

Abstract
A variety of genetic alterations and gene expression changes are involved in the pathogenesis of bladder tumors. To explore expression changes in 4-hydroxybutyl(butyl)nitrosamine-induced rat bladder tumors, microarray analysis was performed. Analysis yielded 1,138 known genes and 867 expressed sequence tags that were changed when comparing tumors to normal rat epithelia. Altered genes included cell cycle-related genes, EGFR-Ras signaling genes, apoptosis genes, growth factors, and oncogenes. Using the pathway visualization tool GenMAPP, we found that these genes can be grouped along several pathways that control apoptosis, cell cycle, and integrin-mediated cell adhesion. When comparing current data with previous mouse bladder tumor data, we found that > 280 of the same known genes were differentially expressed in both mouse and rat bladder tumors, including cell cycle-related genes, small G proteins, apoptosis genes, oncogenes, tumor-suppressor genes, and growth factors. These results suggest that multiple pathways are involved in rat bladder tumorigenesis, and a common molecular mechanism was found in both rat and mouse bladder tumors.
Keywords: Rat bladder tumors, expression profile, microarray, chemical carcinogen, pathways
Introduction
Bladder cancer is the fifth most common cancer in the United States and is associated with exposure to cigarette smoke. Approximately 15% of bladder tumors evolve into invasive tumors after infiltration through the basement membrane. Patients with muscle-invasive disease are at high risk for recurrence, progression, and metastasis. Although early-stage bladder cancer can be treated surgically, the rate of recurrence is quite high [1]. Significant progress has been made in understanding the underlying molecular and genetic events in bladder cancer. Numerous markers have been described to correlate, to some extent, tumor stage and the prognosis of patients with bladder cancer [2]. Although a number of markers have been identified, there remains a need for the development of reliable additional markers that can provide information regarding diagnosis and prognosis. In addition, the identification of specific proteins that might be favorable targets for treatment is of some interest. Expression profiling with high-throughput DNA microarrays has the potential of providing critical clues. Our previous study on mouse bladder tumors revealed that activation of the EGFR-Ras, G13, and TGF-β pathways, and increased cell proliferation appear to play important roles during mouse bladder tumorigenesis.
There are two primary chemically induced models of urinary bladder cancers in rodents. Both employ repeated intragastric administration of 4-hydroxybutyl(butyl)nitrosamine (OH-BBN) to induce bladder cancers in either mice or rats [3,4]. Bladder cancers typically have a mixed histology, showing elements of both transitional and squamous cells. Investigators have found a relatively low frequency of ras mutation in these cancers [5]. However, roughly 50% of these tumors develop p53 mutations [6]_a percentage similar to that found in humans. There has been further characterization of these tumors for various gene products, including mutations in the epidermal growth factor receptor (EGFR) kinase activation loop [7]. Similar to human bladder tumors, these tumors tend to show overexpression of EGFR and amphiregulin. Other genetic changes include ras, erb-B2, and EGFR. The transforming potential of ras is due to mutation, whereas EGFR and erb-B2 are overexpressed in transformed cells. Reported frequencies of H-ras point mutations with a glycine-to-valine substitution in codon 12 in bladder neoplasms vary widely from 0% to 45% between studies [8–11]. Recently, several means of suppressing ras activity, including inhibitors of ras signal transduction and a ras-suppressor mutant, have been reported [12]. Overexpression of EGFR or erb-B2 and ras mutations results in constitutive MAPK activation [13], and this correlates with muscular invasion and extent of tumor invasion [2]. Almost all advanced bladder carcinomas exhibit alterations in cell cycle genes (e.g., decreases in pRb or p16INK4a, or increases in cyclin D1 expression preferentially occurring in earlier stages) [14,15].
In this study, we employed Affymetrix GeneChips (Affymetrix, Santa Clara, CA) representing > 30,000 genes and expressed sequence tags (ESTs) to identify differentially expressed genes in rat bladder tumors. The objectives of the study were: 1) to detect and identify differential gene expression profiles in rat bladder tumors; 2) to help elucidate the underlying mechanisms of rat bladder tumorigenesis; and 3) to compare the present results with our previous data on mouse bladder tumors to identify common genes and pathways that may be particularly relevant to the mechanism of carcinogenesis in the bladder.
Materials and Methods
Rat Bladder Tumors
Rats were obtained from Harlan Sprague-Dawley, Inc. (Indianapolis, IN), at 28 days of age and were housed in polycarbonate cages (five per cage). The animals were kept in a lighted room 12 hours each day and maintained at 22 ± 0.5°C. Teklad 4% mash diet (Harlan Teklad, Madison, WI) and tap water were provided ad libitum. At 56 days of age, mice received the first of 12 weekly gavage treatments with OH-BBN (TCI America, Portland, OR). Each 7.5-mg dose was dissolved in 0.1 ml of ethanol/water (25:75). Rats (unless sacrificed early because of a large palpable bladder mass) were sacrificed 8 months following the first OH-BBN treatment. Bladder tumors were removed and frozen for subsequent molecular assays. A portion of each tumor was fixed and processed for routine paraffin embedding, cut into 5-µm sections, and mounted for hematoxylin-eosin staining for histopathology. All bladder tumors used in this study were diagnosed as bladder carcinomas, with a mixed histology showing elements of both transitional and squamous cells. Both bladder tissues and normal bladder epithelia came from age-matched controls.
RNA Isolation and Amplification
To isolate bladder epithelia, we separated the epithelia from the stroma and muscle tissues by cutting the bladder into half and scraping off the epithelium. Total RNA from normal bladder epithelia and bladder tumors were isolated by Trizol (Invitrogen, Carlsbad, CA) and purified using the RNeasy Mini Kit and RNase-free DNase Set (QIAGEN, Valencia, CA) according to the manufacturer's protocols. In vitro transcription-based RNA amplification was then performed on each sample. cDNA for each sample was synthesized using a Superscript cDNA Synthesis Kit (Invitrogen) and a T7-(dT)24 primer, 5′-GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGG-(dT)24-3′. cDNA were cleaned using phase-lock gels (Fisher cat ID E0032005101) and phenol/chloroform extraction. Then, biotin-labeled cRNA were transcribed in vitro from cDNA using a BioArray High Yield RNA Transcript Labeling Kit (ENZO Biochem, New York, NY) and purified again using the RNeasy Mini Kit.
Affymetrix GeneChip Probe Array and Quantitative Confirmation by Real-Time Polymerase Chain Reaction (PCR)
The labeled cRNA were applied to Affymetrix Rat 230 2.0 GeneChips (Affymetrix) according to the manufacturer's recommendations. Every gene or EST is represented by a probe set consisting of approximately 16 probe pairs (oligonucleotides) of 25-mer oligonucleotides. One sequence of a probe pair represents the complementary strand of the target sequence, whereas the other has a 1-bp mismatch at the central basepair position. This mismatch sequence serves as an internal control for the specificity of hybridization. To evaluate the reliability of array results, genes were randomly selected from the genes detected in the microarray assay for further confirmation by real-time PCR, as previously described [16]. The large number of differentially expressed genes led us to take a further quality control step in which the distribution of fold changes was examined.
Cluster and GenMAPP
Array normalization and gene expression estimates were obtained using Affymetrix Microarray Suite 5.0 software (MAS5). Array mean intensities were scaled to 1500. These estimates formed the basis for statistical testing. Differential expression was determined using the combined basis of t-test with P < .05 and fold changes (either up or down) of > 2-fold. Genes meeting both criteria were called positive for differential expression. Hierarchical clustering was then performed as follows. For selected genes, expression indexes were transformed across samples to an N(0,1) distribution using a standard statistical Z-transform. These values were put into the GeneCluster program of Eisen et al. [17], and genes were clustered using average linkage and correlation dissimilarity. Signal transduction pathways, metabolic pathways, and other functional groupings of genes were evaluated for differential regulation using the visualization tool GenMAPP [18]. We imported the statistical results of our data set into the program and used GenMAPP to illustrate pathways containing differentially expressed genes.
Protein Isolation and Two-Dimensional (2D) Protein Gel
Protein was isolated from epithelia and tumors, and proteomic analysis was performed using 2D differential gel electrophoresis. Protein samples from five normal and five tumor-bearing animals were paired. The protein samples (50 µg) were labeled substoichiometrically with one of two N-hydroxysuccinimide cyanine dyes (GE Healthcare Biosciences, Piscataway, NJ). Immobilized pH gradient strips (24 cm; pH 3–11, nonlinear) were rehydrated with the samples (pooled normal and tumor). The first dimension of isoelectric focusing was performed for 75 kV h in Protean IEF Cell System (Bio-Rad, Hercules, CA). The strips were equilibrated and positioned on a 10% to 20% gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel. Resolved protein images were acquired on a Typhoon 9400 scanner (GE Healthcare Biosciences). Relative quantification of matched gel features was performed using Decyder-DIA software (GE Healthcare Biosciences). Selected gel features were excised and digested in situ with trypsin. The resulting peptide pools were analyzed by tandem mass spectrometry using both matrix assisted layer desorption/ionization tandem time-of-flight mass spectrometer (MALDI-TOF/TOF) (Proteomics 4700; Applied Biosystems, Framingham, MA, and Toronto, Ontario, Canada) and liquid chromatography-tandem mass spectrometer (LC-MS/MS) (LTQ-FTMS; Thermolelectron, San Jose, CA) instruments. Peptide fragmentation spectra were processed using Data Explorer v. 4.5 and Analyst software (Applied Biosystems, Foster City, CA), and MASCOT v. 1.9 (Matrix Sciences, London, UK).
Immunohistochemistry (IHC) Staining
Briefly, paraffin sections of rat normal and tumor bladders (n = 5) were antigen-retrieved in citrate buffer for 20 minutes in a microwave. This was followed by blocking in normal horse serum, and sections were incubated in primary antibody cyclin D1 (sc-450, 1:500; Santa Cruz Biotech, Santa Cruz, CA) or annexin I (sc-12740, 1:50; Santa Cruz Biotech) overnight at 4°C. The corresponding biotinylated secondary IgG (1:500) was used, and the sections were developed by the ABC method (Vector Laboratories, Burlingame, CA) with 3,3′-diaminobenzidine HCl as substrate.
Results
Gene Expression Profile in Bladder Tumors
Microarray data were compared for five rat bladder tumors and their age-matched normal rat bladder epithelia. The fold changes of gene expression were based on the ratios of mean values between tumors and epithelium controls. Two thousand five known genes and ESTs were found to be differentially expressed in rat bladder tumors with a fold change of ≥ 2 and P < .05. Among them, 1,138 genes were known genes, with 770 genes overexpressed and 368 genes underexpressed in bladder tumors (Figure 1). Many of the overexpressed genes were cancer-related genes belonging to EGFR-Ras signaling, cell cycle, and apoptosis (Table 1). Underexpressed genes included the Rab subfamily of genes, tumor-suppressor genes, and genes encoding casein kinases, cytochrome P450s, and RAR-related orphan receptors (Table 2). These genes are involved in a broad range of different pathways, including control of cell proliferation, differentiation, cell cycle, signal transduction, and apoptosis. Tables 1 and 2 list selected genes that were changed in rat bladder tumors. The Ras superfamily is a diverse group of small G proteins participating in many cellular processes and widely involved in tumorigenesis. In this study, many Ras superfamily members were found to be abnormally expressed in bladder tumors. Interestingly, Rab subfamily genes were under-expressed (Table 2). All other Ras-related genes, such as Ras, Rin, Rem, Rap, Rac, Rad, and Rho, were overexpressed in bladder tumors (Table 1). Many overexpressed genes in tumors were cell cycle-related genes that promote entry into cell cycle and mitosis. These include the following: Cdc2A, Cdc20, and Cdc25B; cyclins A2, B1, B2, and D1; Cdkn2a, Cdkn2c, Cdkn2d, and Cdkn3; MAD2; Polo-like kinases 1 and 4; and Gadd45 β and γ (Table 1). A more limited number of cell cycle-related genes (e.g., cyclin M1, p57, and JunDP1) were underexpressed in rat bladder tumors (Table 2). Interestingly, these genes play important roles in cell cycle arrest and G1/S and G2 checkpoints. The Kit, Maf, Fes, Myc, and Fyn oncogenes were overexpressed in rat bladder tumors (Table 1), whereas the WT1, BRCA2, Mycl1, and Pak genes were under-expressed in rat bladder tumors (Table 2). Survivin, TNF, and Bcl-2 were also overexpressed in rat bladder tumors (Table 1).
Figure 1.

Comparison of bladder epithelia and whole bladder tissues as controls to bladder cancers. Among the 1138 known genes found to be differentially expressed in rat bladder tumors compared with epithelia, 770 genes were overexpressed and 368 genes were underexpressed. Green indicates an expression below the mean value for the gene, black indicates an expression near the mean, and red indicates an expression above the mean.
Table 1.
Selected Genes Whose Expression Is Upregulated in Rat Bladder Tumors Compared with Normal Bladder Epithelia Identified by Microarray.
| Gene | Accession Number | Description | Fold Change | P |
| Cell cycle-related genes | ||||
| Cdc2a | NM_019296 | CDC2 homolog A* | 2.7* | .0250 |
| Cdc20 | U05341 | CDC20 homolog* | 4.6* | .0040 |
| Cdc23 | BE111697 | CDC23 (cell division cycle 23, yeast, homolog) (predicted)* | 33.3* | .0170 |
| Cdc25B | NM_133572 | CDC25 homolog B | 2.4 | .0007 |
| Cdc42ep5 | AI599324 | CDC42 effector protein (rho GTPase binding) 5 (predicted) | 2.6 | .0027 |
| Cdca1 | BG375704 | Cell division cycle-associated 1 (predicted) | 5.5 | .0125 |
| Cdca2 | AW532628 | Cell division cycle-associated 2 (predicted) | 5.6 | .0456 |
| Cdca3 | BF417638 | Cell division cycle-associated 3 | 6.1 | .0298 |
| Ccna2 | AA998516 | Cyclin A2* | 15.3* | .0028 |
| Ccnb1 | X64589 | Cyclin B1* | 4.8* | .0135 |
| Ccnb2 | AW253821 | Cyclin B2* | 6.5* | .0041 |
| Ccnd1 | X75207 | Cyclin D1* | 8.8* | .0151 |
| Cdkn2a | AF474976 | Cyclin-dependent kinase inhibitor 2A (p16INK4a)* | 8.7* | .0310 |
| Cdkn2c | NM_131902 | Cyclin-dependent kinase inhibitor 2C (p18, inhibits CDK4)* | 3.7* | .0005 |
| Cdkn2d | BI290067 | Cyclin-dependent kinase inhibitor 2D* | 2.2* | .0053 |
| Cdkn3 | BE113362 | Cyclin-dependent kinase inhibitor 3 (predicted)* | 21.3* | .0136 |
| Mad2l1 | AW143296 | MAD2 (mitotic arrest deficient, homolog)-like 1 (yeast)* | 2.0* | .0050 |
| Plk1 | U10188 | Polo-like kinase 1 (Drosophila)* | 3.7* | .0218 |
| Plk4 | BE109322 | Polo-like kinase 4 (Drosophila) (predicted)* | 2.5* | .0059 |
| Gadd45b | BI287978 | Growth arrest and DNA damage- inducible 45 β (predicted)* | 2.7* | .0007 |
| Gadd45g | AI599423 | Growth arrest and DNA damage- inducible 45 γ (predicted) | 2.4 | .0106 |
| Gas5 | BF287008 | Growth arrest-specific 5 | 2.3 | .0367 |
| Gas7 | AJ131902 | Growth arrest-specific 7 | 2.3 | .0305 |
| Gap43 | NM_017195 | Growth-associated protein 43 | 2.1 | .0167 |
| Gdf1 | BI289525 | Growth differentiation factor 1 (predicted) | 6.1 | .0769 |
| Bub1 | BF388785 | Budding uninhibited by benzimidazoles 1 homolog* | 11.5* | .0000 |
| Bub1b | BF557145 | Budding uninhibited by benzimidazoles 1 homolog, β | 7.9 | .0074 |
| Myc | NM_012603 | Myelocytomatosis viral oncogene homolog (avian) | 2.5 | .0077 |
| Jundp | NM_053894 2 | Jun dimerization protein 2 | 3.8 | .0017 |
| Dusp6 | NM_053883 | Dual-specificity phosphatase 6* | 2.4* | .0245 |
| Mki67 | AI714002 | Antigen identified by monoclonal antibody Ki-67* | 20.8* | .0010 |
| Small G proteins | ||||
| Racgap1 | AI409259 | Rac GTPase-activating protein 1* | 17.8* | .0146 |
| Rad51 | BI303370 | RAD51 homolog (Saccharomyces cerevisiae)* | 5.1* | .0048 |
| Rem2 | BI296482 | Rad- and gem-related GTP-binding protein 2 | 10.4 | .0045 |
| Rap2a | AW251376 | Rap2A-like protein | 3.6 | .0190 |
| Rap2b | NM_133410 | RAP2B, member of RAS oncogene family | 2.9 | .0185 |
| Rap2ip | AI535169 | Rap2-interacting protein* | 4.8* | .0221 |
| Rin3 | AI706777 | Ras and Rab interactor 3 (predicted) | 4.5 | .0045 |
| Rasgrp1 | BI282819 | RAS guanyl-releasing protein 1* | 2.3* | .0129 |
| Rasgrp2 | AW532114 | RAS guanyl-releasing protein 2 (predicted) | 2.1 | .0247 |
| Arhc | AA891940 | Ras homolog gene family, member C (predicted)* | 2.5* | .0003 |
| Arhd | AA955648 | Ras homolog gene family, member D (predicted)* | 2.1* | .0020 |
| Arhe | AI103572 | Ras homolog gene family, member E | 2.8 | .0158 |
| Rnd1 | AI144754 | Rho family GTPase 1 (predicted) | 5.1 | .0405 |
| Arhgap4 | BE111827 | Rho GTPase-activating protein 4 | 3.3 | .0260 |
| Arhgap8 | AA945062 | Rho GTPase-activating protein 8 | 2.3 | .0025 |
| BG377320 | Rho guanine nucleotide exchange factor (GEF) 17 (predicted) | 2.5 | .0003 | |
| Arhgdib | BF285771 | Rho, GDP dissociation inhibitor (GDI) β (predicted) | 3.7 | .0040 |
| Rock2 | BI303031 | Rho-associated coiled coil forming kinase 2 | 2.7 | .0242 |
| BG378261 | Rhophilin, Rho GTPase binding protein 1 (predicted) | 3.9 | .0128 | |
| Oncogenes and tumor-suppressor genes | ||||
| Akt1 | NM_033230 | V-akt murine thymoma viral oncogene homolog 1 | 1.5 | .0077 |
| Akt3 | NM_031575 | Thymoma viral proto-oncogene 3 | 2.4 | .0255 |
| Nf1 | BM386570 | Neurofibromatosis 1 | 3.1 | .0168 |
| Kit | AI454052 | V-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene | 4.2 | .0079 |
| Maf | NM_019318 | V-maf musculoaponeurotic fibrosarcoma (avian) oncogene | 2.7 | .0024 |
| Mafb | AA900536 | V-maf musculoaponeurotic fibrosarcoma oncogene family, protein B (avian) | 3.1 | .0004 |
| Ect2 | AI578135 | Ect2 oncogene (predicted)* | 12.0* | .0122 |
| Fes | BI289400 | Feline sarcoma oncogene (predicted)* | 3.4* | .0068 |
| Fyn | AI230396 | Fyn proto-oncogene | 2.1 | .0266 |
| Myc | NM_012603 | Myelocytomatosis viral oncogene homolog (avian) | 2.5 | .0077 |
| Ndr4 | BG666709 | N-myc downstream regulated 4 | 2.5 | .0031 |
| BM390283 | Large tumor suppressor 2 (predicted) | 2.3 | .0010 | |
| Apoptosis | ||||
| Birc2 | NM_023987 | Inhibitor of apoptosis protein 1* | 3.3* | .0077 |
| Birc5 | NM_022274 | Baculoviral IAP repeat-containing 5 (Survivin) | 3.6* | .0500 |
| Pawr | U05989 | PRKC, apoptosis, WT1, regulator | 2.0 | .0003 |
| Casp1 | D85899 | Caspase 1* | 2.7* | .0173 |
| Casp11 | NM_053736 | Caspase 11* | 4.7* | .0025 |
| Casp12 | NM_130422 | Caspase 12 | 5.7 | .0010 |
| Pycard | BI282953 | Apoptosis-associated speck-like protein containing a CARD | 2.1 | .0047 |
| Bcl11b | BE116855 | B-cell leukemia/lymphoma 11B (predicted) | 4.0 | .0006 |
| Bcl2a1 | NM_133416 | B-cell leukemia/lymphoma 2 related protein A1* | 5.6* | .0033 |
| Bcl3 | AI411774 | B-cell leukemia/lymphoma 3 (predicted)* | 5.8* | .0059 |
| Bcl6 | AI237606 | B-cell leukemia/lymphoma 6 (predicted)* | 3.6* | .0014 |
| Bok | AI227742 | Bcl-2-related ovarian killer protein* | 2.2* | .0040 |
| Cebpd | NM_013154 | CCAAT/enhancer binding protein (C/EBP), δ | 2.9 | .0002 |
| Tnfsf13 | AA800814 | Tumor necrosis factor (ligand) superfamily, member 13 | 2.5 | .0000 |
| Tnfip6 | AF159103 | Tumor necrosis factor α induced protein 6 | 2.4 | .0135 |
| Tnfrsf11b | NM_012870 | Tumor necrosis factor receptor superfamily, member 11b* | 3.9* | .0012 |
| Tnfrsf12a | BI303379 | Tumor necrosis factor receptor superfamily, member 12a | 5.9 | .0000 |
| BI278479 | Tumor necrosis factor, α-induced protein 2 (predicted) | 2.8 | .0024 | |
| Growth factors and related genes | ||||
| Ctgf | NM_022266 | Connective tissue growth factor* | 7.9* | .0002 |
| BF284634 | Epidermal growth factor-containing fibulin-like extracellular matrix protein 1 | 3.9 | .0015 | |
| Fgf13 | NM_053428 | Fibroblast growth factor 13 | 3.6 | .0007 |
| Fgfbp1 | NM_022603 | Fibroblast growth factor binding protein 1* | 30.2* | .0027 |
| Hgf | NM_017017 | Hepatocyte growth factor | 4.3 | .0022 |
| Hgfac | BE119649 | Hepatocyte growth factor activator | 2.1 | .0130 |
| Hdgfrp3 | BI283829 | Hepatoma-derived growth factor, related protein 3 | 3.5 | .0019 |
| Igf1 | M15481 | Insulin-like growth factor 1 | 21.4 | .0010 |
| Igfbp3 | NM_012588 | Insulin-like growth factor binding protein 3* | 3.2* | .0000 |
| Igfbp4 | BE108969 | Insulin-like growth factor binding protein 4 | 2.1 | .0261 |
| Igfbp7 | AI233246 | Insulin-like growth factor binding protein 7 | 3.0 | .0000 |
| Pdgfrb | BM389426 | Platelet-derived growth factor receptor, β polypeptide* | 2.9* | .0017 |
| Pdgfa | BE100812 | Platelet-derived growth factor, α* | 2.1* | .0044 |
| Pdgfc | NM_031317 | Platelet-derived growth factor, C polypeptide | 3.0 | .0054 |
| Scgf | AI576758 | Stem cell growth factor | 2.2 | .0055 |
| Tgfb1 | NM_021578 | Transforming growth factor, β 1 | 6.6 | .0014 |
| Tgfb2 | NM_031131 | Transforming growth factor, β 2* | 5.9* | .0021 |
| Vegfc | NM_053653 | Vascular endothelial growth factor C | 2.5 | .0039 |
Most of the upregulated genes were small G proteins, apoptosis genes, cell cycle-related genes, oncogenes, and growth factors. Fold change is the ratio of the mean gene expression values of tumors to the mean gene expression values of epithelia from the microarray.
The genes were also found to be differentially expressed in mouse bladder tumors.
Table 2.
Selected Genes Whose Expression Is Downregulated in Rat Bladder Tumors Compared with Normal Bladder Epithelia Identified by Microarray.
| Gene | Accession Number | Description | Fold Change | P |
| Cell cycle-related genes | ||||
| BE120410 | Cyclin M1 (predicted) | -2.7 | .0110 | |
| Cdkn1c | AI013919 | Cyclin-dependent kinase inhibitor 1C, p57 | -5.8 | .0000 |
| Jundp1 | NM_021865 | Jun dimerization protein 1 | -2.1 | .0024 |
| Ppara | NM_013196 | Peroxisome proliferator-activated receptor α* | -2.2* | .0153 |
| Pparg | NM_013124 | Peroxisome proliferator-activated receptor, γ* | -2.3* | .0003 |
| Small G proteins | ||||
| Rab14 | AA875010 | RAB14, member RAS oncogene family | -2.1 | .0126 |
| Rab27b | NM_053459 | RAB27B, member RAS oncogene family | -3.3 | .0000 |
| Rab3c | NM_133536 | RAB3C, member RAS oncogene family | -6.7 | .0002 |
| Rab40b | AA924620 | RAB40b, member RAS oncogene family (predicted) | -3.2 | .0001 |
| BF284067 | RAP1, GTPase-activating protein 1 (predicted) | -3.6 | .0000 | |
| Rasa3 | AI237779 | RAS p21 protein activator 3 | -2.6 | .0014 |
| RICS | BE097238 | RhoGAP involved in β-catenin-N-cadherin and NMDA receptor signaling (predicted) | -2.1 | .0005 |
| AI547942 | RhoGEF (Arhgef) and pleckstrin domain protein 1 | -2.2 | .0009 | |
| Oncogenes and tumor-suppressor genes | ||||
| AA900477 | Vav2 oncogene (predicted) | -2.3 | .0002 | |
| Mycl1 | BI300996 | V-myc myelocytomatosis viral oncogene homolog 1, lung carcinoma-derived (avian)* | -2.2* | .0001 |
| Ndrg2 | NM_133583 | N-myc downstream-regulated gene 2* | -3.0* | .0000 |
| BE115673 | Metastasis suppressor 1 (predicted) | -4.7 | .0013 | |
| Pak3 | NM_019210 | P21 (CDKN1A)-activated kinase 3 | -14.3 | .0000 |
| Pak4 | BF404920 | P21 (CDKN1A)-activated kinase 4 (predicted) | -3.3 | .0130 |
| Wt1 | NM_031534 | Wilms tumor 1 | -2.7 | .0278 |
| Brca2 | BF396613 | Breast cancer 2 | -3.5 | .0161 |
| AI548958 | HRAS-like suppressor (predicted) | -2.5 | .0015 | |
| Fhit | NM_021774 | Fragile histidine triad gene | -1.6* | .0107 |
| Apoptosis | ||||
| Dffa | NM_053679 | DNA fragmentation factor, α subunit* | -2.0* | .0060 |
| LOC64171 | NM_022303 | Caspase recruitment domain protein 9 | -2.0 | .0012 |
| Ntrk1 | NM_021589 | Neurotrophic tyrosine kinase, receptor, type 1 | -3.5 | .0391 |
| Growth factors | ||||
| BF418373 | Epidermal growth factor-like protein 6 | -8.3 | .0000 | |
| Fgf1 | BI289840 | Fibroblast growth factor 1* | -2.8* | .0006 |
| FGFR2 | L19107 | Fibroblast growth factor receptor 2 | -2.9 | .0384 |
| BE112403 | Fibroblast growth factor receptor substrate 2 (predicted) | -2.0 | .0042 | |
| BF396448 | Insulin-like growth factor binding protein-like 1 | -2.2 | .0036 | |
| BM384311 | Platelet-derived growth factor receptor-like (predicted) | -4.4 | .0000 | |
| Others | ||||
| Csnk1g1 | AA957549 | Casein kinase 1, γ 1 | -2.1 | .0058 |
| Csnk1g3 | AI176776 | Casein kinase 1, γ 3 | -2.5 | .0147 |
| BE107780 | Casein kinase II, α 2, polypeptide (predicted) | -2.3 | .0012 | |
| Clpx | BG371721 | Caseinolytic protease X (Escherichia coli) (predicted) | -2.7 | .0000 |
| Cyp3a18 | D38381 | Cytochrome P450, 3a18 | -5.6 | .0001 |
| Cyp1a1 | X00469 | Cytochrome P450, family 1, subfamily a, polypeptide 1 | -22.1 | .0053 |
| Cyp11a1 | NM_017286 | Cytochrome P450, family 11, subfamily a, polypeptide 1 | -3.6 | .0000 |
| Cyp4a14 | AA893326 | Cytochrome P450, family 4, subfamily a, polypeptide 14 | -5.2 | .0487 |
| Lcmt1 | BG381002 | Leucine carboxyl methyltransferase 1 | -2.1 | .0001 |
| BE107055 | Leucine-rich repeat-containing 28 (predicted) | -2.0 | .0001 | |
| AI716087 | Leucine zipper transcription factor-like 1 (predicted) | -2.0 | .0000 | |
| BF412229 | Leucine-rich and death domain-containing (predicted) | -2.2 | .0001 | |
| Lgi1 | AI229354 | Leucine-rich, glioma-inactivated 1 | -3.7 | .0007 |
| AI235414 | RAR-related orphan receptor α (predicted) | 2.4 | .0000 | |
| BE110171 | RAR-related orphan receptor γ (predicted) | 2.0 | .0125 | |
Downregulated genes include Rab subfamily genes, tumor-suppressor genes, casein kinases, cytochrome P450s, and RAR-related orphan receptor genes. Fold change is the ratio of the mean gene expression values of tumors to the mean gene expression values of epithelia from the microarray.
The genes were also found to be differentially expressed in mouse bladder tumors.
Confirmation of Differentially Expressed Genes for Both RNA and Protein Levels
With such a large number of differentially expressed genes, we examined the distribution of fold changes to detect if any large skew could account for the results. The distribution of fold changes for differentially expressed genes is shown in Figure 2A, and its symmetry suggests that no skew artifact is present. We validated the differential expression of 15 genes by real-time PCR. Thirteen of 15 genes were confirmed by real-time PCR. The confirmation rate is > 86% at a cutoff of two-fold change and (P < .05). The real-time PCR results of these 13 genes agreed well with microarray data (Figure 2B). We also examined several genes at protein level by 2D protein gel electrophoresis and IHC. The genes annexin A1 and cyclin D1 were chosen for this analysis, and results are presented in Figure 3. Both RNA and protein level changes for these genes agreed well with initial microarray changes.
Figure 2.
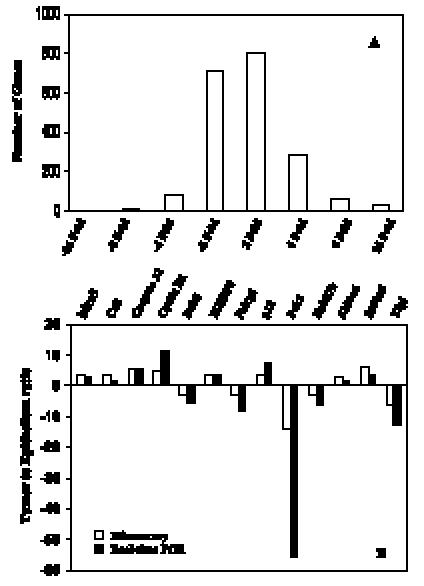
Distribution of 2005 differentially expressed genes and ESTs by microarray analysis and real-time PCR confirmation for selected genes. (A) An overview of the number of genes reveals fold changes different from those in normal bladder epithelia. (B) Comparison of fold change produced by microarray with the relative expression ratio obtained from real-time PCR, with good concordance.
Figure 3.
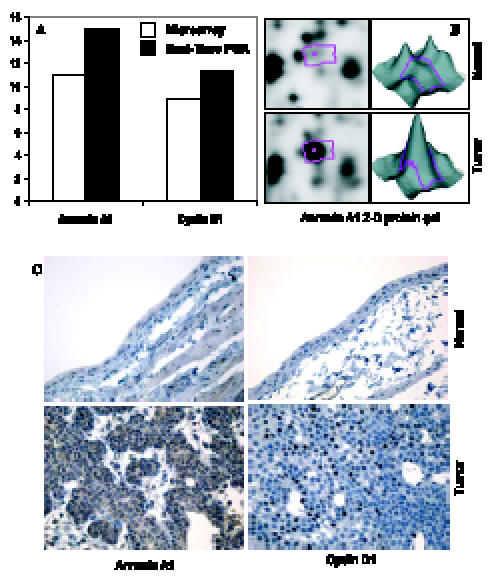
Confirmation of differentially expressed genes for both RNA and protein levels. (A) Comparison of fold change produced by microarray with the relative expression ratio obtained from RT-PCR. (B) 2D protein gel electrophoresis indicates that annexin A1 protein is overexpressed in rat bladder cancers. (C) IHC suggests that both annexin A1 and cyclin D1 are overexpressed in rat bladder cancers.
Differentially Expressed Genes Interpreted by GenMAPP
We introduced the differentially expressed genes found in microarrays into GenMAPP. GenMAPP search revealed that apoptosis, cell cycle, and integrin-mediated cell adhesion are actively involved in bladder tumorigenesis, with expression changes in multiple genes, each of which may play important roles in the pathogenesis of bladder cancers. Figure 4 represents the genes differentially expressed in rat bladder tumors involved in these signaling pathways.
Figure 4.
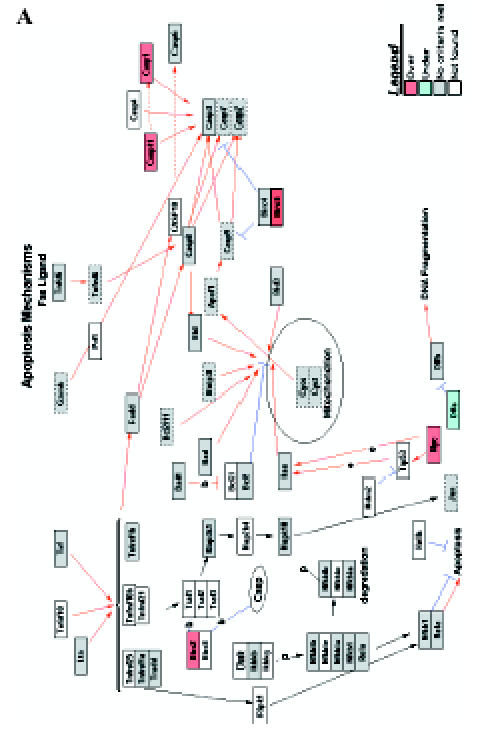
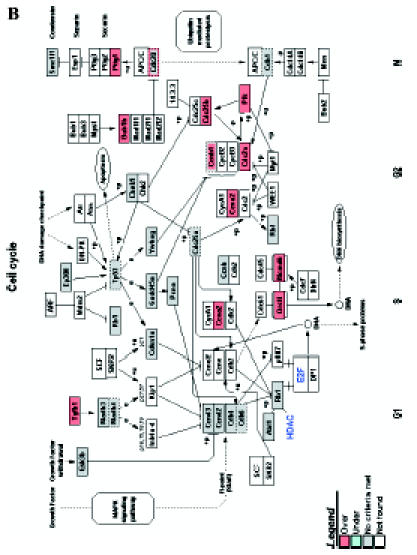
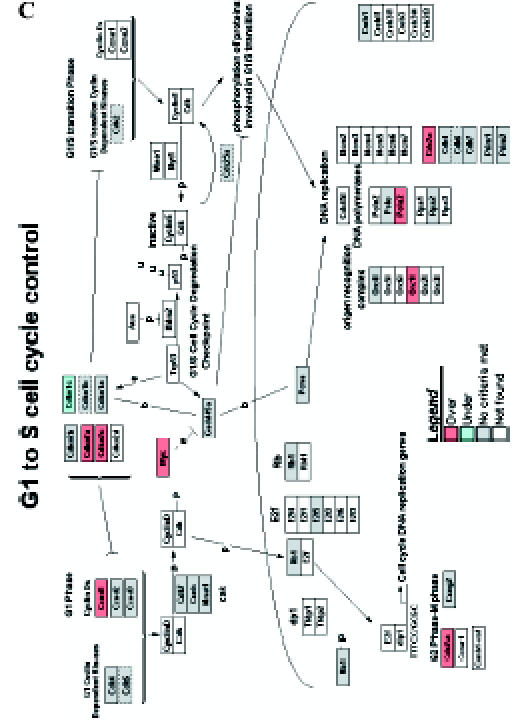
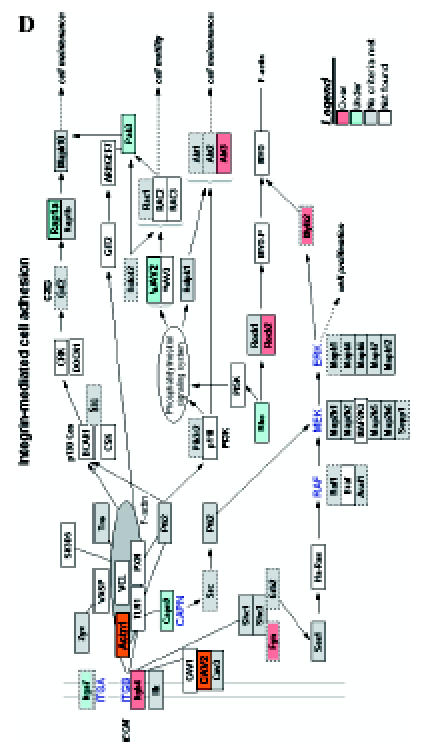
GenMAPP signaling pathways integrated into rat bladder tumorigenesis with a cutoff fold change of ≥ 1.5 and P < .05. Yellow and blue indicates overexpressed and underexpressed genes in tumor samples, respectively. Grey indicates that selection criteria were not met but the gene was represented in the array. White boxes indicate that the gene was not present in the chip. (A) Apoptosis. (B) Cell cycle. (C) G1-to-S cell cycle control. (D) Integrin-mediated cell adhesion.
Comparison of Differentially Expressed Genes Between Rat and Mouse Bladder Tumors
We compared the differentially expressed genes from both rat and mouse bladder tumors using gene expression data from mouse bladder tumors [19]. Of 860 comparable genes between rats and mice, there were 280 genes that had the same tendency of expression changes in both mouse and rat bladder tumors. Selected genes were listed in Tables 1 and 2 and marked with asterisks. The majority of these genes are cell cycle-related genes, ras small G proteins, and apoptosis-related and growth factor-related genes.
Discussion
In this study, we have shown that microarray can be used to enhance the search for the molecular pathogenesis of tumors. We found that inappropriate regulation of ras, cell cycle, and apoptosis pathways may be the three major steps in the tumorigenesis of rat bladder malignancy. In addition, we were able to identify a variety of genes whose expression was highly increased independent of whether they are directly involved in the mechanism of tumorigenesis in this model. These highly modulated genes were also proved to be changed at the protein level and may prove highly useful in identifying early lesions and tumors in samples from urine or serum. In addition, both these highly overexpressed genes and many of the genes that are along the mechanistic pathway may prove to be modulated by effective preventive or therapeutic agents.
One group of genes found to be differentially expressed in bladder tumors comprises cell cycle-related genes. Tumor proliferation depends on the derangement of normal cell cycle progression and control. Cell cycle-associated protein complexes composed of cyclins and cyclin-dependent kinases (CDKs) regulate normal cellular proliferation. Different CDK-cyclin complexes cooperate to drive cells through different phases of the cell cycle. Activation of CDK4 and CDK6 by D-type cyclins is thought to be involved in progression through early G1. We observed an increased expression of a number of CDK-related phosphatases. Thus, a variety of cell cycle commitment genes, including cyclins, CDCs, Cdca, GADD45s, Gas, Plks, Mad2, and Bub1, were found to be overexpressed in bladder tumors. Increased levels of cyclin D1 are associated with a wide variety of cancers, including breast, colon, and lung cancers. We confirmed the increase in cyclin D1 observed in microarrays both by reverse transcription (RT) PCR and by IHC (Figure 3). These results help to demonstrate the potential use of array analysis in determining biomarkers that might be useful in the identification of early lesions. Overall, the results are in agreement with our finding of a relatively high proliferative index in lesions derived from this model.
Annexin A1 (ANXA1) is a calcium-binding and phospholipid-binding protein of the annexin superfamily that is found in a wide range of organisms, including vertebrates, invertebrates, and plants. Overexpression of ANXA1 was found in breast [20], stomach [21], pancreatic [22], and hepatic cancers [23], whereas underexpression of ANXA1 was recorded in prostate [24,25], esophageal [25], and head and neck [26] cancers. Thus, the role of ANXA1 in carcinogenesis may occur in a tissue-specific manner. The exact function of ANXA1 remains unknown. There are a number of possible functions of ANXA1 in cancer development. For example, ANXA1 serves as a substrate for EGFR [27] and is a steroidregulated protein [28]; thus, it has been linked with cell proliferation and regulation of cell migration through the regulation of the extracellular signal-regulated kinase/mitogen-activated protein kinase (MAPK) signal transduction pathway [29]. ANXA1 is also a critical mediator of apoptosis [30]. Previous studies have also suggested that ANXA1 can serve as a gene target and gene maker for cancer treatment, development, progression, and diagnosis. Expression of ANXA1 significantly correlated with clinicopathological features and survival in esophagus and esophagogastric junction adenocarcinomas [31] and breast cancer [32]. Immunocytochemical detection of ANXA1 represents a simple, inexpensive, highly sensitive, and specific assay for the diagnosis of hairy cell leukemia [33].
The striking increases in ANXA1 levels observed in the microarray, which was confirmed by RT-PCR, were then examined by 2D gel analysis (Figure 3). Finally, based on consistent increases observed in these various studies, we performed IHC and similarly found striking increases in annexin A1 expression. As can be seen in the IHC panel, a strong response should allow us to examine the expression of annexin A1 for identifying earlier lesions (dysplasias and papillomas) or to potentially look for it in the urine of tumor-bearing rats. Thus, the results with cyclin D1 and annexin A1 demonstrate methods that might allow one to combine results from gene expression and proteomic analyses.
We also found that COX-2 expression was increased by roughly five times in RNA expressions in bladder tumors. Interestingly, by IHC, we have shown that the highest expression was not in epithelial cells but rather in endothelial cells in the tumor [3]. Furthermore, we have found that both celecoxib and a wide variety of nonsteroidal anti-inflammatory drugs (NSAIDs) were highly effective in blocking bladder tumor formation in this model [3]. This parallels epidemiologic studies in humans showing the efficacy of NSAIDs against bladder tumors. Interestingly, levels of PPARγ were reduced roughly by 3.5 times in microarrays. We have recently found that PPARγ expression was decreased in tumors, as assessed by IHC procedures (Grubbs and Fischer, data not shown). That indirectly reflects our finding that the PPARγ agonist rosiglitazone is a bladder tumor promoter in this model. Two mechanistically significant genes that we have previously demonstrated to exhibit altered levels in rat bladder tumors are Fhit (fragile histidine triad) and the IAP (inhibition of apoptosis protein) Survivin. Fhit expression is often lost in a wide variety of human tumors (e.g., lung, head and neck, and bladder). We have previously shown that the Fhit gene is methylated in rat bladder tumors and is associated with a decrease in FHIT protein expression [34]. We also previously found an increased expression of the IAP protein in rodent bladder tumors [35], as it is expressed in a variety of human tumors, including bladder. These results reinforce the use of microarrays in looking for the expression of specific “relevant genes,” in addition to its more generalized use in looking for important pathways (Figure 4, A–D) or a much wider variety of genes (Tables 1 and 2).
The ras superfamily regulates many cellular processes, such as cell cycle progression, actin cytoskeletal dynamics, and membrane traffic. The transforming potential of ras is due to a mutation, which, in human bladder tumors, occurs in H-ras [36], although these rat tumors do not appear to have mutations in ras genes. Alternatively, overexpression of H-ras, K-ras, and N-ras transcripts has also been associated with bladder tumor transition [37,38]. Guanine nucleotide exchange factors (GEFs) stimulate Ras superfamily members to exchange bound guanosine 5c-diphosphate (GDP) for guanosine 5c-triphosphate (GTP), thereby increasing the amount of active form [39]. Rho mutations in tumors are quite rare, but overexpression is more common [40]. Dysregulation of Rho family member activity probably also contributes to human cancer in that some RhoGEFs act as oncogenes [41], whereas RhoGAPs [42] act as tumor suppressors. Reduced expression of RhoGDIs has recently been shown to correlate with increasing invasive and metastatic abilities in human bladder carcinoma cell lines [43,44]. We found that RhoGAPs, RhoGEFs, and RhoGDIs were differentially expressed in rat bladder tumors.
Our results implicated many members of the integrin-mediated cell adhesion pathway in bladder tumorigenesis. Integrin α7, calpain 9, Pak3, rhodopsin (Rho), Rap1a, and Vav2 underexpressed rat bladder tumors, whereas integrin β4, actin α1, caveolin-2, Fyn, Rock2, Mylk2, and Akt3 overexpressed in rat bladder tumors. The small GTPase Rap1 is involved in several aspects of cell adhesion, including integrin-mediated cell adhesion and cadherin-mediated cell junction formation [45]. Rap1 regulates all integrins that are associated with the actin cytoskeleton, such as integrins β1, β2, and β3 [46]. Active Rap1 binds to a subset of Rac GEFs, including Vav2 and Tiam1. Overexpressed Vav2 and Tiam1 specifically require Rap1 to promote spreading, even though Rac1 is activated independently of Rap1 [47]. Rac is both required and sufficient to mediate Rap1-induced cell spreading. Thus, integrin-mediated cell adhesion appears to play a role in rat bladder tumorigenesis and progression.
Increased activity of another Ras effector, PI3-kinase, is similarly associated with many types of human cancer. Because PI3-kinase is an immediate downstream effector of Ras and EGFR, multiple pathways may contribute to the increases in PI3-kinase activity observed in many bladder cancers. PI3-kinase consistently prevents apoptosis in many cell systems through activation of the Rac GTPase, possibly through activation of NF-κB [48]. Thus, the activation of PI3-kinase associated with excessive Ras activity may promote oncogenesis by blunting apoptosis-inducing stimuli associated with oncogenic transformation. RacGAPs stimulate intrinsic GTPase activity, thus leading to Rac inactivation. Rac and Rac-GEFs play key roles in the control of various aspects of malignant transformation and metastatic cascade in various models [49], as well as in the control of mitogenesis through its ability to regulate G1/S transition and cyclin D1 expression [50–52]. Ect2 and RacGAP also regulate the activation and function of Cdc42 in mitosis [53].
Finally, the differentially expressed genes between mouse and rat bladder tumors were compared.We found that, among the 860 comparable genes between rats and mice, there were 280 genes that had the same tendency of expression in both mouse and rat bladder tumors, accounting for about one third of the genes. The majority of these genes are cell cycle-related genes, ras small G proteins, and apoptosis-related and growth factor-related genes. These results strongly indicated that both in mice and in rats, similar mechanisms are involved in bladder tumorigenesis.
In human bladder tumors, low-grade papillary tumors frequently show constitutive activation of the receptor tyrosine kinase/Ras pathway, exhibiting activating mutations in H-ras and fibroblast growth factor receptor 3 (FGFR3) genes [54,55]. In contrast, carcinoma in situ and invasive tumors frequently show alterations in p53 and Rb genes and pathways [56,57]. The cell cycle is controlled by the p53 and Rb pathways, with cells receiving extracellular growth signals through the Ras/MAPK pathway. Previous studies have shown that chemically induced rat bladder cancers typically have a mixed histology showing elements of both transitional and squamous cells [3,4], with a relatively low frequency of H-ras mutations and roughly 50% of these tumors developing p53 mutations [58,59], similar to the percentage found in humans.
In recent years, genomewide expression profiling by the use of microarray technology has provided new insights into the gene expression patterns and dysregulation of genes during bladder tumorigenesis. Gene expression profiles can be used not only to elucidate the underlying molecular mechanisms and pathways involved in bladder tumorigenesis but also to make distinctions among different histologic subtypes and to predict tumor recurrence and patient survival. Gene expression array studies on human bladder cancers have revealed a broad range of genes that are differentially expressed during bladder tumorigenesis, progression, and invasion. Kawakami et al. [60] indicated that genes involved in metabolism, transcription, cell adhesion/surface, and cytoskeleton/cell membrane were significantly differentially expressed in superficial and invasive bladder tumors. Kim et al. examined gene expression patterns in the development of bladder cancer from preneoplasia along papillary and nonpapillary pathways and identified alterations in seven gene clusters controlling proliferation, differentiation, and apoptosis that were common for both papillary and nonpapillary cancers. In contrast, genes controlling cellular and stromal interactions were altered in nonpapillary cancer [61]. Elsamman et al. also identified the significant upregulation of 40 genes in cell differentiation and keratinization, cell cycle, cell adhesion, transcription, and apoptosis associated with superficial noninvasive bladder tumors, and the significant upregulation of 34 genes related to extracellular matrix degradation, immune responses, cell cycling, and angiogenesis was associated with invasive bladder tumors [62]. Dyrskjot et al. identified a 45-gene signature of bladder tumor progression that was involved in regulating apoptosis, cell differentiation, and cell cycle. BNIP3L, BIRC4, NCKAP1, and BIRC6 genes, which are involved in the apoptotic cell death pathway, were upregulated in the nonprogressing group. Cdc25B, Cdc20, and MCM7 genes, which are involved in regulating cell cycle and cell proliferation, were upregulated in the progressing group [63]. Modlich et al. also identified genes encoding transcription factors, protein synthesis, and metabolism; cell cycle progression and differentiation were overexpressed in superficial bladder tumors, whereas transcripts for immune, extracellular matrix, adhesion, peritumoral stroma, and muscle tissue components; proliferation; and cell cycle controllers were upregulated in invasive tumors [64]. In concordance with human bladder tumors, in this study, we confirmed chemically induced rat bladder cancers undergoing similar molecular mechanisms and pathways during tumorigenesis and, maybe, in progression. Genes involved in cell cycle, apoptosis, cell adhesion, transcription factors, and ras gene pathway were significantly differentially expressed in rat bladder cancers (Tables 1 and 2). These results suggested that chemically induced rat bladder cancers can be used to represent human bladder cancer. It is a good model for studying the pathogenesis, progression, treatment, and prevention of human bladder cancer.
In summary, we have determined the expression profiles of genes differentially expressed during rat bladder tumorigenesis. Our results suggest that EGFR-Ras pathway, cell cycle, apoptosis, and integrin-mediated cell adhesion are involved in bladder tumorigenesis. Our results also suggest that common mechanisms play important roles in both rat and mouse tumorigeneses. Furthermore, certainty on identified genes may suggest potential target molecules for preventing cancer in this model.
References
- 1.Amling CL. Diagnosis and management of superficial bladder cancer. Curr Probl Cancer. 2001;25:219–278. doi: 10.1067/mcn.2001.117539. [DOI] [PubMed] [Google Scholar]
- 2.Brandau S, Bohle A. Bladder cancer: I. Molecular and genetic basis of carcinogenesis. Eur Urol. 2001;39:491–497. doi: 10.1159/000052494. [DOI] [PubMed] [Google Scholar]
- 3.Grubbs CJ, Lubet RA, Koki AT, Leahy KM, Masferrer JL, Steele VE, Kelloff GJ, Hill DL, Seibert K. Celecoxib inhibits N-butyl-N-(4-hydroxybutyl)-nitrosamine-induced urinary bladder cancers in male B6D2F1 mice and female Fischer-344 rats. Cancer Res. 2000;60:5599–5602. [PubMed] [Google Scholar]
- 4.Grubbs CJ, Moon RC, Squire RA, Farrow GM, Stinson SF, Goodman DG, Brown CC, Sporn MB. 13-cis-Retinoic acid: inhibition of bladder carcinogenesis induced in rats by N-butyl-N-(4-hydroxybutyl)nitrosamine. Science. 1977;198:743–744. doi: 10.1126/science.910158. [DOI] [PubMed] [Google Scholar]
- 5.Ogawa K, Uzvolgyi E, St John MK, de Oliveira ML, Arnold L, Cohen SM. Frequent p53 mutations and occasional loss of chromosome 4 in invasive bladder carcinoma induced by N-butyl-N-(4-hydroxybutyl)nitrosamine in B6D2F1 mice. Mol Carcinog. 1998;21:70–79. doi: 10.1002/(sici)1098-2744(199801)21:1<70::aid-mc9>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto S, Chen T, Murai T, Mori S, Morimura K, Oohara T, Makino S, Tatematsu M, Wanibuchi H, Fukushima S. Genetic instability and p53 mutations in metastatic foci of mouse urinary bladder carcinomas induced by N-butyl-N-(4-hydroxybutyl)nitrosamine. Carcinogenesis. 1997;18:1877–1882. doi: 10.1093/carcin/18.10.1877. [DOI] [PubMed] [Google Scholar]
- 7.el-Marjou A, Delouvee A, Thiery JP, Radvanyi F. Involvement of epidermal growth factor receptor in chemically induced mouse bladder tumour progression. Carcinogenesis. 2000;21:2211–2218. doi: 10.1093/carcin/21.12.2211. [DOI] [PubMed] [Google Scholar]
- 8.Czerniak B, Cohen GL, Etkind P, Deitch D, Simmons H, Herz F, Koss LG. Concurrent mutations of coding and regulatory sequences of the Ha-ras gene in urinary bladder carcinomas. Hum Pathol. 1992;23:1199–1204. doi: 10.1016/0046-8177(92)90285-b. [DOI] [PubMed] [Google Scholar]
- 9.Olderoy G, Daehlin L, Ogreid D. Low-frequency mutation of Ha-ras and Ki-ras oncogenes in transitional cell carcinoma of the bladder. Anticancer Res. 1998;18:2675–2678. [PubMed] [Google Scholar]
- 10.Buyru N, Tigli H, Ozcan F, Dalay N. Ras oncogene mutations in urine sediments of patients with bladder cancer. J Biochem Mol Biol. 2003;36:399–402. doi: 10.5483/bmbrep.2003.36.4.399. [DOI] [PubMed] [Google Scholar]
- 11.Knowles MA. Molecular genetics of bladder cancer. Br J Urol. 1995;1:57–66. [PubMed] [Google Scholar]
- 12.Shinohara N, Koyanagi T. Ras signal transduction in carcinogenesis and progression of bladder cancer: molecular target for treatment? Urol Res. 2002;30:273–281. doi: 10.1007/s00240-002-0275-0. [DOI] [PubMed] [Google Scholar]
- 13.Swiatkowski S, Seifert HH, Steinhoff C, Prior A, Thievessen I, Schliess F, Schulz WA. Activities of MAP-kinase pathways in normal uroepithelial cells and urothelial carcinoma cell lines. Exp Cell Res. 2003;282:48–57. doi: 10.1006/excr.2002.5647. [DOI] [PubMed] [Google Scholar]
- 14.Orntoft TF, Wolf H. Molecular alterations in bladder cancer. Urol Res. 1998;26:223–233. doi: 10.1007/s002400050050. [DOI] [PubMed] [Google Scholar]
- 15.Knowles MA. The genetics of transitional cell carcinoma: progress and potential clinical application. BJU Int. 1999;84:412–427. doi: 10.1046/j.1464-410x.1999.00217.x. [DOI] [PubMed] [Google Scholar]
- 16.Yao R, Wang Y, Lubet RA, You M. Differentially expressed genes associated with mouse lung tumor progression. Oncogene. 2002;21:5814–5821. doi: 10.1038/sj.onc.1205422. [DOI] [PubMed] [Google Scholar]
- 17.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- 19.Yao R, Lemon WJ, Wang Y, Grubbs CJ, Lubet RA, You M. Altered gene expression profile in mouse bladder cancers induced by hydroxybutyl(butyl)nitrosamine. Neoplasia. 2004;6:569–577. doi: 10.1593/neo.04223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pencil SD, Toth M. Elevated levels of annexin I protein in vitro and in vivo in rat and human mammary adenocarcinoma. Clin Exp Metastasis. 1998;16:113–121. doi: 10.1023/a:1021917017109. [DOI] [PubMed] [Google Scholar]
- 21.Sinha P, Hutter G, Kottgen E, Dietel M, Schadendorf D, Lage H. Increased expression of annexin I and thioredoxin detected by two-dimensional gel electrophoresis of drug resistant human stomach cancer cells. J Biochem Biophys Methods. 1998;37:105–116. doi: 10.1016/s0165-022x(98)00020-7. [DOI] [PubMed] [Google Scholar]
- 22.Bai XF, Ni XG, Zhao P, Liu SM, Wang HX, Guo B, Zhou LP, Liu F, Zhang JS, Wang K, et al. Overexpression of annexin 1 in pancreatic cancer and its clinical significance. World J Gastroenterol. 2004;10:1466–1470. doi: 10.3748/wjg.v10.i10.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masaki T, Tokuda M, Ohnishi M, Watanabe S, Fujimura T, Miyamoto K, Itano T, Matsui H, Arima K, Shirai M, et al. Enhanced expression of the protein kinase substrate annexin in human hepatocellular carcinoma. Hepatology. 1996;24:72–81. doi: 10.1053/jhep.1996.v24.pm0008707286. [DOI] [PubMed] [Google Scholar]
- 24.Patton KT, Chen HM, Joseph L, Yang XJ. Decreased annexin I expression in prostatic adenocarcinoma and in high-grade prostatic intraepithelial neoplasia. Histopathology. 2005;47:597–601. doi: 10.1111/j.1365-2559.2005.02300.x. [DOI] [PubMed] [Google Scholar]
- 25.Paweletz CP, Ornstein DK, Roth MJ, Bichsel VE, Gillespie JW, Calvert VS, Vocke CD, Hewitt SM, Duray PH, Herring J, et al. Loss of annexin 1 correlates with early onset of tumorigenesis in esophageal and prostate carcinoma. Cancer Res. 2000;60:6293–6297. [PubMed] [Google Scholar]
- 26.Garcia Pedrero JM, Fernandez MP, Morgan RO, Herrero Zapatero A, Gonzalez MV, Suarez Nieto C, Rodrigo JP. Annexin A1 down-regulation in head and neck cancer is associated with epithelial differentiation status. Am J Pathol. 2004;164:73–79. doi: 10.1016/S0002-9440(10)63098-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pepinsky RB. Phosphorylation of lipocortin-1 by the epidermal growth factor receptor. Methods Enzymol. 1991;198:260–272. doi: 10.1016/0076-6879(91)98027-4. [DOI] [PubMed] [Google Scholar]
- 28.Buckingham JC, Flower RJ. Lipocortin 1: a second messenger of glucocorticoid action in the hypothalamo-pituitary-adrenocortical axis. Mol Med Today. 1997;3:296–302. doi: 10.1016/S1357-4310(97)88908-3. [DOI] [PubMed] [Google Scholar]
- 29.Alldridge LC, Harris HJ, Plevin R, Hannon R, Bryant CE. The annexin protein lipocortin 1 regulates the MAPK/ERK pathway. J Biol Chem. 1999;274:37620–37628. doi: 10.1074/jbc.274.53.37620. [DOI] [PubMed] [Google Scholar]
- 30.Solito E, de Coupade C, Canaider S, Goulding NJ, Perretti M. Transfection of annexin 1 in monocytic cells produces a high degree of spontaneous and stimulated apoptosis associated with caspase-3 activation. Br J Pharmacol. 2001;133:217–228. doi: 10.1038/sj.bjp.0704054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang KL, Wu TT, Resetkova E, Wang H, Correa AM, Hofstetter WL, Swisher SG, Ajani JA, Rashid A, Hamilton SR, et al. Expression of annexin A1 in esophageal and esophagogastric junction adenocarcinomas: association with poor outcome. Clin Cancer Res. 2006;12:4598–4604. doi: 10.1158/1078-0432.CCR-06-0483. [DOI] [PubMed] [Google Scholar]
- 32.Shen D, Nooraie F, Elshimali Y, Lonsberry V, He J, Bose S, Chia D, Seligson D, Chang HR, Goodglick L. Decreased expression of annexin A1 is correlated with breast cancer development and progression as determined by a tissue microarray analysis. Hum Pathol. 2006;37:1583–1591. doi: 10.1016/j.humpath.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 33.Falini B, Tiacci E, Liso A, Basso K, Sabattini E, Pacini R, Foa R, Pulsoni A, Dalla Favera R, Pileri S. Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1) Lancet. 2004;363:1869–1870. doi: 10.1016/S0140-6736(04)16356-3. [DOI] [PubMed] [Google Scholar]
- 34.Han SY, Iliopoulos D, Druck T, Guler G, Grubbs CJ, Pereira M, Zhang Z, You M, Lubet RA, Fong LY, et al. CpG methylation in the Fhit regulatory region: relation to Fhit expression in murine tumors. Oncogene. 2004;23:3990–3998. doi: 10.1038/sj.onc.1207526. [DOI] [PubMed] [Google Scholar]
- 35.Lubet RA, Huebner K, Fong LY, Altieri DC, Steele VE, Kopelovich L, Kavanaugh C, Juliana MM, Soong SJ, Grubbs CJ. 4-Hydroxybutyl(butyl)nitrosamine-induced urinary bladder cancers in mice: characterization of FHIT and survivin expression and chemopreventive effects of indomethacin. Carcinogenesis. 2005;26:571–578. doi: 10.1093/carcin/bgh352. [DOI] [PubMed] [Google Scholar]
- 36.Kroft SH, Oyasu R. Urinary bladder cancer: mechanisms of development and progression. Lab Invest. 1994;71:158–174. [PubMed] [Google Scholar]
- 37.Theodorescu D, Cornil I, Fernandez BJ, Kerbel RS. Overexpression of normal and mutated forms of HRAS induces orthotopic bladder invasion in a human transitional cell carcinoma. Proc Natl Acad Sci USA. 1990;87:9047–9051. doi: 10.1073/pnas.87.22.9047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vageli D, Kiaris H, Delakas D, Anezinis P, Cranidis A, Spandidos DA. Transcriptional activation of H-ras, K-ras and N-ras protooncogenes in human bladder tumors. Cancer Lett. 1996;107:241–247. doi: 10.1016/0304-3835(96)04372-8. [DOI] [PubMed] [Google Scholar]
- 39.Cherfils J, Chardin P. GEFs: structural basis for their activation of small GTP-binding proteins. Trends Biochem Sci. 1999;24:306–311. doi: 10.1016/s0968-0004(99)01429-2. [DOI] [PubMed] [Google Scholar]
- 40.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 41.Zheng Y. Dbl family guanine nucleotide exchange factors. Trends Biochem Sci. 2001;26:724–732. doi: 10.1016/s0968-0004(01)01973-9. [DOI] [PubMed] [Google Scholar]
- 42.Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- 43.Seraj MJ, Harding MA, Gildea JJ, Welch DR, Theodorescu D. The relationship of BRMS1 and RhoGDI2 gene expression to metastatic potential in lineage related human bladder cancer cell lines. Clin Exp Metastasis. 2000;18:519–525. doi: 10.1023/a:1011819621859. [DOI] [PubMed] [Google Scholar]
- 44.Gildea JJ, Seraj MJ, Oxford G, Harding MA, Hampton GM, Moskaluk CA, Frierson HF, Conaway MR, Theodorescu D. RhoGDI2 is an invasion and metastasis suppressor gene in human cancer. Cancer Res. 2002;62:6418–6423. [PubMed] [Google Scholar]
- 45.Bos JL. Linking Rap to cell adhesion. Curr Opin Cell Biol. 2005;17:123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 46.Enserink JM, Price LS, Methi T, Mahic M, Sonnenberg A, Bos JL, Tasken K. The cAMP- Epac -Rap1 pathway regulates cell spreading and cell adhesion to laminin-5 through the alpha3beta1 integrin but not the alpha6beta4 integrin. J Biol Chem. 2004;279:44889–44896. doi: 10.1074/jbc.M404599200. [DOI] [PubMed] [Google Scholar]
- 47.Arthur WT, Quilliam LA, Cooper JA. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J Cell Biol. 2004;167:111–122. doi: 10.1083/jcb.200404068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Joneson T, Bar-Sagi D. Suppression of Ras-induced apoptosis by the Rac GTPase. Mol Cell Biol. 1999;19:5892–5901. doi: 10.1128/mcb.19.9.5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature. 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- 50.Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- 51.Joyce D, Bouzahzah B, Fu M, Albanese C, D'Amico M, Steer J, Klein JU, Lee RJ, Segall JE, Westwick JK, et al. Integration of Racdependent regulation of cyclin D1 transcription through a nuclear factor-kappaB- dependent pathway. J Biol Chem. 1999;274:25245–25249. doi: 10.1074/jbc.274.36.25245. [DOI] [PubMed] [Google Scholar]
- 52.Mettouchi A, Klein S, Guo W, Lopez-Lago M, Lemichez E, Westwick JK, Giancotti FG. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol Cell. 2001;8:115–127. doi: 10.1016/s1097-2765(01)00285-4. [DOI] [PubMed] [Google Scholar]
- 53.Oceguera-Yanez F, Kimura K, Yasuda S, Higashida C, Kitamura T, Hiraoka Y, Haraguchi T, Narumiya S. Ect2 and MgcRacGAP regulate the activation and function of Cdc42 in mitosis. J Cell Biol. 2005;168:221–232. doi: 10.1083/jcb.200408085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Billerey C, Chopin D, Aubriot-Lorton MH, Ricol D, Gil Diez de Medina S, Van Rhijn B, Bralet MP, Lefrere-Belda MA, Lahaye JB, Abbou CC, et al. Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol. 2001;158:1955–1959. doi: 10.1016/S0002-9440(10)64665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Rhijn BW, Vis AN, van der Kwast TH, Kirkels WJ, Radvanyi F, Ooms EC, Chopin DK, Boeve ER, Jobsis AC, Zwarthoff EC. Molecular grading of urothelial cell carcinoma with fibroblast growth factor receptor 3 and MIB-1 is superior to pathologic grade for the prediction of clinical outcome. J Clin Oncol. 2003;21:1912–1921. doi: 10.1200/JCO.2003.05.073. [DOI] [PubMed] [Google Scholar]
- 56.Lamy A, Gobet F, Laurent M, Blanchard F, Varin C, Moulin C, Andreou A, Frebourg T, Pfister C. Molecular profiling of bladder tumors based on the detection of FGFR3 and TP53 mutations. J Urol. 2006;176:2686–2689. doi: 10.1016/j.juro.2006.07.132. [DOI] [PubMed] [Google Scholar]
- 57.Cote RJ, Dunn MD, Chatterjee SJ, Stein JP, Shi SR, Tran QC, Hu SX, Xu HJ, Groshen S, Taylor CR, et al. Elevated and absent pRb expression is associated with bladder cancer progression and has cooperative effects with p53. Cancer Res. 1998;58:1090–1094. [PubMed] [Google Scholar]
- 58.Sugiura S, Ogawa K, Hirose M, Takeshita F, Asamoto M, Shirai T. Reversibility of proliferative lesions and induction of nonpapillary tumors in rat urinary bladder treated with phenylethyl isothiocyanate. Carcinogenesis. 2003;24:547–553. doi: 10.1093/carcin/24.3.547. [DOI] [PubMed] [Google Scholar]
- 59.Masui T, Dong Y, Yamamoto S, Takada N, Nakanishi H, Inada K, Fukushima S, Tatematsu M. p53 mutations in transitional cell carcinomas of the urinary bladder in rats treated with N-butyl-N-(4-hydroxybutyl)-nitrosamine. Cancer Lett. 1996;105:105–112. doi: 10.1016/0304-3835(96)04268-1. [DOI] [PubMed] [Google Scholar]
- 60.Kawakami K, Enokida H, Tachiwada T, Gotanda T, Tsuneyoshi K, Kubo H, Nishiyama K, Takiguchi M, Nakagawa M, Seki N. Identification of differentially expressed genes in human bladder cancer through genome-wide gene expression profiling. Oncol Rep. 2006;16:521–531. [PubMed] [Google Scholar]
- 61.Kim JH, Tuziak T, Hu L, Wang Z, Bondaruk J, Kim M, Fuller G, Dinney C, Grossman HB, Baggerly K, et al. Alterations in transcription clusters underlie development of bladder cancer along papillary and nonpapillary pathways. Lab Invest. 2005;85:532–549. doi: 10.1038/labinvest.3700250. [DOI] [PubMed] [Google Scholar]
- 62.Elsamman E, Fukumori T, Ewis AA, Ali N, Kajimoto K, Shinohara Y, Ishikawa M, Takahashi M, Nishitani MA, Baba Y, et al. Differences in gene expression between noninvasive and invasive transitional cell carcinoma of the human bladder using complementary deoxyribonucleic acid microarray: preliminary results. Urol Oncol. 2006;24:109–115. doi: 10.1016/j.urolonc.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 63.Dyrskjot L, Zieger K, Kruhoffer M, Thykjaer T, Jensen JL, Primdahl H, Aziz N, Marcussen N, Moller K, Orntoft TF. A molecular signature in superficial bladder carcinoma predicts clinical outcome. Clin Cancer Res. 2005;11:4029–4036. doi: 10.1158/1078-0432.CCR-04-2095. [DOI] [PubMed] [Google Scholar]
- 64.Modlich O, Prisack HB, Pitschke G, Ramp U, Ackermann R, Bojar H, Vogeli TA, Grimm MO. Identifying superficial, muscleinvasive, and metastasizing transitional cell carcinoma of the bladder: use of cDNA array analysis of gene expression profiles. Clin Cancer Res. 2004;10:3410–3421. doi: 10.1158/1078-0432.CCR-03-0134. [DOI] [PubMed] [Google Scholar]