

Abstract
DNA methylation is among the most stable epigenetic marks, ensuring tissue-specific gene expression in a heritable manner throughout development. Here we report that differentiated mesodermal somatic cells can confer tissue-specific changes in DNA methylation on epidermal progenitor cells after fusion in stable multinucleate heterokaryons. Myogenic factors alter regulatory regions of genes in keratinocyte cell nuclei, demethylating and activating a muscle-specific gene and methylating and silencing a keratinocyte-specific gene. Because these changes occur in the absence of DNA replication or cell division, they are mediated by an active mechanism. Thus, the capacity to transfer epigenetic changes to other nuclei is not limited to embryonic stem cells and oocytes but is also a property of highly specialized mammalian somatic cells. These results suggest the possibility of directing the reprogramming of readily available postnatal human progenitor cells toward specific tissue cell types.
Keywords: nuclear reprogramming
The hallmark of nuclear reprogramming is erasure of epigenetic identity. Stable epigenetic modifications control the expression of cell type-specific repertories of genes and are critical for maintaining differentiated cell phenotypes. One of the most prominent epigenetic modifications is methylation of DNA at CpG dinucleotides, which is associated with transcriptional repression (1–3). Changes in DNA methylation states are critical for the nuclear reprogramming that underlies the remarkable reversion of a somatic cell nucleus to a totipotent state after nuclear transfer and the subsequent development into an adult animal (4, 5). Embryonic stem cells also appear to have the capacity to impose global changes in gene expression and transfer epigenetic changes associated with pluripotency to more specialized cells to which they are fused (6–8). However, differentiated cells are not thought to have the capacity to reverse epigenetic states of other nuclei. After fusion of two differentiated somatic cells in mitotic hybrids, regulated changes in gene expression have previously been observed (9–14). Such nuclear reprogramming also occurs in differentiated cells fused in nondividing multinucleate heterokaryons (15–19). In this system, the direction of reprogramming is thought to be dictated by the ratio of the nuclei derived from each cell type (20–22). Here we demonstrate that, contrary to expectation, specialized somatic skeletal muscle cells confer epigenetic information to epidermal progenitor nuclei by directing the methylation and demethylation of DNA of the genes they activate or silence. These methylation changes are targeted in a tissue-specific manner, with keratinocyte-specific gene methylation accompanied by muscle-specific gene demethylation. Because the cell hybrids did not undergo mitosis or DNA replication, DNA demethylation occurs by an active mechanism. An implication of these results is that the methylation state of specific loci is continuously maintained in differentiated cells. The changes in epigenetic control cross germ layers, with mesodermal cells resetting the methylation status of genes of epidermal lineage. These results demonstrate that differentiated nuclei can impart tissue-specific epigenetic changes in a dynamic manner leading to the targeted reprogramming of the nuclei of readily available postnatal human progenitor cells.
Results
Transcriptional Reprogramming After Heterokaryon Formation.
To test the hypothesis that epigenetic reprogramming occurs in somatic cell heterokaryons, we investigated changes in DNA methylation at the regulatory regions of tissue-specific genes after fusion of mouse muscle cells with human primary keratinocytes derived from neonatal foreskin. In this heterokaryon system, differentiating myoblasts exit the cell cycle and fuse with each other to form multinucleated myotubes, to which human keratinocytes are fused by using PEG. Cultures are treated with the agents cytosine arabinoside (ara-c) and ouabain, which eliminate remaining cycling or unfused human cells, respectively (15). The resulting interspecies hybrids are stable: the nuclei remain distinct and intact, and mitosis does not occur (Fig. 1A). The use of cell types from different species permits gene expression and epigenetic alterations to be examined in a specific manner at the loci of interest in the non-muscle nuclei. Individual cells are documented to be heterokaryons by their Hoechst nuclear staining patterns; human nuclei stain diffusely whereas murine nuclei are punctate (Fig. 1B).
Fig. 1.

Reprogramming of gene expression in heterokaryons formed between differentiating murine myoblasts and human primary keratinocytes. (A) Keratinocyte populations uniformly expressing keratin-5 (K-5) were fused with myotubes using PEG, and resulting cultures were grown in low-serum medium supplemented with ara-c and ouabain (K-5, green; Hoechst, blue). Skeletal muscle NCAM is expressed by keratinocyte nuclei only after fusion with muscle cells (NCAM, red). (B) Individual cells are determined to be heterokaryons by the presence of nuclei derived from both human (uniform staining pattern) and mouse (punctate staining pattern) nuclei. (C Lower Left) Frequency of reprogramming is scored as the percentage of individual heterokaryons that express human NCAM. Error bars indicate the standard error of the proportion calculated from the binomial equation. (C Upper) Reprogramming of gene expression over time after fusion is shown. RT-PCR using human-specific primers for MyoD, keratin-14 (K-14), β-globin, and GAPDH. PCR was performed on control samples to test primer specificity and absence of amplification in mouse muscle cells. C2, C2C12 myoblasts; Rh, Rh30 human rhabdomyosarcoma cell line; K, keratinocytes; E, TF-1 human erythroleukemia cell line. (C Lower Right) Time course of gene expression by keratinocyte nuclei before and at daily time points after fusion (MyoD, 35 cycles; K-14, 26 cycles; β-globin, 35 cycles; GAPDH, 29 cycles).
In heterokaryons, non-muscle nuclei activate transcription of muscle genes and repress non-muscle genes in response to an excess of muscle-specific factors that are diffusible in the cytoplasm. A classical measure of reprogramming frequency in heterokaryons is immunostaining using the antibody 5.1H11, which is specific for skeletal muscle neural cell adhesion molecule (NCAM) and reacts only with the human and not the mouse muscle isoform (23). During muscle differentiation, expression of skeletal muscle NCAM is controlled by the myogenic basic helix–loop–helix transcription factor MyoD (24). Reprogramming is scored on a cell-by-cell basis by using NCAM expression as the indicator. The population of keratinocytes used in this study is pure based on uniform expression of keratin-5 in all cells (Fig. 1A Right). Although keratinocytes do not express NCAM, after fusion with excess muscle cells to form heterokaryons, keratinocyte nuclei activate NCAM at a frequency of 50% and 61% after 3 and 6 days in culture, respectively (Fig. 1C Lower Left), similar to results from previously published experiments (20).
We reasoned that NCAM expression is a downstream indicator of the well defined sequence of events that characterizes muscle differentiation, namely initiation by the myogenic basic helix–loop–helix transcription factors that drive expression of structural muscle genes. Therefore we chose to investigate activation of MyoD transcription as a focal point for studies of epigenetic reprogramming. In addition, as reprogramming would be expected to involve repression of the keratinocyte program, a keratinocyte-specific gene, keratin-14, was evaluated. Human-specific, intron-spanning PCR primers were designed for each of these genes, as well as the housekeeping gene, GAPDH, such that in total RNA preparations from heterokaryon cultures, mouse transcripts would not be detected (Fig. 1C Upper). The β-globin gene was chosen as a control because neither keratinocyte nor muscle expression profiles involve β-globin. RT-PCR analysis was performed on RNA isolated from cultures before fusion and daily thereafter (Fig. 1C Lower Right). Whereas keratinocytes do not express MyoD before fusion, low- level expression of this gene was detectable 1 day after heterokaryon formation. Expression was significantly increased by days 3, 5, and 7. By contrast, keratin-14 transcript levels, although highly abundant in keratinocytes, steadily declined after fusion. As expected, heterokaryons do not express significant levels of β-globin transcripts compared with the positive control, TF-1 erythroleukemia cells. Thus, MyoD activation and keratin-14 repression occur in the tissue-specific manner expected of this heterokaryon system.
Identification of Cell Type-Specific Patterns of CpG Methylation.
We postulated that reprogramming involves reversal of epigenetic silencing mechanisms at regulatory regions to allow activation of MyoD, and the converse for keratin-14. DNA methylation patterns in differentiated cells are generally thought to be stable epigenetic modifications. To determine whether DNA methylation is specifically altered as part of reprogramming in heterokaryons, a combination of bisulfite conversion and restriction analysis (COBRA) (25) was used [supporting information (SI) Fig. 5]. We focused on individual CpG dinucleotides, as their methylation status has been shown to be important for tissue and physiological context-specific gene expression in normal development and postnatal life (26–30) as well as during nuclear reprogramming using oocytes (31–33). To identify candidate CpG sites in the MyoD regulatory regions, we surveyed the methylation status of conserved sites between mouse and man in muscle and non-muscle tissues and cells. The MyoD regulatory regions consist of the promoter, distal regulatory region, and a 258-bp core enhancer located 20 kb upstream from the promoter. The core enhancer is highly conserved between mouse and man and is essential for appropriate expression of MyoD during development (34, 35). Of sites that could be evaluated by using the COBRA technique, our analysis of the MyoD core enhancer revealed one region containing two CpG sites at which methylation status exhibits a strong inverse correlation with gene expression (Fig. 2A). In mouse tissue samples comprised of somewhat heterogeneous populations of cells, the MyoD core enhancer is primarily unmethylated in skeletal muscle. This is evidenced by the predominance of uncleaved PCR product after exposure to the enzyme HpyCH4IV that recognizes the two ACGT sites within the amplified region. By comparing the intensity of the cleaved with the uncleaved bands, the relative amounts of methylated and unmethylated DNA can be assessed. By contrast, in samples from brain, liver, and hematopoietic cells, cleavage products predominate, indicative of methylation in these tissues (77%, 62%, and 73%, respectively). Similarly, analysis of the MyoD core enhancer in purified cell lines showed an absence of methylation in mouse and human muscle cells (C2 in Fig. 2A and Rh in Fig. 2B) and a high degree of methylation in primary human keratinocytes (76%) (Fig. 2B). For human cells, one ACTG site is contained within the amplified region as opposed to two in the mouse, providing the expected distinct pattern of bands in samples from each species after enzyme digestion. Evaluation of CpG sites in the keratin-14 promoter and β-globin HS2 locus also revealed tissue-specific methylation patterns, establishing these sites as relevant candidates for studying methylation changes during reprogramming (Fig. 2 C and D).
Fig. 2.

Analysis of tissue-specific DNA methylation in mouse and human tissues and cell lines. (A–D Top) Schematic drawings of regulatory regions of the genes. (A–D Middle) The regions analyzed, enlarged to show the HpyCH4IV recognition sites (ACGT) examined (black circles), bisulfite-specific primers for PCR (black arrows), and the sizes of HpyCH4IV cleaved PCR products (above the line) and uncleaved PCR products (below the line). (A–D Bottom) COBRA methylation analysis of the ACGT sites of the genes studied: uncleaved and cleaved PCR products visualized by gel electrophoresis (labeled as UM and M, respectively), and the bar graphs showing the percentage of methylation in the corresponding lane above. (A) The mouse MyoD core enhancer. Mu, mouse muscle; Br, brain; L, liver; Bl, blood; C2, C2 myoblasts. (B) The human MyoD core enhancer. (C) The human keratin-14 promoter. (D) The human β-globin HS2 locus. Rh, human rhabdomyosarcoma muscle cells (RH30); K, primary keratinocytes; E, erythroblasts (TF-1 cells).
Detection of Cell Type-Specific Methylation and Demethylation in Heterokaryons.
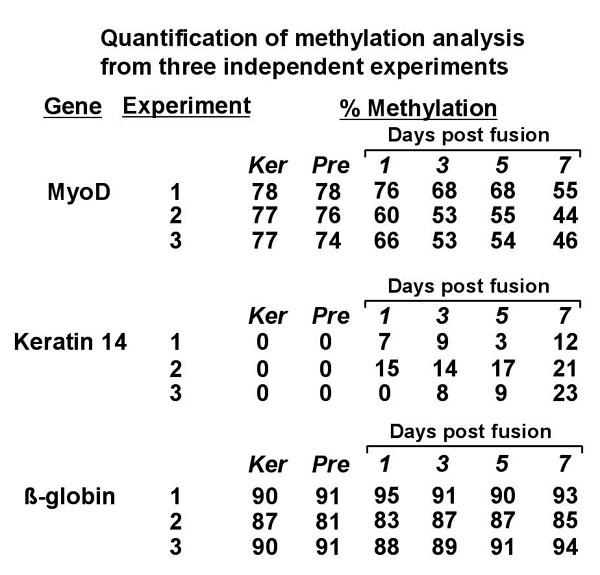
For studies of methylation changes in keratinocyte nuclei in multinucleate heterokaryons, the COBRA technique had to be modified. To distinguish human from mouse regulatory regions, nested primers were designed that specifically amplify only human, bisulfite converted DNA. Note the absence of amplification in lanes containing samples of DNA not treated with bisulfite or in those containing mouse DNA (Fig. 3A). Before PCR amplification, the amount of total human DNA was measured in each mixed mouse and human culture sample by quantitative PCR, and equal amounts of human DNA were used for each bisulfite reaction and PCR (SI Fig. 6). These conditions provided precise, reproducible analysis of methylation in heterokaryons. PCR amplification produced a band of 554 bp that is only present in samples containing bisulfite-treated keratinocyte DNA. After amplification, DNA samples were digested with the restriction enzyme HpyCH4IV. Cleavage at the single site within the amplified 554-bp product produces two bands of 218 and 336 bp, respectively. Methylation of the keratinocyte MyoD core enhancer was evaluated over time after fusion in three independent heterokaryon experiments. A representative time course from one experiment is shown together with quantitative analysis (Fig. 3B). Demethylation of the core enhancer was apparent within 24 hours and maximal 7 days after fusion. Notably, similar to the time course seen for demethylation of the Oct-4 promoter in Xenopus nuclear transfer experiments, the time course was on the order of days, not hours (32). On day 7, the site examined was unmethylated 56% of the time (Fig. 3B), in good agreement with the frequency of human NCAM activation (61 ± 6% at day 6). In addition, despite the use of ouabain, a small proportion of unfused keratinocytes remains in cultures even at 7 days, and these contribute to a portion of the analyzed DNA. As would be predicted if demethylation were important for MyoD transcription during reprogramming, the time course of demethylation of the core enhancer correlated with that of MyoD expression (Fig. 1C). To assess the specificity of demethylation, analysis was also performed at conserved CpG sites in the keratin-14 promoter and the β-globin HS locus at which methylation levels increased and remained constant, respectively (Fig. 3 C–E). As an independent verification of the COBRA analysis, bisulfite sequencing was performed on two samples, before fusion and 7 days after, with similar results (P = 0.0083, Fisher's exact test) (Fig. 3F). Sequencing provided additional information relating to two other core enhancer CpG sites not analyzable by restriction analysis, one that was significantly demethylated (from 94% to 79%, P = 0.014) and one at which demethylation was apparent (from 54% to 42%) but was not statistically significant. All sequences obtained were human, providing additional confirmation of the species specificity of the PCR amplification process used in these experiments. Time course analyses were also performed for keratin-14 and β-globin. Methylation levels, undetectable before fusion, steadily increased at the keratin-14 site, to 21% at 7 days, in accordance with the decrease in transcript levels (Fig. 3 C and E). By contrast, the β-globin HS2 locus digestion profile remained unchanged (Fig. 3 D and E). A compilation of data from three independent time course experiments is shown (Fig. 3E and SI Fig. 7). Together, these data indicate that changes in methylation states occur in a dynamic, tissue-specific pattern in accordance with gene expression.
Fig. 3.

Dynamic and gene-specific changes in DNA methylation during nuclear reprogramming. (A) Confirmation of the specificity of bisulfite-specific, human-specific primers: both untreated and bisulfite-treated DNA from human keratinocytes (K), RH30 muscle cells (M), mouse C2 myoblasts (C2), and liver tissue (L) were used, and only bisulfite-treated human DNA showed specific amplification. (B–D) Representative gels of COBRA methylation analysis of keratinocyte DNA during nuclear reprogramming in heterokaryons. (B) MyoD core enhancer. (C) Keratin-14 promoter. (D) β-Globin HS2 locus. Sample order is as follows: differentiating mouse myoblasts (C2), keratinocytes (K), keratinocytes before fusion with C2 (Pre), and heterokaryons cultured for 1, 3, 5, and 7 days after fusion. (E) Average and SD of DNA methylation status of the keratinocyte MyoD core enhancer, keratin-14 promoter, and β-globin HS2 locus in heterokaryons across three independent experiments (see SI Fig. 7). (F) Bisulfite sequencing analysis performed on the keratinocyte MyoD core enhancer: before fusion (Pre) and in heterokaryons cultured for 7 days after fusion. This region contains three CpG sites. Arrows indicate the CpG site analyzed in COBRA assay. Each sample had 48 clones sequenced. The numbers at the bottom indicate the percentage of DNA methylation at each CpG site before and after nuclear reprogramming. ∗, P < 0.01; ∗∗, P < 0.05.
DNA Is Demethylated by an Active Mechanism During Reprogramming.
Demethylation of DNA can occur passively by failure to methylate newly synthesized DNA during replication or actively in the absence of DNA replication. Active demethylation is known to occur during reprogramming after oocyte nuclear transfer (36), during gametogenesis, and during T cell activation (36–40). The postmitotic nature of heterokaryons and reprogramming observed in the presence of ara-c suggested that the demethylation of the MyoD core enhancer might occur by an active process. The existence of a mechanism for active DNA demethylation in muscle cells has been previously suggested (41, 42), but the absence of DNA replication in cells was not conclusively shown. To confirm the absence of DNA replication in heterokaryons, cultures were labeled continuously with BrdU starting at the time of fusion. Whereas growing myoblast or keratinocyte nuclei readily incorporate BrdU, heterokaryon nuclei remained uniformly negative when scored daily throughout the 7-day time course of labeling, determined by examination of >5,000 individual nuclei (Fig. 4). Cultures stained at daily time points exhibited rare (<0.6%) BrdU-positive keratinocyte nuclei, but none of these nuclei were in heterokaryons. Thus, to the extent detectable by BrdU incorporation, epigenetic reprogramming in heterokaryons occurs in the absence of DNA replication.
Fig. 4.

Assessment of DNA replication in heterokaryon cultures. Cells were labeled with BrdU in the medium, and incorporation was detected by immunofluorescence (green). (A) Representative images of growing myoblasts used as a positive control (upper six images) and heterokaryons (lower six images). In each case the white box indicates the region shown at higher magnification in the three panels below. (Right) Hoechst 33258 was used to label all nuclei. (B) The percentage of BrdU-positive myoblast and human nuclei is shown for various time points. GM indicates cells in growth medium (12 h of BrdU labeling). “Pre fusion” indicates cells in differentiation medium during coculture before PEG (4 h of BrdU labeling). For all other points BrdU was added to the medium at the time of PEG treatment and replaced daily, and cells were fixed and stained at the indicated time points. At each time point, at least 700 nuclei were analyzed.
Discussion
Epigenetic reprogramming has previously been considered an attribute particular to gametogenesis, early embryogenesis, and cloning (1, 43). However, the findings presented in this article show that stable, heritable epigenetic modifications are readily reversible by mechanisms that exist in differentiated nonreplicating somatic cells. In addition to activators and repressors of transcription, instructional information in the form of DNA methylation patterns can be transferred via the cytoplasm from the nuclei of one cell type to those of another in heterokaryons. Because DNA demethylation occurs in the absence of DNA replication, it entails an active mechanism. Moreover, the preexisting DNA methylation and demethylation machinery present in the cytoplasm are targeted to distinct genetic loci in recipient nuclei in accordance with the reprogrammed state.
This regulation of methylation status appears to be ongoing. Demethylation of the MyoD core enhancer accompanies muscle cell specialization during development (44). Thus, the methylation patterns of the regulatory regions studied here were already well established in the muscle cells before use in heterokaryons. It therefore appears surprising that differentiated myotubes would still contain the factors necessary to execute the observed changes. We propose that specialized methylation patterns, although stable, may involve continuous regulation based on tissue-specific feedback among other chromatin modifiers and signaling pathways (22). Such a scenario would provide an explanation for the demethylation of muscle regulatory regions and methylation of keratinocyte regulatory regions in keratinocyte nuclei after transfer into the multinucleate muscle environment, where they would be subject to an excess of muscle regulators and ongoing epigenetic maintenance.
For the purposes of cellular therapy, elucidating molecular mechanisms responsible for reprogramming nuclei toward a muscle fate in heterokaryons merits further study. Cell type, cell cycle phase, differentiation state, and age of the non-muscle nucleus may all influence the efficiency of nuclear reprogramming, and the effects of these differences need to be eluciated. Additionally, if the challenge of extracting reprogrammed nuclei from heterokaryons and expanding the resultant cells can be overcome, in vitro reprogramming may provide an alternative to nuclear transfer or ES cell fusion for the in vitro generation of therapeutic cells. This method would obviate the need to direct differentiation through the use of genetic alterations by using intrinsic factors to reprogram cells to the desired phenotype. Finally, the heterokaryon approach could be extended to examine the feasibility of reprogramming cells not only to the muscle fate, but also to phenotypes useful for treating other degenerative diseases.
Materials and Methods
Cells and Tissue Culture.
Mouse C2C12 muscle cells were grown in DMEM supplemented with 20% FCS and penicillin/streptomycin. To induce myotube formation, myoblasts were seeded at near confluent densities on collagen-coated plates in DMEM supplemented with 2% horse serum and antibiotics (DM). After 24 h the medium was changed and ara-c (10−5 M; Sigma–Aldrich, St. Louis, MO) was added to eliminate dividing myoblasts. Medium supplemented with ara-c was replaced daily thereafter. After 48 h, myotubes formed in this manner were used for the production of heterokaryons.
Human primary keratinocytes isolated from neonatal foreskin were generously provided by Sally Pennypacker (Veterans Affairs Medical Center, San Francisco, CA). Keratinocytes were grown in medium 154-CF (Cascade Biologics, Portland, OR) and supplemented with human keratinocyte growth supplement (Cascade Biologics) and 0.07 mM calcium. Keratinocytes were used at passage three or less for heterokaryon formation.
Human erythroblasts (TF-1) were grown in 10% FBS and 90% RPMI 1640 medium with 2 mM l-glutamine adjusted to contain 1.5 g/liter sodium bicarbonate, 4.5 g/liter glucose, 10 mM Hepes, and 1.0 mM sodium pyruvate supplemented with 2 ng/ml recombinant human GM-CSF as described by the American Type Culture Collection (Manassas, VA).
Human rhabdomyosarcoma cells (RH30) were grown in DMEM supplemented with 10% FCS and penicillin/streptomycin. Mouse tissues were harvested from an adult C57BL mouse.
PEG Fusion.
Heterokaryons were produced by fusing myotubes and keratinocytes using PEG. Keratinocytes were resuspended in DM and plated onto 2-day-old myotube cultures. Four hours later, cultures were treated with PEG 1500 (50% wt/vol in Hepes) (Roche, Indianapolis, IN) for 60 sec at 37°C, followed by three successive washes with DMEM. DM was replaced after washing and then again at 4–6 h after PEG treatment, along with the addition of ara-c and ouabain (10−5 M; Sigma).
Immunofluorescence.
Heterokaryons were stained with the mouse monoclonal antibody 5.1H11 (Developmental Studies Hybridoma Bank, University of Iowa). Live cells were exposed to the antibody undiluted in mouse ascites fluid for 30 min at 37°C. Cells were washed three times in DM for 5 min each, followed by incubation with the secondary antibody, Alexa Fluor 488 goat anti-mouse (Molecular Probes, Carlsbad, CA), at a 1:500 dilution in DM for 30 min at 37°C. Cells were rinsed with DM twice, followed by PBS, and fixed in 1.5% formaldehyde in PBS for 10–15 min at room temperature. After permeabilization with ice-cold methanol for 20 min at room temperature, cells were rinsed three times in PBS and nuclei were stained with Hoechst 33258 (0.12 μM) in PBS for 15 min, followed by four PBS washes.
For keratin-5 immunostaining, cells were fixed and permeabilized as above. Blocking was performed with 20% FCS in PBS, followed by incubation with the primary antibody rabbit anti keratin-5 (Covance, Berkeley, CA) at 1:1,000 dilution in blocking buffer for 30 min at room temperature. Cells were rinsed three times in PBS and then incubated in secondary antibody, Alexa Fluor 488 goat anti-rabbit (Molecular Probes), at 1:500 dilution for 30 min, rinsed, stained with Hoechst, and processed as above.
For BrdU labeling, cells were exposed to BrdU labeling reagent (Roche BrdU labeling and detection kit I) for the indicated times and processed according to the manufacturer's protocol.
Cells were visualized by using a Zeiss Axioplan 2 fluorescence microscope equipped with a 40× water immersion objective (Carl Zeiss, Thornwood, NY). Images were acquired by using OpenLab software (Improvision, Coventry, UK).
Analysis of Gene Expression.
RNA was prepared from cells by using the RNeasy mini kit (Qiagen, Valencia, CA). One microgram of total RNA was reverse-transcribed by using oligo dT primers and the First Strand cDNA synthesis kit (Roche). Two microliters of cDNA was amplified with gene-specific primers by using Invitrogen PCR supermix (Carlsbad, CA). Primer sequences were as follows: MyoD forward, 5′-AGCACTACAGCGGCGACT-3′; MyoD reverse, 5′-GCGACTCAGAAGGCACGTC-3′; keratin-14 forward, 5′-GTGGCCTTGGTGCTGGCT-3′; keratin-14 reverse, 5′-CATTGTCCACTGTGGCTGTG-3′; β-globin forward: 5′-CCTGA-GGAGAAGTCTGCCGTTAC-3′; β-globin reverse, 5′-ATTAGCCACACCAGCCACCACTTT-3′; GAPDH forward, 5′-GCCCCAGCAAGAGCACAAGAGG-3′; GAPDH reverse, 5′-ACAAGGTGCGGCTCCCTA-3′.
Genomic DNA Extraction.
Cells and heterokaryons grown in 6-cm dishes were trypsinized and pelleted, and DNA was extracted by using the DNeasy Tissue Kit (Qiagen). DNA was eluted into 200 μl of AE buffer, and the concentration was measured by A260 using a ND-1000 Spectrometer (NanoDrop, Wilmington, DE). DNA from various mouse tissues was extracted by using the DNeasy Tissue Kit (Qiagen) according to the manufacturer's protocol.
Quantification of Human DNA in Heterokaryons.
To determine the amount of human DNA present in heterokaryon DNA samples, quantitative PCR was carried out by using Platinum SYBR Green qPCR Supermix UDG (Invitrogen) in a Rotor-Gene (RG-3000) real-time thermal cycler (Corbett Research, Sydney, Australia). Human-specific GAPDH primers were 5′-GCC CCA GCA AGA GCA CAA GAG G (forward) and 5′-ACA AGG TGC GGC TCC CTA (reverse). The PCR amplification was carried out as follows: 94°C for 3 min, 40× (94°C for 30 sec, 59°C for 30 sec, and 72°C for 30 sec).
COBRA Assay of DNA Methylation. Bisulfite treatment of DNA.
Bisulfite treatment was done by using the EZ DNA Methylation kit (ZYMO Research, Orange, CA) according to the manufacturer's protocol with the following modifications. DNA samples were calculated to contain 40 ng of human DNA based on quantitative PCR. Mouse C2 DNA was added to samples to achieve the same amount of total DNA (1.5 μg). The DNA solution was mixed with 0.5 μl of EcoRI (20 units/μl; NEB, Ipswitch, MA), 4.5 μl of EcoRI buffer (NEB), and ddH2O, to reach 45 μl of total volume. Enzyme digestion was carried out at 37°C for 4 h. Next, the samples were heated at 70°C for 25 min followed by heating at 97°C for 5 min to denature the DNA. After cooling to room temperature, 5 μl of M-dilution buffer was added and mixed, and then incubated at 42°C for 30 min. Then, 100 μl of CT conversion reagent was added, and DNA samples were wrapped in aluminum foil and incubated at 55°C for 15 h. The resulting bisulfite-treated DNA was purified according to the kit's protocol. The DNA was eluted in 20 μl of M-elution buffer, and 4 μl of this was used in the bisulfite-specific PCR amplification.
Designing bisulfite-specific human-specific primers.
Gene-specific primers were designed to be both bisulfite-specific and human-specific. Specially, the PCR primers were designed to be complementary to one of the converted DNA strands with no CpG dinucleotides in the original sequences. To increase the sensitivity of PCR amplification, two sets of primers were designed for nested PCR. To analyze the human MyoD core enhancer, the two external primers were 5′-TTA ATA TTT GTT ATT TAG GTA AGG AAG GTT (forward) and 5′-TAC TAA CTC ACT TCA ACC ACC ATA CAC AAA (reverse), and the two internal primers were 5′-GGA ATT ATG GGG TTT TAA GTG GTA GGT ATT (forward) and 5′-AAA AAA CCA AAA CCT TCT AAT CAT TTC TAA (reverse). For the human β-globin HS2 locus, the two external primers were 5′-TGA TTA AAT AAG TTT TAG TTT TTT TTT AGT TTT (forward) and 5′-CTA ATA CAC CAA AAC TAA CCT AAA ATA (reverse), and the two internal primers were 5′-TAA GTA TGA GTA GTT TTG GTT AGG TTT (forward) and 5′-TTC CAT ATC CTT ATT TCA TAT TAA TAC ATA TA (reverse). For the human keratin-14 promoter, the two external primers were 5′-TTT TGG TTT TTA TGA GGT TAT TTG TTT (forward) and 5′-CAC TAA ACT TAA CCA AAA TAA CAA AAA A (reverse), and the two internal primers were 5′-GG ATG GGA TAG ATT TTT TAG TAT AGT (forward) and 5′-CCA AAA TAA CAA AAA ACA AAA CCT CTA (reverse).
Bisulfite-specific PCR and restriction enzyme digestion.
Both first and second rounds of bisulfite-specific PCR amplifications were carried out as follows: 94°C for 3 min, 30× (94°C for 30 sec, 60°C for 30 sec, and 68°C for 30 sec) and 68°C for 5 min. For keratin-14, the annealing temperature was 64°C instead of 60°C. One microliter of first-round PCR product was used as a template for second-round PCR amplification. The amplified PCR products were purified by the QIAquick PCR purification kit (Qiagen) and eluted in 40 μl of EB buffer. DNA concentration was measured by NanoDrop, and 600 ng of PCR product was digested with 1 μl of HpyCH4IV (10 units/μl) in 20-μl volume at 37°C for 2 h. The digested PCR products were separated by agarose gel electrophoresis. The digital gel images were acquired by using a BioDoc-It system or Imagestore 7500 UV transilluminator (UVP) and were analyzed by 1D Image Analysis software (version 4.0.3) from Kodak (Rochester, NY). The values of the total intensity minus the background of all of the bands were exported to Microsoft Excel. The percentage of methylation in a given sample is calculated as the ratio of the intensity of cleaved PCR products to that of all PCR products.
Bisulfite Sequencing.
PCR products from second-round bisulfite-specific PCR amplification were ligated with the vector by using the pGEM-T Easy Vector system (Promega, Madison, WI). Ligations were carried out at room temperature for 2 h and were transformed into DH5α subcloning efficiency-competent cells (Invitrogen). Bacterial colonies were grown on LB/Amp/X-Gal plates overnight, and white colonies were picked randomly and inoculated into 5 ml of LB/Amp liquid culture. Bacterial cultures were shaken overnight at 37°C, and plasmid DNA was extracted by using Qiaprep Spin Miniprep Kit (Qiagen). One microgram of DNA was sent for sequencing by using SP6 primers (3′ to the insert). One hundred plasmid DNA samples were sequenced in total (50 for each sample).
Statistical Analysis.
Bisulfite sequencing results were analyzed by using a one-tailed Fisher's exact test, a nonparametric test for categorical variables appropriate for small and medium sample sizes.
Supplementary Material
Acknowledgments
We thank Chen Liu for technical assistance, Mark Hammer for critical reading of the manuscript, and other members of the laboratory for helpful suggestions and advice. This work was supported by National Institutes of Health National Research Service Award AF051678 (to J.H.P.); a Howard Hughes Medical Institute fellowship (to G.S.); a National Science Foundation fellowship (to A.T.P.); National Institutes of Health Grants HD018179, AG009521, AG024987, and AG020961 (to H.M.B.); the Ellison Medical Foundation; and the Baxter Foundation (H.M.B.).
Abbreviations
- NCAM
neural cell adhesion molecule
- ara-c
cytosine arabinoside
- COBRA
combined bisulfite conversion and restriction analysis.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0700181104/DC1.
References
- 1.Li E. Nat Rev Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 2.Bird A. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 3.Jones PA, Takai D. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 4.Rhind SM, Taylor JE, De Sousa PA, King TJ, McGarry M, Wilmut I. Nat Rev Genet. 2003;4:855–864. doi: 10.1038/nrg1205. [DOI] [PubMed] [Google Scholar]
- 5.Blelloch R, Wang Z, Meissner A, Pollard S, Smith A, Jaenisch R. Stem Cells. 2006;24:2007–2013. doi: 10.1634/stemcells.2006-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silva J, Chambers I, Pollard S, Smith A. Nature. 2006;441:997–1001. doi: 10.1038/nature04914. [DOI] [PubMed] [Google Scholar]
- 7.Cowan CA, Atienza J, Melton DA, Eggan K. Science. 2005;309:1369–1373. doi: 10.1126/science.1116447. [DOI] [PubMed] [Google Scholar]
- 8.Tada M, Takahama Y, Abe K, Nakatsuji N, Tada T. Curr Biol. 2001;11:1553–1558. doi: 10.1016/s0960-9822(01)00459-6. [DOI] [PubMed] [Google Scholar]
- 9.Ephrussi B, Davidson RL, Weiss MC, Harris H, Klein G. Nature. 1969;224:1314–1316. doi: 10.1038/2241314a0. [DOI] [PubMed] [Google Scholar]
- 10.Davidson RL. Annu Rev Genet. 1974;8:195–218. doi: 10.1146/annurev.ge.08.120174.001211. [DOI] [PubMed] [Google Scholar]
- 11.Peterson JA, Weiss MC. Proc Natl Acad Sci USA. 1972;69:571–575. doi: 10.1073/pnas.69.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darlington GJ, Bernard HP, Ruddle FH. Science. 1974;185:859–862. doi: 10.1126/science.185.4154.859. [DOI] [PubMed] [Google Scholar]
- 13.Shay JW. Mol Cell Biochem. 1983;57:17–26. doi: 10.1007/BF00223521. [DOI] [PubMed] [Google Scholar]
- 14.Fenyo EM, Wiener F, Klein G, Harris H. J Natl Cancer Inst. 1973;51:1865–1875. doi: 10.1093/jnci/51.6.1865. [DOI] [PubMed] [Google Scholar]
- 15.Blau HM, Chiu CP, Webster C. Cell. 1983;32:1171–1180. doi: 10.1016/0092-8674(83)90300-8. [DOI] [PubMed] [Google Scholar]
- 16.Wright WE. Exp Cell Res. 1984;151:55–69. doi: 10.1016/0014-4827(84)90355-0. [DOI] [PubMed] [Google Scholar]
- 17.Baron MH, Maniatis T. Cell. 1986;46:591–602. doi: 10.1016/0092-8674(86)90885-8. [DOI] [PubMed] [Google Scholar]
- 18.Spear BT, Tilghman SM. Mol Cell Biol. 1990;10:5047–5054. doi: 10.1128/mcb.10.10.5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terranova R, Pereira CF, Du Roure C, Merkenschlager M, Fisher AG. J Cell Sci. 2006;119:2065–2072. doi: 10.1242/jcs.02945. [DOI] [PubMed] [Google Scholar]
- 20.Blau HM, Pavlath GK, Hardeman EC, Chiu CP, Silberstein L, Webster SG, Miller SC, Webster C. Science. 1985;230:758–766. doi: 10.1126/science.2414846. [DOI] [PubMed] [Google Scholar]
- 21.Pavlath GK, Blau HM. J Cell Biol. 1986;102:124–130. doi: 10.1083/jcb.102.1.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blau HM, Baltimore D. J Cell Biol. 1991;112:781–783. doi: 10.1083/jcb.112.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walsh FS, Ritter MA. Nature. 1981;289:60–64. doi: 10.1038/289060a0. [DOI] [PubMed] [Google Scholar]
- 24.Kawahigashi H, Harada Y, Asano A, Nakamura M. Biochim Biophys Acta. 1998;1397:305–315. doi: 10.1016/s0167-4781(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 25.Xiong Z, Laird PW. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomassin H, Flavin M, Espinas ML, Grange T. EMBO J. 2001;20:1974–1983. doi: 10.1093/emboj/20.8.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Condorelli DF, Nicoletti VG, Barresi V, Caruso A, Conticello S, de Vellis J, Giuffrida Stella AM. J Neurosci Res. 1994;39:694–707. doi: 10.1002/jnr.490390610. [DOI] [PubMed] [Google Scholar]
- 28.Young HA, Ghosh P, Ye J, Lederer J, Lichtman A, Gerard JR, Penix L, Wilson CB, Melvin AJ, McGurn ME, et al. J Immunol. 1994;153:3603–3610. [PubMed] [Google Scholar]
- 29.Lee DU, Agarwal S, Rao A. Immunity. 2002;16:649–660. doi: 10.1016/s1074-7613(02)00314-x. [DOI] [PubMed] [Google Scholar]
- 30.White GP, Watt PM, Holt BJ, Holt PG. J Immunol. 2002;168:2820–2827. doi: 10.4049/jimmunol.168.6.2820. [DOI] [PubMed] [Google Scholar]
- 31.Dean W, Santos F, Reik W. Semin Cell Dev Biol. 2003;14:93–100. doi: 10.1016/s1084-9521(02)00141-6. [DOI] [PubMed] [Google Scholar]
- 32.Simonsson S, Gurdon J. Nat Cell Biol. 2004;6:984–990. doi: 10.1038/ncb1176. [DOI] [PubMed] [Google Scholar]
- 33.Barton SC, Arney KL, Shi W, Niveleau A, Fundele R, Surani MA, Haaf T. Hum Mol Genet. 2001;10:2983–2987. doi: 10.1093/hmg/10.26.2983. [DOI] [PubMed] [Google Scholar]
- 34.Goldhamer DJ, Brunk BP, Faerman A, King A, Shani M, Emerson CP., Jr Development (Cambridge, UK) 1995;121:637–649. doi: 10.1242/dev.121.3.637. [DOI] [PubMed] [Google Scholar]
- 35.Chen JC, Love CM, Goldhamer DJ. Dev Dyn. 2001;221:274–288. doi: 10.1002/dvdy.1138. [DOI] [PubMed] [Google Scholar]
- 36.Dean W, Santos F, Stojkovic M, Zakhartchenko V, Walter J, Wolf E, Reik W. Proc Natl Acad Sci USA. 2001;98:13734–13738. doi: 10.1073/pnas.241522698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Nature. 2000;403:501–502. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 38.Bruniquel D, Schwartz RH. Nat Immunol. 2003;4:235–240. doi: 10.1038/ni887. [DOI] [PubMed] [Google Scholar]
- 39.Kersh EN, Fitzpatrick DR, Murali-Krishna K, Shires J, Speck SH, Boss JM, Ahmed R. J Immunol. 2006;176:4083–4093. doi: 10.4049/jimmunol.176.7.4083. [DOI] [PubMed] [Google Scholar]
- 40.Murayama A, Sakura K, Nakama M, Yasuzawa-Tanaka K, Fujita E, Tateishi Y, Wang Y, Ushijima T, Baba T, Shibuya K, et al. EMBO J. 2006;25:1081–1092. doi: 10.1038/sj.emboj.7601012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lucarelli M, Fuso A, Strom R, Scarpa S. J Biol Chem. 2001;276:7500–7506. doi: 10.1074/jbc.M008234200. [DOI] [PubMed] [Google Scholar]
- 42.Weiss A, Keshet I, Razin A, Cedar H. Cell. 1996;86:709–718. doi: 10.1016/s0092-8674(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 43.Surani MA, Ancelin K, Hajkova P, Lange UC, Payer B, Western P, Saitou M. Cold Spring Harbor Symp Quant Biol. 2004;69:1–9. doi: 10.1101/sqb.2004.69.1. [DOI] [PubMed] [Google Scholar]
- 44.Brunk BP, Goldhamer DJ, Emerson CP., Jr Dev Biol. 1996;177:490–503. doi: 10.1006/dbio.1996.0180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}