

Abstract
Processing of the N-terminal initiator methionine is an essential cellular process conserved from prokaryotes to eukaryotes. The enzymes that remove N-terminal methionine are known as methionine aminopeptidases (MetAPs). Human MetAP2 has been shown to be required for the proliferation of endothelial cells and angiogenesis. The physiological function of MetAP1, however, has remained elusive. In this report we demonstrate that a family of inhibitors with a core structure of pyridine-2-carboxylic acid previously developed for the bacterial and yeast MetAP1 is also specific for human MetAP1 (HsMetAP1), as confirmed by both enzymatic assay and high-resolution x-ray crystallography. Treatment of tumor cell lines with the MetAP1-specific inhibitors led to an accumulation of cells in the G2/M phase, suggesting that HsMetAP1 may play an important role in G2/M phase transition. Overexpression of HsMetAP1, but not HsMetAP2, conferred resistance of cells to the inhibitors, and the inhibitors caused retention of N-terminal methionine of a known MetAP substrate, suggesting that HsMetAP1 is the cellular target for the inhibitors. In addition, when HsMetAP1 was knocked down by gene-specific siRNA, cells exhibited slower progression during G2/M phase, a phenotype similar to cells treated with MetAP1 inhibitors. Importantly, MetAP1 inhibitors were able to induce apoptosis of leukemia cell lines, presumably as a consequence of their interference with the G2/M phase checkpoint. Together, these results suggest that MetAP1 plays an important role in G2/M phase of the cell cycle and that it may serve as a promising target for the discovery and development of new anticancer agents.
Keywords: aminopeptidase inhibitors, angiogenesis, apoptosis
Protein synthesis is initiated with a methionine residue in eukaryotic cells or a formylated methionine in prokaryotes, mitochondria, and chloroplasts. For a large subset of proteins, the initiator methionine is cotranslationally removed before further posttranslational modification. The proteolytic removal of N-terminal methionine is catalyzed by a family of enzymes known as methionine aminopeptidases (MetAPs). The functions of these enzymes are evolutionally conserved and essential, as demonstrated by the lethal phenotype of the map null mutant in bacteria. Although only one MetAP gene is present in the genome of most, but not all, prokaryotes, at least two types of MetAPs, type I and type II, are known in eukaryotic cells. In budding yeast Saccharomyces cerevisiae, deletion of either ScMetAP1 or ScMetAP2 resulted in a slow-growth phenotype compared with the wild-type strain, whereas the double mutant is nonviable, indicating the redundant yet essential functions of both types of MetAP (1, 2). In multicellular organisms MetAP2 has been shown to be essential for the proliferation and development of specific tissues (3, 4).
Human MetAP2 has been identified as the primary target of the fumagillin family of natural products that potently inhibit angiogenesis (5, 6). A synthetic analog of fumagillin, TNP-470, with higher potency and lower toxicity, has entered clinical trials for a variety of cancers (7, 8). Much evidence now exists supporting the notion that HsMetAP2 plays an important role in endothelial cell proliferation and is likely to mediate inhibition of endothelial cells by fumagillin and related analogs (5, 6, 9).
In contrast to HsMetAP2, little is known about the physiological function of human MetAP1, although genetic studies in yeast have suggested a more dominant role for ScMetAP1, as evidenced by the more severe growth defect observed in ScMetAP1 knockout strain than that in ScMetAP2 knockout strain (10). There has also been circumstantial evidence implicating a role of HsMetAP1 in tumor cell proliferation. By using a proteomics-based approach, both human MetAPs were identified as the binding targets of bengamides, a class of marine natural products that inhibit tumor growth in vitro and in vivo (11). Recently, pyridinyl pyrimidines have also been identified as nonselective inhibitors for MetAPs, and inhibit the proliferation of tumor cell lines (12). Because most tumor cell lines are refractory to the fumagillin family of HsMetAP2 inhibitors due likely to the defects in p53 pathway, the antiproliferative effects of bengamides and pyridinyl pyrimidines could have arisen from inhibition of HsMetAP1. These studies suggested an important function of MetAP1 in human cell proliferation. Despite all of the previous studies, however, the physiological function of HsMetAP1 has remained largely unknown.
We have taken two parallel approaches, chemical and genetic, to assess the function of human MetAP1 in cell proliferation. In the chemical approach we attempted to identify small chemical compounds that selectively inhibit the enzymatic activity of HsMetAP1 over HsMetAP2. These compounds would then be used to assess the consequence of inhibition of HsMetAP1 on cell proliferation. Pyridine-2-carboxylic acid–amide derivatives, including compound 1, were previously reported to inhibit both the bacterial and yeast MetAP1 (13, 14). We found this class of compounds has >100-fold selectivity for HsMetAP1 over HsMetAP2. Determination of the x-ray crystal structure of HsMetAP1 in complex with 1 helped to confirm the interaction between 1 and the enzyme, in addition to revealing a metal-dependent inhibitory mechanism. These compounds inhibited HsMetAP1 in cells and blocked proliferation of tumor cell lines. Unlike fumagillin, which arrests cell cycle at G1/S phase, compound 1 caused a significant cell cycle delay during G2/M phase. For the genetic approach, gene-specific silencing of HsMetAP1 also led to a delay in G2/M phase cell cycle progression, corroborating the observations with the specific chemical inhibitors of HsMetAP1. Together, these findings suggested a pivotal role of HsMetAP1 in cell division, which may be exploited in the development of anticancer agents.
Results
HsMetAP1-Specific Inhibitors Block Proliferation of Tumor Cell Lines.
It has been reported that pyridine-2-carboxylic acid–amide derivatives are inhibitory for Escherichia coli MetAP (EcMetAP) and ScMetAP1 (13). Analogs from the same class were synthesized and tested against both human MetAP1 and MetAP2 (12) in the presence of cobalt (II) ions. As shown in Table 1, lead compounds were able to potently inhibit human MetAP1 enzymatic activity with IC50 values in the low micromolar range. However, none of them inhibited HsMetAP2 activity up to their solubility limits (300–1,000 μM). Because manganese (II) has been suggested as the physiological metal ion for HsMetAP2 (15), we also tested these analogs in the presence of manganese ion. HsMetAP2 remained unaffected by the highest concentrations of each compounds in the presence of manganese ion (data not shown). Taken together, these results suggested that pyridine-2-carboxylic acid–amide derivatives are highly specific for HsMetAP1, rendering them useful molecular probes to elucidate the cellular function of HsMetAP1.
Table 1.
Structure of pyridine-2-carboxylic acid inhibitors and their inhibition of HsMetAP1, HsMetAP2, and cell proliferations
| Compound |  |
IC50, μM |
Ratio |
IC50, μM |
||
|---|---|---|---|---|---|---|
| R | hMetAP1 | hMetAP2 | MetAP1/MetAP2 | HeLa | HT1080 | |
| 1 | 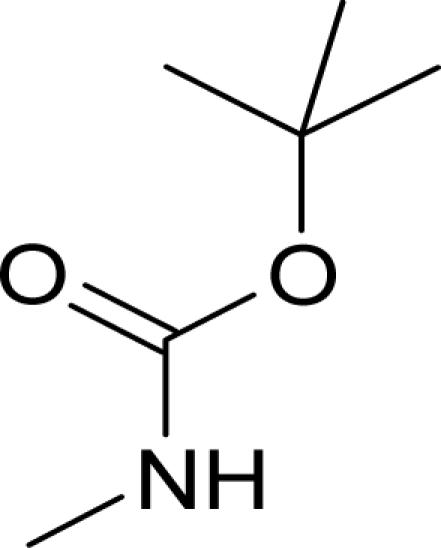 |
1.5 ± 0.2 | >300 | <0.005 | 0.6 ± 0.1 | 0.3 ± 0.1 |
| 2 | 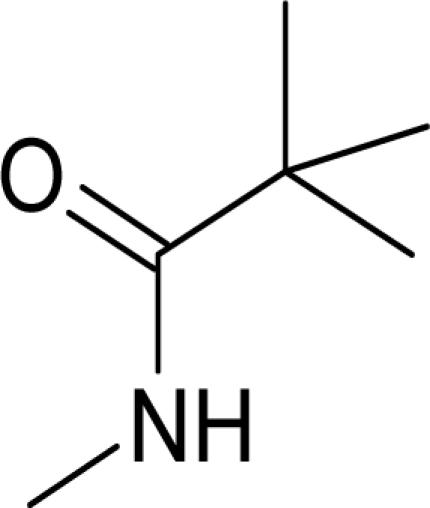 |
2.9 ± 0.1 | >500 | <0.006 | 3.9 ± 0.2 | 2.5 ± 0.3 |
| 3 | 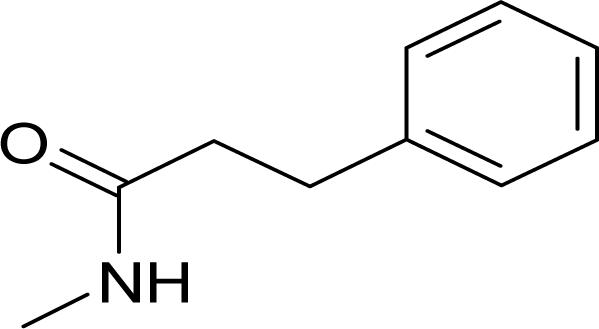 |
3.5 ± 0.2 | >500 | <0.007 | 5.1 ± 0.3 | 4.3 ± 0.1 |
| 4 | 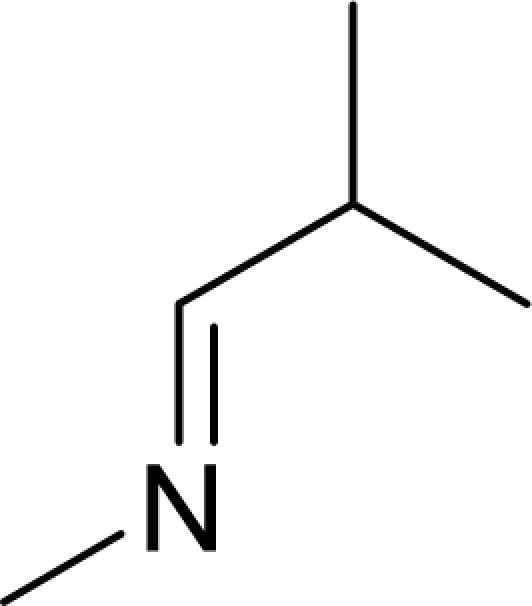 |
6.4 ± 0.6 | >1,000 | <0.006 | 8.9 ± 0.5 | 9.5 ± 0.8 |
| 5 |  |
>500 | >500 | N.A. | >50 | >50 |
Results from cobalt (II) supplied enzymatic assay are shown. Each experiment was conducted in triplicate. N.A., not applicable.
We next determined the effects of HsMetAP1 inhibitors on cell proliferation using a [3H]thymidine incorporation assay (12). Both HeLa and HT-1080 cells were inhibited with IC50 values in the low micromolar range. There is a correlation between cellular inhibitory effects and HsMetAP1 inhibition. It is noteworthy that compound 5, which is inactive against both HsMetAP1 and HsMetAP2, also has no effect on the proliferations of either cell line.
Pyridine-2-Carboxylic Acids Inhibit MetAP1 Inside Cells.
Although we have shown that pyridine-2-carboxylic acid derivatives selectively inhibit HsMetAP1 in vitro and block cell proliferation in culture, the causative relationship between these two effects remained to be established. As the first step to assess this relationship, we determined whether pyridine-2-carboxylic acid derivatives are capable of entering cells and inhibiting HsMetAP1 activity in vivo by examining the N-terminal initiator methionine status of a known protein substrate, 14-3-3γ (11). HeLa cells were incubated with various concentrations of compound 1 for 24 h before they were harvested for Western blot with a monoclonal antibody (clone HS23) specific for the methionylated N-terminal fragment of 14-3-3γ protein (11). As shown in Fig. 1, treatment with 1 resulted in a dose-dependent increase in the amounts of N-terminal methionine-containing 14-3-3γ protein, compared with vehicle control, suggesting that 1 is capable of inhibiting HsMetAP1 activity inside cells.
Fig. 1.

Inhibition of MetAP by compound 1. Shown is SDS/PAGE Western blot analysis of HeLa cells exposed to compound 1 at the indicated concentrations for 24 h. The membrane was probed with a monoclonal antibody specific for the methionylated 14-3-3γ (Upper) and tubulin (Lower).
If the inhibition of cell proliferation by the pyridine-2-carboxylic acid derivatives is due to the inhibition of HsMetAP1, it is expected that overexpression of HsMetAP1 should lead to a gain of resistance to the inhibitors. HeLa cells were thus transfected with expression vectors for HsMetAP1, or HsMetAP2 as well as an empty vector as control. Overexpression of HsMetAPs was confirmed by Western blots (Fig. 6, which is published as supporting information on the PNAS web site). The growth of HeLa cells was not affected by different transfections. However, cells overexpressing HsMetAP1 (≈6-fold of control determined by Western blot) showed an ≈5-fold decrease in the potency for compound 1 in comparison to cells transfected with the vector (Table 2). In contrast, HeLa cells overexpressing HsMetAP2 remained as sensitive to 1 as control cells, suggesting that HsMetAP1 plays a unique role in HeLa cell proliferation that could not be compensated for by HsMetAP2. When the same cells were treated with paclitaxel known to target tubulin, which is mechanistically unrelated to MetAP inhibitors, all three cell populations exhibited similar sensitivity (Table 2), further supporting the notion that compound 1 inhibits cell growth by inhibiting the cellular MetAP1 enzyme.
Table 2.
Cells overexpressing targeted HsMetAP1 resisted compound 1 in cell proliferation assay
| Compound | HeLa proliferation IC50, μM |
||
|---|---|---|---|
| Vector | MetAP1 overexpression | MetAP2 overexpression | |
| Paclitaxel | 0.0023 ± 0.0003 | 0.002 ± 0.0003 | 0.0022 ± 0.0003 |
| 1 | 0.6 ± 0.1 | 2.9 ± 0.4 | 0.7 ± 0.2 |
Each experiment was conducted in triplicate.
Pyridine-2-Carboxylic Acids Inhibit Cell Growth by Delaying the Cell Cycle Progression Through G2/M Phase.
To understand the mechanism of cell proliferation inhibition by HsMetAP1 inhibitors, we examined their effects on cell cycle progression using flow cytometry (16). Unsynchronized HeLa cells treated with vehicle control showed canonic distribution in G1, S, and G2/M phases. However, treatment with compound 1 led to a significant increase in cell populations at the G2/M phase (Fig. 2A). When HeLa cells were synchronized by double thymidine at G1/S check point and released in the presence or absence of compound 1, respectively, no difference was observed at the 6-h time point for these two populations of cells (Fig. 2B). However, 9–12 h after resumption of cell cycle, HeLa cells treated with 1 exhibited a significantly slower progression through G2/M phase, even though cells treated with 1 eventually were able to complete mitosis at 16 h after thymidine release (data not shown).
Fig. 2.

Function of HsMetAP1 is required for accurate cell cycle progression through G2/M phase. (A) FACS cell cycle analysis for unsynchronized HeLa cells treated with 1 for 24 h. (B) Synchronized HeLa cells showed delayed G2/M progression in the presence of compound 1. Double thymidine synchronized cells were released for 6, 9, and 12 h, respectively, before being collected for FACS analysis. (C) Western blot analysis from HeLa cells treated with respective siRNA duplexes for 48 h. Blots were sequentially probed with anti-HsMetAP1, HsMetAP2, and 14-3-3γ proteins; β-actin is the gel-loading control. (D) HsMetAP1 siRNA duplexes delayed cell cycle progression during G2/M phase. Synchronized HeLa cells were harvested for FACS analysis at different time points from double thymidine release.
In addition to 1, we also tested compounds 2 and 5, the active and inactive inhibitors of HsMetAP1, respectively. Compound 2 is capable of inhibiting HsMetAP1 in HeLa cells as evidenced by the increase in methionylated 14-3-3γ protein, whereas 5 showed no effect relative to the vehicle control (Fig. 7, which is published as supporting information on the PNAS web site). When synchronized HeLa cells were released from G1/S check point upon double thymidine blockade, cells treated with 2 advanced through G2/M phases more slowly than those treated with vehicle control. Nonetheless, cells treated with the inactive analog 5 showed no difference from control cells (Fig. 7), indicating that the G2/M phase delay observed with compounds 1 and 2 was due to inhibition of HsMetAP1.
HsMetAP1 Is Required for Timely Cell Cycle Progression Through G2/M Phase.
The G2/M delay caused by the active inhibitors 1 and 2 suggested that HsMetAP1 may be essential for timely progression through this phase of the cell cycle. To further verify this possibility, we took a complementary approach using selective siRNA duplexes to down-regulate the expression of HsMetAP1 and determined its consequence on cell cycle progression. HeLa cells were transfected with 100 nM siRNA duplexes for either HsMetAP1 or HsMetAP2, and the cellular protein levels were determined 48 h after transfection by Western blot analysis. As shown in Fig. 2C, HsMetAP1- and HsMetAP2-specific siRNA duplexes were able to down-regulate targeted proteins significantly in comparison with scrambled-duplex control without interfering with the expression of nontargeted MetAP or β-actin. In agreement with the down-regulation of either HsMetAP1 or HsMetAP2 by their respective siRNAs, corresponding increases in methionylated 14-3-3γ proteins were observed, as expected (Fig. 2C).
We next used these siRNA duplexes to transfect HeLa cells 6 h before the initiation of double thymidine synchronization of cell cycle. Cells were harvested at 0, 4, 8, and 12 h after the second thymidine release, followed by cell cycle analysis with FACS. As shown in Fig. 2D, all cells were synchronized on G1/S checkpoint at 0 h and launched their genome replication at the 4-h time point with comparable speed. However, HsMetAP1 siRNA-treated cells showed significant delay in progression through G2/M phase at 8 h, in comparison with cells transfected with scrambled or HsMetAP2 siRNA duplexes (Fig. 2D). Eventually, all of the cells were able to complete mitosis for a new cycle of cell proliferation at the 12-h time point. Similar effects were observed in HT1080 cells during their G2/M phase progression (data not shown). Thus, HsMetAP1 is required for the precise progression through G2/M phase.
HsMetAP1-Specific Inhibition Delayed Cyclin B Protein Degradation During Mitosis.
Proper cell cycle progression is regulated by different cyclin proteins and cyclin-dependent kinases (CDKs). Cdk1/cyclin B is the universal cell cycle regulator implicated in the G2/M phase transition. Exit from mitosis involves inactivation of CDK kinase through cyclin B degradation (17). To understand the molecular mechanism of HsMetAP1-specific inhibition, we examined the protein levels of cyclin B1 and cdc2/Cdk1 kinase during mitosis. When synchronized HeLa cells were released from thymidine arrest, cyclin B1 decreased dramatically between the 8-h and 10-h time points for vehicle control cells. However, cells treated with 1 still have significant amount of cyclin B1 protein at 10-h time point (Fig. 3A). Cyclin B1 protein level eventually dropped at the 12-h time point (data not shown). Interestingly, cdc2/Cdk1 protein level did not change under compound 1 treatment (Fig. 3A).
Fig. 3.

Inhibition of HsMetAP1 resulted in delayed degradation of cyclin B protein. (A) A significant delay of cyclin B1 degradation in the presence of 1 is shown by Western blot. Cell lysates were harvested at different time points from double thymidine release. (B) RT-PCR analysis of the cyclin B1 mRNA level was carried out from total RNA isolated at different time points with appropriate primers specific for cyclin B1 and β-actin.
Cyclin B1 protein expression is regulated at both transcriptional and posttranscriptional levels. To dissect the regulatory mechanisms of cyclin B1 protein by HsMetAP1 inhibition, we determined the mRNA level of cyclin B1 by RT-PCR. As shown in Fig. 3B, cyclin B1 mRNA decreased during mitosis. Neither compound 1, the HsMetAP1-specific inhibitor, nor TNP-470, the HsMetAP2-specific inhibitor, altered the decrease of cyclin B1 mRNA level between 8 and 10 h. These results indicated that delayed degradation of cyclin B1 protein is likely to be regulated at the posttranscriptional level.
HsMetAP1-Specific Inhibition Induces Cellular Apoptosis.
The G2/M phase transition is critical for proper cell division, and a G2/M checkpoint disruption has been shown to cause apoptosis in a number of tumor cell lines (18). To test whether HsMetAP1-specific inhibition could cause apoptosis, we determined their effect on Jurkat T cells. As shown in Fig. 4A, treatment of Jurkat T cells with compound 1 resulted in fragmentation of nucleosomal DNA, judged by DNA ladder pattern, the hallmark of apoptosis. In addition to DNA laddering, we also examined the proteolytic cleavages of poly(ADP-ribose) polymerase (PARP) and pro-caspase-3. Treatment with compound 1 for 24 h resulted in a dosage-dependent cleavage of PARP protein. Concurrently, the full-length pro-caspase-3 decreased in a similar dose-dependent fashion, complemented by the appearance of the active fragment of caspase-3 (Fig. 4B). Similar results have been observed in a B cell non-Hodgkin's lymphoma cell line (Karpas 1106) (data not shown). Thus, by causing a delay in G2/M phase, inhibitors of HsMetAP1 are capable of inducing apoptosis among cancer cells, suggesting that HsMetAP1 may be a useful target for anticancer agents.
Fig. 4.

HsMetAP1 inhibition induces cellular apoptosis. (A) Ethidium bromide-stained genomic DNA isolated from Jurkat T cells in the absence (−) and presence (+) of 10 μM compound 1, respectively, after 16 h of treatment. M, marker. (B) Western blot analysis of protein lysate isolated from vehicle- and compound 1-treated Jurkat T cells for 24 h. Blots were sequentially probed with antibodies specific to PARP, procaspase-3, and active caspase-3. An arrow indicates the release of the 89-kDa fragment from PARP protein. β-Actin is the gel-loading control.
Metal-Mediated Interaction Between the Inhibitors and the HsMetAP1.
Crystal structures of compounds 1 and 2 in complex with the (Δ1–89) truncated human MetAP1 were determined at 1.5- and 1.6-Å resolution, respectively (Fig. 5A). In addition to the presence of two active site CoII ions, a third CoII ion was observed that mediates the interaction between His-212 of the enzyme and the basic pyridine carboxylic acid scaffold of the inhibitors. N1 and N8 of the inhibitors and Nε2 of the His-212 form a part of the octahedral coordination around the third cobalt ion with the other three sites filled by water molecules. The thiazole ring of the inhibitor is buried deep in the active site pocket surrounded by several hydrophobic residues (Pro-192, Tyr-195, Phe-198, and Phe-309) in addition to Cys-203 (Fig. 5B). N13 of the thiazole ring points in the direction of the third metal center and forms a hydrogen bond with one of the water molecules from its coordination sphere. This hydrogen bond between the N13 and the water molecule locks the only free rotatable dihedral angle (C12–N13–C14–S15) into a cis conformation. The other two water molecules from the third metal coordination center interact with the active site bridging water/μ hydroxo anion. In addition, each of these two water molecules forms hydrogen bonds with active site residues (Nε2 of His-310 and Oε2 of Glu-336). Based on modeling (data not shown), the sulfur atom of the substrate methionine and the sulfur atom of the inhibitor are placed within 1.2 Å, with the latter farther from catalytic center. The side chain containing the tert-butoxycarbonylamino group in 1 is in plane with the pyridine ring. Because there is a slight bend in the main scaffold in compound 1, the side chain is offset from the position of the tert-butylcarbonylamino group in 2 (Fig. 5B). The tert-butyl group in 2 is pointed into the hydrophobic depression on the surface of the protein formed by Tyr-195, Tyr-196, and Trp-353. In compound 1 the tert-butyl group points away from this depression and has only one methyl group pointed toward Tyr-196 with other two solvent-exposed. The tert-butyl groups in both the molecules are separated by 2.3 Å from each other. There is no difference in the structure of the protein molecules between the two complexes within the error of the models except for a slight movement of the Tyr-196 (O… O distance 0.4 Å).
Fig. 5.

Crystal structure of truncated HsMetAP1 in complex with 1 and 2. (A) Superposed is the “omit” electron density map shown in the inhibitor binding region of compounds 1 and 2. Coefficients are (Fo − Fc), where the Fo are the observed structure amplitudes. The calculated amplitudes Fc and phases are obtained from the refined model with the inhibitors removed. The maps calculated are at 1.5 Å (contoured at 3.6 σ) for 1 and at 1.6 Å (contoured at 3.6 σ) for 2. (B) Stereo diagram showing the superposition of enzyme-inhibitor complexes of compounds 1 (green) and 2 (magenta) in the active site pocket of the truncated HsMetAP1 (cyano). Note that both the compounds use a third metal ion (CoII) in binding to the protein. Except for the contact through the metal ion, there are no obvious hydrogen bond contacts between the protein and the inhibitors, although they share several hydrophobic interactions. (C) Stereo diagram of the superposed structures of HsMetAP1 in complex with compounds 1 and 2 and HsMetAP2 (silver). Note that Tyr-444 of the latter enzyme experiences a severe steric clash with the side chains of compounds 1 and 2, explaining the lower affinity of these compounds for HsMetAP2.
Discussion
Small molecules have played an important part in the elucidation of functions of genes. Early examples include the use of the immunosuppressant drugs CyclosporinA and FK506 to reveal the important role of the protein phosphatase calcineurin mediating intracellular calcium signaling pathway (19). The application of another immunosuppressive natural product, rapamycin, has shed significant light on the TOR kinases in myriad signaling processes from cytokine signaling to nutrient sensing (20). Similarly, the identification of MetAP2 as the cellular target for the fumagillin family of antiangiogenic natural products has helped to reveal a unique function of this otherwise universally expressed general-processing enzyme in endothelial cells (5, 6) and T cells (21). However, in contrast to MetAP2, study of the cellular functions of MetAP1 has been hampered in part by the lack of an inhibitor with sufficient selectivity.
Following the identification of HsMetAP2 as a target of fumagillin and ovalicin, the MetAP family of enzymes has been under increasing scrutiny as targets for developing antibacterial, antifungal, and anticancer drugs. Among the MetAP inhibitors reported, the pyridine-2-carboxylic acid thiazole-2-ylamide class has emerged as potent inhibitors of both E. coli and yeast MetAP1. Moreover, members of this class of compounds have been subsequently shown to inhibit recombinant HsMetAP1 (14, 22). It remained unknown, however, whether this family of MetAP1 inhibitors also cross-interact with MetAP2. We synthesized several analogs of this class of inhibitors and determined their specificity for the two isoforms of HsMetAPs. Gratifyingly, all compounds synthesized (1–4) exhibited exquisite specificity for HsMetAP1, with ratios in IC50 values for the two enzymes >200-fold, rendering these inhibitors useful probes for the cellular function of HsMetAP1 without the complication of cross-inhibition on HsMetAP2 at relatively low concentrations.
The effects of HsMetAP1 inhibitors on G2/M phase transition appear to be significant, yet different from those seen with other cytotoxic anticancer drugs such as paclitaxel or colchicine that also inhibit cell cycle at the G2/M phase. Rather than a sustained blockade of cells through the G2/M phase, compound 1 and its analogs caused a 3- to 4-h delay of cell cycle progression through the G2/M phase and allowed cells to eventually reach G1 phase to resume another round of cell cycle. Although this delay, not blockade, in G2/M phase by HsMetAP1 inhibitors is plausibly overcome by some cancer cell lines, it led to apoptosis of both Jurkat and Karpas 1106 lymphoma cell lines. It is possible that, in both Jurkat and Karpas 1106 lymphoma cell lines, the delay in G2/M phase progression by HsMetAP1 inhibitors is not compatible with the preexisting disruption of the G2/M checkpoint control and causes cells to undergo apoptosis (23). As such, HsMetAP1 inhibitors represent a novel mechanistic class of G2/M phase inhibitors that may be useful for treatment of cancer.
Timely degradation of cyclin B protein is critical for exit from mitosis during cell cycle progression (24). Our results suggested that inhibition of HsMetAP1 regulates cyclin B protein level through a posttranscriptional mechanism. The penultimate residue for cyclin B protein is alanine, which qualifies cyclin B as a substrate for MetAP; however, it remains unclear whether N-terminal methionine retention would directly account for the delayed degradation of cyclin B protein. It is equally reasonable that the delayed-degradation effect on cyclin B protein is indirect, as a consequence of the N-terminal methionine retention of another protein. Our further experiments have demonstrated that the lead compound is capable of promoting cellular apoptosis by the activation of caspase-3 and cleavage of PARP protein. We therefore propose that inhibition of HsMetAP1 slows down cell cycle progression, activates the G2/M checkpoint, and eventually leads to apoptosis.
The high-resolution crystal structures of the complexes between HsMetAP1 and compounds 1 and 2 threw significant new light on the highly specific molecular interaction between the enzyme and the inhibitors. Compounds 1 and 2 differ by only one oxygen atom. The orientation of the tert-butyl group is different in both the compounds with respect to the rest of the molecule. In 2 the tert-butyl group points in a direction close to the scaffold, which we refer it as a syn conformation, whereas in 1 it points away in a trans conformation (Fig. 5A). This directionality of the tert-butyl groups seems to play an important role in the selectivity as suggested by the biochemical data, both from our present study and from the results of Luo et al. (13). Structural data suggest that the overall direction of the tert-butyl group is away from the protein surface in the ester-based side chain of 1 whereas it points into the hydrophobic depression on the protein surface in the ketone-based compound 2. Compound 3 also follows the similar trend as 2 although it has slightly lower affinity. It is possible that the aromatic side chain of compound 3 forms π–π stacking interactions with either or both aromatic rings of Tyr-195 and Tyr-196. The double bond in the side chain of 4 probably imposes extra rigidity and limits the freedom of orientation that is necessary for interaction with protein side chains, thus explaining the lower affinity. In addition, it is also possible that the two methyl groups in 4 may play a role in the lowering of affinity. The compounds described here have 100- to 200-fold lower affinity toward the type 2 human enzyme compared with that with type 1 enzyme (Table 1). Structure alignment of complexes of 1 and 2 HsMetAP1 and the holo form of HsMetAP2 (Protein Data Bank ID code 1BN5) is demonstrated in Fig. 5C. The side chain of compound 2, which is close to the surface of the protein in the HsMetAP1 complex, seems to have a more severe steric clash with the side chain of Tyr-444 of the HsMetAP2 compared with that of the compound 1. Such steric interactions provide the clue to the difference in affinity of 1 and 2 toward HsMetAP2. A similar observation was made for pyridinylpyrimidine compounds described in our previous study (12). Our in vitro enzymatic assay suggested that pyridine-2-carboxylic acid derivatives are more selective inhibitors than pyridinylpyrimidines.
Sequence alignment of type 1 MetAPs suggests that Tyr-195 and Trp-353, two of the three hydrophobic residues that form the surface depression, are ≈80% and ≈98% conserved, respectively. However, Tyr-196 is only ≈20% conserved and is replaced with a variety of amino acids with histidine next in frequency (≈18%). Note that Tyr-196 is the only common residue that is in direct contact with side chains of 1 and 2. Together these data suggest that one can design an organism-specific type 1 MetAP inhibitor that does not inhibit the human type 2 MetAP. Together, the structural information on the two enzyme-inhibitor complexes should facilitate the design of more potent inhibitors of HsMetAP1.
Processing of the initiator methionine residue is an evolutionarily conserved prerequisite step for different posttranslational protein modifications, such as N-terminal acetylation (25), protein subcellular localizations (26), and protein half-lives (27). Identification of HsMetAP1-specific inhibitors has shed light on the functions of type I MetAP in mammalian cells. These small molecules could also serve as lead compounds for the development of therapeutic reagents. Future applications of proteomics technology should facilitate the identifications of downstream substrates for either HsMetAP1 or HsMetAP2. Such information will greatly assist in elucidating the relationship between removal of the initiating methionine and cell proliferations.
Materials and Methods
Materials.
[3H]Thymidine was obtained from PerkinElmer (Wellesley, MA). MetAP1 polyclonal antibodies were a generous gift from Y.-H. Chang (Saint Louis University School of Medicine, St. Louis, MO). A MetAP2 monoclonal antibody was generated with the help of J. E. K. Hildreth (Department of Pharmacology, Johns Hopkins University School of Medicine). 14-3-3γ monoclonal antibody (clone HS23) was obtained from Novus Biologicals (Littleton, CO). PARP and active caspase-3 monoclonal antibodies were obtained from BD Bioscience (San Diego, CA). Tubulin, Cyclin B1, cdc2/Cdk1 monoclonal antibodies, and pro-caspase-3 antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). DMSO, paclitaxel, thymidine, propidium iodide, DNase-free RNase, and β-actin monoclonal antibody were obtained from Sigma–Aldrich (St. Louis, MO). SuperFect reagents were obtained from Qiagen (Valencia, CA). Oligofectamine and TRIzol reagent were purchased from Invitrogen (Carlsbad, CA).
MetAP Enzyme Assay.
Full-length and truncated HsMetAP1 were generated as previously described (28). Recombinant HsMetAP2 was produced according to Turk et al. (29). MetAP enzymatic assay was carried out as described previously (30).
Double Thymidine Synchronization.
Cultured HeLa and HT-1080 cells were synchronized according to Hirota et al. (31). Briefly, 1.5 × 105 cells were seeded in a six-well plate and treated with 2 mM thymidine for 20 h before release with fresh medium for 8 h. Thymidine (2 mM) was then added as the second arrest for 14 h before release by fresh medium with respective compounds.
Cell Cycle Analysis.
Cultured cells were trypsinized and fixed with 70% ethanol at 4°C overnight before being stained with propidium iodide by using Staining solution [20 μg/ml propidium iodide, 200 μg/ml DNase-free RNase A, and 0.1% (vol/vol) Triton X-100 in PBS] prepared freshly. DNA contents were analyzed by using the FACScan (Becton Dickinson, San Jose, CA) as described previously (16). Data were analyzed by CellQuest software (Becton Dickinson).
siRNA Transfection.
siRNAs duplexes were obtained from Dharmacon (Lafayette, CO). The following siRNA targeting (sense) sequences were selected: MetAP1 siRNA, 5′-GGCCAGUGCCAAGUUAUAU-dTdT-3′, corresponding to bases 317–336 in the ORF of the MetAP1 mRNA. MetAP2 and scrambled control siRNA duplexes were adopted from Bernier et al. (32). MetAP2 siRNA, 5′-GAAGAGAUUUGGAAUGAUU-dTdT-3′, corresponding to bases 521–540 in the ORF of the MetAP2 mRNA. The scrambled control siRNA duplex sequence was 5′-AUUAGACUCUUCAUGGAAA-dTdT-3′. A total of 1.5 × 105 HeLa cells were seeded into six-well plates before transfection by Oligofectamine (Invitrogen) according to the manufacturer's instructions for 6 h. The final siRNA concentration was 100 nM. Double thymidine synchronization was then initiated.
Other Methods.
Details regarding the synthesis of pyridine-2-carboxylic acid–amide compounds, cell culture, cell proliferation assay, RT-PCR, and protein expression and crystallization are provided in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
We are grateful for reagents and help from Dr. Y.-H. Chang and Dr. J. E. K. Hildreth. We also thank all the members of the J.O.L. laboratory for their help. This work was supported by the National Institutes of Health, the Keck Center (J.O.L.), and Howard Hughes Medical Institute (B.W.M.). X.H. is a Predoctoral Fellow with the Department of Defense Breast Cancer Program.
Abbreviations
- MetAP
methionine aminopeptidase
- CDK
cyclin-dependent kinase
- PARP
poly(ADP-ribose) polymerase.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2NQ6 and 2NQ7).
References
- 1.Chang YH, Teichert U, Smith JA. J Biol Chem. 1992;267:8007–8011. [PubMed] [Google Scholar]
- 2.Li X, Chang YH. Proc Natl Acad Sci USA. 1995;92:12357–12361. doi: 10.1073/pnas.92.26.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boxem M, Tsai CW, Zhang Y, Saito RM, Liu JO. FEBS Lett. 2004;576:245–250. doi: 10.1016/j.febslet.2004.08.077. [DOI] [PubMed] [Google Scholar]
- 4.Cutforth T, Gaul U. Mech Dev. 1999;82:23–28. doi: 10.1016/s0925-4773(99)00006-4. [DOI] [PubMed] [Google Scholar]
- 5.Griffith EC, Su Z, Turk BE, Chen S, Chang YH, Wu Z, Biemann K, Liu JO. Chem Biol. 1997;4:461–471. doi: 10.1016/s1074-5521(97)90198-8. [DOI] [PubMed] [Google Scholar]
- 6.Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. Proc Natl Acad Sci USA. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ingber D, Fujita T, Kishimoto S, Sudo K, Kanamaru T, Brem H, Folkman J. Nature. 1990;348:555–557. doi: 10.1038/348555a0. [DOI] [PubMed] [Google Scholar]
- 8.Satchi-Fainaro R, Mamluk R, Wang L, Short SM, Nagy JA, Feng D, Dvorak AM, Dvorak HF, Puder M, Mukhopadhyay D, et al. Cancer Cell. 2005;7:251–261. doi: 10.1016/j.ccr.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 9.Yeh JR, Ju R, Brdlik CM, Zhang W, Zhang Y, Matyskiela ME, Shotwell JD, Crews CM. Proc Natl Acad Sci USA. 2006;103:10379–10384. doi: 10.1073/pnas.0511313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen S, Vetro JA, Chang YH. Arch Biochem Biophys. 2002;398:87–93. doi: 10.1006/abbi.2001.2675. [DOI] [PubMed] [Google Scholar]
- 11.Towbin H, Bair KW, DeCaprio JA, Eck MJ, Kim S, Kinder FR, Morollo A, Mueller DR, Schindler P, Song HK, et al. J Biol Chem. 2003;278:52964–52971. doi: 10.1074/jbc.M309039200. [DOI] [PubMed] [Google Scholar]
- 12.Hu X, Addlagatta A, Matthews BW, Liu JO. Angew Chem Int Ed Engl. 2006;45:3772–3775. doi: 10.1002/anie.200600757. [DOI] [PubMed] [Google Scholar]
- 13.Luo QL, Li JY, Liu ZY, Chen LL, Li J, Qian Z, Shen Q, Li Y, Lushington GH, Ye QZ, et al. J Med Chem. 2003;46:2631–2640. doi: 10.1021/jm0300532. [DOI] [PubMed] [Google Scholar]
- 14.Li JY, Chen LL, Cui YM, Luo QL, Gu M, Nan FJ, Ye QZ. Biochemistry. 2004;43:7892–7898. doi: 10.1021/bi0360859. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Sheppard GS, Lou P, Kawai M, Park C, Egan DA, Schneider A, Bouska J, Lesniewski R, Henkin J. Biochemistry. 2003;42:5035–5042. doi: 10.1021/bi020670c. [DOI] [PubMed] [Google Scholar]
- 16.Kanzawa T, Germano IM, Kondo Y, Ito H, Kyo S, Kondo S. Br J Cancer. 2003;89:922–929. doi: 10.1038/sj.bjc.6601193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murray AW. Cell. 2004;116:221–234. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 18.Rieder CL, Maiato H. Dev Cell. 2004;7:637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 20.Wullschleger S, Loewith R, Hall MN. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Turk BE, Su Z, Liu JO. Bioorg Med Chem. 1998;6:1163–1169. doi: 10.1016/s0968-0896(98)00078-9. [DOI] [PubMed] [Google Scholar]
- 22.Cui YM, Huang QQ, Xu J, Chen LL, Li JY, Ye QZ, Li J, Nan FJ. Bioorg Med Chem Lett. 2005;15:4130–4135. doi: 10.1016/j.bmcl.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Taylor WR, Stark GR. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 24.Page AM, Hieter P. Annu Rev Biochem. 1999;68:583–609. doi: 10.1146/annurev.biochem.68.1.583. [DOI] [PubMed] [Google Scholar]
- 25.Polevoda B, Sherman F. J Biol Chem. 2000;275:36479–36482. doi: 10.1074/jbc.R000023200. [DOI] [PubMed] [Google Scholar]
- 26.Boutin JA. Cell Signal. 1997;9:15–35. doi: 10.1016/s0898-6568(96)00100-3. [DOI] [PubMed] [Google Scholar]
- 27.Varshavsky A. Nat Cell Biol. 2003;5:373–376. doi: 10.1038/ncb0503-373. [DOI] [PubMed] [Google Scholar]
- 28.Addlagatta A, Hu X, Liu JO, Matthews BW. Biochemistry. 2005;44:14741–14749. doi: 10.1021/bi051691k. [DOI] [PubMed] [Google Scholar]
- 29.Turk BE, Griffith EC, Wolf S, Biemann K, Chang YH, Liu JO. Chem Biol. 1999;6:823–833. doi: 10.1016/s1074-5521(99)80129-x. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Y, Guo XC, Yi T, Yoshimoto T, Pei D. Anal Biochem. 2000;280:159–165. doi: 10.1006/abio.2000.4513. [DOI] [PubMed] [Google Scholar]
- 31.Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, Hatakeyama K, Saya H. Cell. 2003;114:585–598. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- 32.Bernier SG, Taghizadeh N, Thompson CD, Westlin WF, Hannig G. J Cell Biochem. 2005;95:1191–1203. doi: 10.1002/jcb.20493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
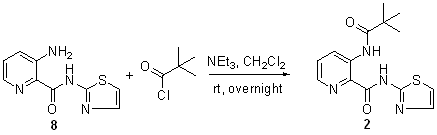{kind=link}
{kind=link}
{kind=link}
{kind=link}