotydal serine; ScavR, Scavenger receptor(s) which bind PS; TSP, thrombospondin.
Other immune cell types, that have been reported to exhibit an increased incidence of apoptosis in sepsis, are CD8+ lymphoid-derived dendritic cells of the spleen after CD3+CD4+ T cell activation (Hotchkiss et al. 2003) (Figure 2). Interestingly, the potential impact of the loss of dendtritic cells, via apoptosis or some other process, on the septic animals ability to survive experimental sepsis, is illustrated by the increased mortality seen in conditionally-induced dendritic cell deficient mice (CD11c-diptheria toxin receptor transgenic mice) (Scumpia et al. 2005). While a little more controversial (because of the capacity to phagocytes to engulf apoptotic material), macrophages have in some cases also been reported to undergo apoptosis during sepsis (Williams et al. 1998;Iwata et al. 2003). Here again, indirect evidence from Iwata et al. (Iwata et al. 2003) show that mice over-expressing Bcl-2 under a myeloid as opposed to a lymphoid restricted fashion, not only produced a survival advantage but also found that adoptive transferred of blood monocytes from these animals to septic rodents improved their survival. This implies that protecting the myeloid compartment from apoptosis is also a valuable asset to the host’s immune response to sepsis. These results, however, extend the concept from simply the loss of lymphocytes to include the loss of dendritic cells and myeloid cells as contributors to the septic host’s inability to ward off the lethal effects of sepsis.
In contrast, it is worth noting that in vivo neutrophils, unlike lymphocytes or the other cell types mentioned above, undergo constitutive induction of apoptosis within 1-2 days normally (Figure 2). Also unlike the lymphocytes, in response to a septic insult they react with a decrease in apoptosis, which appears to be linked to a decrease in caspase-9 and caspase-3 activity and a prolonged maintenance of the mitochondrial transmembrane potential (Taneja et al. 2004). Studies investigating neutrophil apoptosis during septic peritonitis in mice have revealed that while blood neutrophils react with a decrease in apoptosis, which is TNF-α regulated, after immigration into the peritoneal cavity they exhibit increased apoptosis (Wesche et al. 2005). However, in some cases this may also be detrimental. As an example, experimental hemorrhage also primes neutrophils into delaying apoptosis and increasing their respiratory burst (Ayala et al. 2002) in a fashion similar to that seen in trauma patients’ blood neutrophils (Jimenez et al. 1997). Transfer of these experimentally primed neutrophils to a septic environment increases their recruitment into the lungs and leads to acute lung injury (Ayala et al. 2002).
That said, it is now becoming more evident that non-immune cells may also be exhibiting apoptotic changes that were not initially clear because they were not rigorously looked for or were not obvious based on the techniques available (Figure 2). In this respect, increased apoptosis of the gut mucosal epithelial cells (Coopersmith et al. 2002;Perl et al. 2005), hepatocytes (Kim et al. 2000) and to a lesser extent endothelial cells (Mutunga et al. 2001) have been reported in clinically comparable models of sepsis (Zhou et al. 2004). While the significance of these apoptotic events in sepsis is yet to be clarified, it is tempting to speculate how this potentiated loss of cells might contribute not only to loss of innate host defense/barrier function but also organ to dysfunction/damage.
What regulates these changes in septic immune cell apoptosis during sepsis? Interestingly, this appears, at least with respect to the lymphoid compartment, to vary somewhat with the cell and/or tissue type being examined. Also while high doses of endotoxin can induce this process in vitro and/or in vivo, studies with more clinically comparable models of sepsis indicate that much of the immune cell apoptosis seen occurs independent of the capacity to sense/respond to endotoxin through TLR4 (Wesche et al. 2005;Hotchkiss et al. 2003). Lymphocyte apoptosis in the thymus appears to be effected by glucocorticoids (Wesche et al. 2005) nitric oxide as well as by the 5a fragment of complement (Guo et al. 2000), but not by endotoxin or death receptors (Wesche et al. 2005). Alternatively, in the bone marrow and lamina propria B cells (Wesche et al. 2005), splenic T cells, intestinal intraepithelial lymphocytes (IELs), and mucosal T and B cells of the Peyer’s patches (Chung et al. 1998;Ayala et al. 2003), apoptosis is mainly death receptor-driven via Fas-FasL activation. Interestingly, evidence of both intrinsic and extrinsic apoptotic pathway activation has been reported in the peripheral blood of septic patients (Hotchkiss et al. 2005).
Lymphocyte apoptosis may be associated with immune dysfunction as a result of decreased proliferation and IFN-γ release capability. IFN-γ is a potent macrophage activator and induces a Th1 response (Docke et al. 1997). As seen in apoptotic and necrotic splenocyte adoptive transfer experiments, necrotic and apoptotic cells exert their effects through variation of IFN-γ levels. Transfer of apoptotic splenocytes retro-orbitally in CLP mice decreased their survival, whereas adoptive transfer of necrotic splenocytes increased splenocyte IFN-γ, and in doing so, improved survival. This survival benefit was blocked in IFN-γ deficient mice or in mice treated with an anti-IFN-γ antibody. These results are interesting as this adoptive transfer study illustrates the potential impact of apoptotic cells in vivo in sepsis and thus points to another mechanism (beside loss/death of functional immune cells (Figure 3B-C)) by which immune suppression might be promulgated in the septic animal (Hotchkiss et al. 2003) (Figure 3Cii). Alternatively, the inability to appropriately clear these dying lymphocytes/cells (due to dysfunction in their phagocytic capacities often seen following sepsis and shock (Rana et al. 1990;Huber-Lang et al. 2002)) may allow them to progress to a state of secondary necrosis, producing localized bystander injury in the tissue (Fig. 3B). Such a scenario has been recently put forward by Vandivier et al (Vandivier et al. 2002) as a possible mechanism for tissue inflammation and the enhanced susceptibility to infection seen in cystic fibrosis patients. It remains to be determined whether such defects in macrophage-mediated clearance of apoptotic cells contribute to the changes seen in septic mice.
Intrinsic and Extrinsics Aspects of the Apoptotic Process as a Therapeutic Target
One of the earliest anti-apoptotic approaches considered for sepsis was the inhibition of caspase activation. Caspase inhibitors usually contain fluoromethyl ketones (fmk) or chloromethyl ketones (cmk) that are derivatives of peptides that imitate cleavage sites of known caspase substrates. The caspase-specific inhibitors that have been used inclde z-DEVD-fmk (caspase-3 and -7) and Ac-YVAD-cmk (caspase-1) (Rouquet et al. 1996). It has also been shown that broad spectrum caspase inhibitors such as z-VAD-fmk can prevent lymphocyte apoptosis in sepsis, and in turn, improve septic animal survival by 40-45% (Hotchkiss et al. 2003). However, at high doses, caspase inhibitors can have non-specific effects and cause cytotoxicity. In this respect, a different kind of pan-caspase inhibitor called Q-VD-Oph has been studied (Caserta et al. 2003), which potently inhibits apoptosis but is less toxic at high doses and may be more efficacious in the clinical setting.
Peptidomimetics represent another potentially useful method of targeting the apoptotic pathway. “Peptidomimetics” are mimics that have similar structure and functional properties of the native parental peptides. This approach was adopted since the use of biologically active peptides as pharmaceutical compound have more or less failed due to their inability to stay bio-available and reach their cellular targets. Synthetic mimics, however, can be generated to be more conformationally stable compounds that resist enzyme degradation, can cross cell membranes, and target specific proteins (Li et al. 2004). We mention this new type of approach here, for while it is being studied at present only for its potential pro-apoptotic effect on Bid (Walensky et al. 2004) activation as well as via antagonism of IAP (Li et al. 2004) in cancer models, it seems possible to speculate that peptidomimetics may also be useful in future therapies that modulate anti-apoptotic protein-protein interactions in the case of sepsis. This idea may also be an alternative to gene therapy, which, in the past, has not done well in the clinical setting due to marked inflammatory sequelae and oncogenesis (Lehrman 1999).
With respect to gene therapy, enhancement of anti-apoptotic proteins, such as Bcl-2, has been shown to produce almost complete protection against T cell apoptosis in mice that over-express Bcl-2. This, in turn, improved their survival after sepsis (Hotchkiss et al. 2003). In addition, adoptive transfer of T cells from Bcl-2 over-expressing mice into wild type septic mice also improved their survival (Hotchkiss et al. 2003). While this clearly illustrates the important role of the lymphocyte in sepsis in controlling infection, it also illustrates a clear therapeutic target, which might also be exploited to restore lymphocytes numbers during this state.
Yet another target that decreases lymphocyte apoptosis is Akt, a regulator of cell proliferation and death. It has been shown that in mice over-expressing Akt that lymphocyte apoptosis is decreased and survival after CLP is improved to 94% (Bommhardt et al. 2004). It may be at this level, (i.e. the activation of Akt) that treatments such as glucose control with insulin therapy or low-dose steroids have an effect on apoptosis in septic individuals. Based on the observation of increased Fas expression in the tissues of septic mice (Chung et al. 2003;Chung et al. 1998;Wesche-Soldato et al. 2005), there has been increasing interest in targeting components of the death receptor/extrinsic pathway in an attempt to ameliorate the effects of sepsis. In this regard, studies from our own lab had initially focused on blocking the pathway at the death receptor itself (Fas). Animals that were treated with Fas fusion protein (FasFP; Amgen Inc., Thousand Oaks, CA), to inhibit the receptor ligation, exhibited a marked survival benefit (Chung et al. 2003), reduced hepatic injury while improving total hepatic, intestinal and cardiac blood flow during sepsis (Wesche et al. 2005;Chung et al. 2001). In addition, the cell survival tyrosine kinase (MET) has been found to sequester Fas on hepatocytes but in a fashion that inhibits Fas self-aggregation and Fas ligand binding, protecting the liver from injury (Wang et al. 2002).
Most recently, we have utilized interfering RNA technology to target gene expression of members of the extrinsic death receptor/Fas pathway. Fas and caspase-8 siRNA given 30 min. after CLP improved survival by 50% and reduced indices of organ damage and apoptosis in both the liver and spleen (Wesche-Soldato et al. 2005). This is in keeping with the findings that both Fas and caspase-8 siRNA have salutary effects in models of fulminate hepatitis (Song et al. 2003). The mechanism of this survival benefit in sepsis is still largely unknown; however, preliminary data from our lab suggest that CD4+ and CD8+ T cells, B cells as well as hepatocytes take up Fas siRNA. In the case of the spleen, it appears that lymphocyte apoptosis is reduced by silencing Fas, therefore enabling the host to maintain innate and/or adaptive immune response to septic challenge. With respect to the liver, preliminary results suggest that Fas siRNA reduces not only the expression of Fas on potential target cells in the liver but also suppresses recruitment of potentially activated cytotoxic lymphocytes. It has been suggested in a model of hepatitis C that Fas ligand expressing CD4+ T cells can induce chronic hepatic inflammation (Cruise et al. 2005), which in the case of sepsis, may be instrumental in initiating multiple organ failure. By the same token, experimental CLP mice lacking CD8+ T cells exhibit improved survival over wild type (Sherwood et al. 2003).
As intriguing as this approach is, several hurdles need to be overcome, beyond cell targeting, before siRNA can be applied clinically. While we saw little evidence of overt toxicity/off-target effects/inflammation, as we observed no marked induction of IFN-α, TNF-α or IL-6 with either targeted siRNAs or the control constructs, these issues will need further consideration as the delivery systems are changed/modified and/or lipid conjugations of siRNA are added (Wesche-Soldato et al. 2005). Probably the biggest hurdle facing the use of siRNA clinically is that of its delivery. While a hydrodynamic-based method (where large volume and rapid rate of injection are critical in the uptake of naked constructs) is easily employed in research, it clearly cannot be used in human therapy in this way. Because naked siRNAs are degraded within seconds following a normal low volume injection (as opposed to hydrodynamic-based delivery), there needs to be a carrier or delivery vehicle that will protect the siRNA. Cationic liposomes encoding anti-TNF siRNA have been studied, and appeared to reduce TNF-α levels after endotoxemia. While this represents a useful alternative method for encapsulating/delivering siRNA, uptake of siRNA was largely limited to vascular endothelial cells following i.v. administration (Sorenson et al. 2003). Other investigators have suggested the use of vectors as another mode of targeted siRNA delivery. However, vectorized gene delivery may cause inflammation by itself (Bridge et al. 2004).
Conclusions
Despite overwhelming research efforts and clinical trials, there has yet to be a therapy offered that significantly modifies the outcome of sepsis. Even though there have been promising candidates for therapeutic intervention, sepsis manifests itself as multiple processes, making this task difficult. Here we have reviewed several experimental studies that have focused not only on the apoptotic process and it’s potential pathological roles in sepsis, but have revealed several targets within the apoptotic pathway, which may be useful going forward in designing individual or adjuvant therapies for this critically ill patient population.
Acknowledgements:
This work was supported in part by funds from NIH-RO1s GM53209 and HL73525 (to A.A.) as well as fellowship support from NIH-T32 GM65085 (for R.S.) and GAANN P200A03100 (for D.E.W-S.).
References:
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- Annane D, Sebille V, Charpentier C, Bollaert P-E, Francois B, Korach J-M, Capellier G, Cohen Y, Azoulay E, Troche G, Chaumet-Riffaut P, Bellissant E. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–871. doi: 10.1001/jama.288.7.862. [DOI] [PubMed] [Google Scholar]
- Ayala A, Chung CS, Lomas JL, Song GY, Doughty LA, Gregory SH, Cioffi WG, LeBlanc BW, Reichner J, Simms HH, Grutkoski PS. Shock induced neutrophil mediated priming for acute lung injury in mice: divergent effects of TLR-4 and TLR-4/FasL deficiency. Amer J Pathol. 2002;161:2283–2294. doi: 10.1016/S0002-9440(10)64504-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala A, Lomas JL, Grutkoski PS, Chung CS. Pathological aspects of apoptosis in severe sepsis and shock? Int J Biochem & Cell Biol. 2003;35:7–15. doi: 10.1016/s1357-2725(02)00099-7. [DOI] [PubMed] [Google Scholar]
- Barinaga M. Is apoptosis key in alzhimer’s disease? Science. 1998;281:1303–1304. doi: 10.1126/science.281.5381.1303. [DOI] [PubMed] [Google Scholar]
- Barr PJ, Tomei LD. Apoptosis and its role in human disease. Bio/Technology. 1994;12:487–493. doi: 10.1038/nbt0594-487. [DOI] [PubMed] [Google Scholar]
- Bommhardt U, Chang KC, Swanson PE, Wagner TH, Tinsley KW, Karl IE, Hotchkiss RS. Akt decreases lymphocyte apoptosis and improves survival in sepsis. J Immunol. 2004;172:7583–7591. doi: 10.4049/jimmunol.172.12.7583. [DOI] [PubMed] [Google Scholar]
- Bridge AJ, Pebernard S, Ducraux A, Nicovlaz AL, Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nature Genet. 2004;34:263–264. doi: 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8:345–352. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- Chung CS, Song GY, Lomas J, Simms HH, Chaudry IH, Ayala A. Inhibition of Fas/Fas ligand signaling improves septic survival: differential effects on macrophage apoptotic and functional capacity. J Leukoc Biol. 2003;74:344–351. doi: 10.1189/jlb.0102006. [DOI] [PubMed] [Google Scholar]
- Chung CS, Xu YX, Wang W, Chaudry IH, Ayala A. Is Fas ligand or endotoxin responsible for mucosal lymphocyte apoptosis in sepsis? Arch Surg. 1998;133:1213–1220. doi: 10.1001/archsurg.133.11.1213. [DOI] [PubMed] [Google Scholar]
- Chung CS, Yang SL, Song GY, Lomas J, Wang P, Simms HH, Chaudry IH, Ayala A. Inhibition of Fas signaling prevents hepatic injury and improves organ blood flow during sepsis. Surgery. 2001;130:339–345. doi: 10.1067/msy.2001.116540. [DOI] [PubMed] [Google Scholar]
- Coopersmith CM, Stromberg PE, Dunne WM, Davis CG, Amiot IDM, Buchman TG, Karl IE, Hotchkiss RS. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA. 2002;287:1716–1721. doi: 10.1001/jama.287.13.1716. [DOI] [PubMed] [Google Scholar]
- Cruise MW, Melief HM, Lukens J, Soguero C, Hahn YS. Increased Fas ligand expression of CD4 +Tcells by HCV core induces T cell-dependent hepatic inflammation. J Leukocyte Biol. 2005;78:412–425. doi: 10.1189/jlb.0105005. [DOI] [PubMed] [Google Scholar]
- Docke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, Volke HD, Kox WJ. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. 1997;3:678–681. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- Guo R-F, Huber-Lang M, Wang X, Sarma V, Padgaonkar VA, Craig RA, Riedemann NC, McClintock SD, Hlaing T, Shi MM, Ward PA. Protective effects of anti-C5a in sepsis-induced thymocyte apoptosis. J Clin Invest. 2000;106:1271–1280. doi: 10.1172/JCI10793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RS, Osmon SB, Chang KC, Wagner TH, Coopersmith CM, Karl IE. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathway. J Immunol. 2005;174 doi: 10.4049/jimmunol.174.8.5110. in press. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Tinsley KW, Karl IE. Role of apoptotic cell death in sepsis. Scand J Infect Dis. 2003;35:585–592. doi: 10.1080/00365540310015692. [DOI] [PubMed] [Google Scholar]
- Huber-Lang MS, Riedemann NC, Sarma JV, Younkin EM, McGuire SR, Laudes IJ, lung KT, Guo RF, Neff TA, Padgaonkar VA, Lambris JD, Spruce L, Mastellos D, Zetoune FS, Ward PA. Protection of innate immunity by C5aR antagonist in septic mice. FASEB J. 2002;16:1567–1574. doi: 10.1096/fj.02-0209com. [DOI] [PubMed] [Google Scholar]
- Iwata A, Stevenson VM, Minard A, Tasch M, Tupper J, Lagasse E, Weissman I, Harlan JM, Winn RK. Over-expression of Bcl-2 provides protection in septic mice by a trans effect. J Immunol. 2003;171:3136–3141. doi: 10.4049/jimmunol.171.6.3136. [DOI] [PubMed] [Google Scholar]
- Jimenez MF, Watson WG, Parodo J, Evans D, Foster D, Steinberg M, Rotstein OD, Marshall JC. Dysregulated expression of neutrophil apoptosis in the systemic inflammatory response syndrome. Arch Surg. 1997;132:1263–1270. doi: 10.1001/archsurg.1997.01430360009002. [DOI] [PubMed] [Google Scholar]
- Kim Y-M, Kim T-H, Chung H-T, Talanian RV, Yin X-M, Billiar TR. Nitric oxide prevents tumor necrosis factor α-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology. 2000;32:770–778. doi: 10.1053/jhep.2000.18291. [DOI] [PubMed] [Google Scholar]
- Lehrman S. Virus treatment questioned after gene therapy death. Nature. 1999;401:517–518. doi: 10.1038/43977. [DOI] [PubMed] [Google Scholar]
- Liles CJ, Friedman DJ, Wang C, Metelev V, Pardee AB. Induction of apoptosis in uninfected lymphocytes by HIV-1 Tat protein. Science. 1995;268:429–431. doi: 10.1126/science.7716549. [DOI] [PubMed] [Google Scholar]
- Liles L, Thomas RM, Suzuki H, DeBrabander JK, Wang X, Harran PG. A small molecule smac mimic potentiates TRAIL- and TNF-alpha-mediated cell death. Science. 2004;305:1471–1474. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- Liles WC. Apoptosis-role in infection and inflammation. Current Opinion in Infectious Diseases. 1997;10:165–170. [Google Scholar]
- Mutunga M, Fulton B, Bullock R, Batchelor A, Gascoigne A, Gillespie JI, Baudouin SV. Circulating endothelial cells in patients with septic shock. Am J Respis Crit Care Med. 2001;163:195–200. doi: 10.1164/ajrccm.163.1.9912036. [DOI] [PubMed] [Google Scholar]
- Perl M, Chung CS, Lomas-Neira J, Rachel TM, Biffl WL, Cioffi WG, Ayala A. Silencing of Fas-but not caspase-8 in lung epithelial cells ameliorates experimental acute lung injury. Am J Pathol. 2005;167:1545–1559. doi: 10.1016/S0002-9440(10)61240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana MW, Ayala A, Dean RE, Chaudry IH. Decreased Fc receptor expression on macrophages following simple hemorrhage as observed by scanning immuno electron microscopy. J Leukocyte Biol. 1990;48:512–518. doi: 10.1002/jlb.48.6.512. [DOI] [PubMed] [Google Scholar]
- Rice TW, Bernard GR. Therapeutic interventions and targets for sepsis. Annual Review Medicine. 2005;56:225–248. doi: 10.1146/annurev.med.56.082103.104356. [DOI] [PubMed] [Google Scholar]
- Rouquet N, Pages JC, Molina T, Briand J, Joulin V. ICE inhibitor YVADcmk is a potent therapeutic agent against in vivo liver apoptosis. Curr Biol. 1996;6:1192–1195. doi: 10.1016/s0960-9822(02)70688-x. [DOI] [PubMed] [Google Scholar]
- Scumpia PO, McAuliffe PF, O’Malley KA, Ungaro R, Uchida T, Matsumoto T, Remick DG, Clare-Salzler MJ, Moldawer LL, Efron PA. CD11c+ dendritic cells are required for survival in murine polymicrobial sepsis. J Immunol. 2005;175:3282–3286. doi: 10.4049/jimmunol.175.5.3282. [DOI] [PubMed] [Google Scholar]
- Sherwood ER, Lin CY, Tao W, Hartmann CA, Dujan JE, French AJ, Varma TK. B2 microglobulin knockout mice are resistant to lethal intra-abdominal sepsis. Am J Respir Crit Care Med. 2003;167:1641–1649. doi: 10.1164/rccm.200208-950OC. [DOI] [PubMed] [Google Scholar]
- Song E, Lee S-K, Wang J, Ince N, Ouyang N, Min J, Chen J, Shankar P, Lieberman J. RNA interference targeting Fas protects mice from fulminant hepatitis. Nature Medicine. 2003;9:347–351. doi: 10.1038/nm828. [DOI] [PubMed] [Google Scholar]
- Sorenson DR, Leirdal M, Sioud M. Gene silencing by systemic delivery of synthetic siRNAs in adult mice. J Mol Biol. 2003;327:761–766. doi: 10.1016/s0022-2836(03)00181-5. [DOI] [PubMed] [Google Scholar]
- Taneja R, Parodo J, Jia SH, Kapus A, Rotstein OD. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med. 2004;32:1460–1469. doi: 10.1097/01.ccm.0000129975.26905.77. [DOI] [PubMed] [Google Scholar]
- van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive Insulin Therapy in Critically Ill Patients. N Engl J Med. 2001;345:1359–1367. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- Vandivier RW, Fadok VA, Hoffmann PR, Bratton DL, Penvari C, Brown KK, Brain JD, Accurso FJ, Henson PM. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest. 2002;109:661–670. doi: 10.1172/JCI13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, DeFrances MC, Dai Y, Pediaditakis P, Johnson C, Bell A, Michalopoulos GK, Zarnegar R. A mechanism of cell survival: sequestration of Fas by the HGF receptor Met. Mol Cell. 2002;9:411–421. doi: 10.1016/s1097-2765(02)00439-2. [DOI] [PubMed] [Google Scholar]
- Wesche-Soldato DE, Chung CS, Lomas-Neira JL, Doughty LA, Gregory SH. In vivo delivery of caspase 8 or Fas siRNA improves the survival of septic mice. Blood. 2005;106:2295–2301. doi: 10.1182/blood-2004-10-4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesche DE, Lomas-Neira JL, Perl M, Chung CS, Ayala A. Leukocyte apoptosis and its significance in sepsis and shock. J Leukocyte Biol. 2005;25:325–337. doi: 10.1189/jlb.0105017. [DOI] [PubMed] [Google Scholar]
- Williams MA, Withington S, Newland AC, Kelsey SM. Monocyte anergy in septic shock is associated with a prediliction to apoptosis and is reversed by granulocyte-macrophage colony stimulating factor ex vivo. J Infect Dis. 1998;178:1421–1433. doi: 10.1086/314447. [DOI] [PubMed] [Google Scholar]
- Zhou M, Simms HH, Wang P. Adrenomedullin and adrenomedullin binding protein-1 attenuate vascular endothelial cell apoptosis in sepsis. Ann Surgery. 2004;240:321–330. doi: 10.1097/01.sla.0000133253.45591.5b. [DOI] [PMC free article] [PubMed] [Google Scholar]