

Abstract
Diverse forms of injury and stress evoke a hypertrophic growth response in adult cardiac myocytes, which is characterized by an increase in cell size, enhanced protein synthesis, assembly of sarcomeres, and reactivation of fetal genes, often culminating in heart failure and sudden death. Given the emerging roles of microRNAs (miRNAs) in modulation of cellular phenotypes, we searched for miRNAs that were regulated during cardiac hypertrophy and heart failure. We describe >12 miRNAs that are up- or down-regulated in cardiac tissue from mice in response to transverse aortic constriction or expression of activated calcineurin, stimuli that induce pathological cardiac remodeling. Many of these miRNAs were similarly regulated in failing human hearts. Forced overexpression of stress-inducible miRNAs was sufficient to induce hypertrophy in cultured cardiomyocytes. Similarly, cardiac overexpression of miR-195, which was up-regulated during cardiac hypertrophy, resulted in pathological cardiac growth and heart failure in transgenic mice. These findings reveal an important role for specific miRNAs in the control of hypertrophic growth and chamber remodeling of the heart in response to pathological signaling and point to miRNAs as potential therapeutic targets in heart disease.
Keywords: calcineurin, myosin heavy chain, thoracic aortic banding, cardiomyocytes
The adult heart is a dynamic organ capable of significant remodeling and hypertrophic growth as a means of adapting function to altered workloads or injury. Hemodynamic stress or neuroendocrine signaling associated with myocardial infarction, hypertension, aortic stenosis, and valvular dysfunction evoke a pathologic remodeling response through the activation of intracellular signaling pathways and transcriptional mediators in cardiac myocytes (reviewed in ref. 1). Activation of these molecular pathways enhances cardiomyocyte size and protein synthesis, induces the assembly of sarcomeres, and causes re-expression of fetal cardiac genes (2, 3). Although aspects of the hypertrophic response after acute and chronic stress may initially augment cardiac output, prolonged hypertrophy is a major predictor of heart failure and sudden death. There have been major advances in the identification of genes and signaling pathways involved in this disease process, but the overall complexity of hypertrophic remodeling suggests that additional regulatory mechanisms remain to be identified (4).
MicroRNAs (miRNAs) are small, noncoding RNAs of 18–25 nt that regulate gene expression in a sequence-specific manner. miRNAs are transcribed by RNA polymerase II as primary transcripts that are usually several thousand bases in length and are subsequently processed into smaller, functional RNA fragments. Primary transcripts are first processed into a 70- to 100-nt hairpin-shaped precursor within the nucleus by the action of the RNase Drosha, after which they are transported to the cytoplasm and cleaved by Dicer, releasing the mature double-stranded miRNA (reviewed in ref. 5). A single strand of the mature miRNA is incorporated into the RNA-induced silencing complex to enable its interaction with target mRNA sequences. Binding of mature miRNAs to mRNAs negatively influences the expression of specific proteins by either degradation of the bound mRNA target or direct translational inhibition. The high sequence conservation of many miRNAs across metazoan species suggests strong evolutionary pressure and participation in essential biologic processes (6, 7). Indeed, miRNAs have been shown to play fundamental roles in diverse biological and pathological processes, including cell proliferation, differentiation, apoptosis, and carcinogenesis in species ranging from Caenorhabditis elegans and Drosophila melanogaster to humans (8–11).
In light of their involvement in modulating cellular phenotypes, we hypothesized that miRNAs might play a role in regulating the response of the heart to cardiac stress, which is known to result in transcriptional and translational changes in gene expression. Here, we describe >12 miRNAs that are modulated in hypertrophic or failing hearts from mice and humans. Overexpression of selected miRNAs in cardiomyocytes in vitro induced cardiac hypertrophy and overexpression of miR-195, a stress inducible miRNA, in transgenic (Tg) mice resulted in pathological cardiac remodeling and heart failure. These findings point to miRNAs as key regulators of cardiac growth and function and potential therapeutic targets in the setting of heart disease.
Results
miRNA Expression During Cardiac Hypertrophy.
To investigate the potential involvement of miRNAs in cardiac hypertrophy, we performed a side-by-side miRNA microarray analysis in two established mouse models of cardiac hypertrophy, using a microarray that represented 186 different miRNAs (Fig. 6, which is published as supporting information on the PNAS web site). Mice that were subjected to thoracic aortic banding (TAB), which induces hypertrophy by increased afterload on the heart (12), were compared with sham-operated animals. In a second model, Tg mice expressing activated calcineurin A (CnA) in the heart, which results in a severe, well characterized form of hypertrophy (13), were compared with WT littermates (Fig. 1A). RNA isolated from hearts of mice subjected to TAB showed increased expression of 27 miRNAs compared with sham-operated controls, and CnA Tg mice showed increased expression of 33 miRNAs compared with non-Tg littermate controls, of which 21 were up-regulated in both models. Similarly, TAB- and CnA-induced hypertrophy were accompanied by reduced expression of 15 and 14 miRNAs, respectively, of which 7 miRNAs were down-regulated in common (Fig. 1B and Table 2, which is published as supporting information on the PNAS web site). Northern blot analysis of these miRNAs (our unpublished data) and previous microarray analyses (14–18) indicate that they are expressed in a wide range of tissues. Based on their relative expression levels (Fig. 7, which is published as supporting information on the PNAS web site), conservation of human, rat, and mouse sequences, and levels of expression during hypertrophy, we focused on 11 up-regulated and 5 down-regulated miRNAs (Fig. 1C).
Fig. 1.

miRNA expression during cardiac hypertrophy. (A) H&E-stained sections of representative hearts from mice after sham and TAB for 21 days (Upper) and from WT and CnA Tg mice (Lower). (Scale bars: 2 mm.) (B) Numbers of miRNAs that were regulated in response to CnA or TAB are indicated. Although some changes were unique for either TAB or CnA-induced hypertrophy, most miRNAs that were induced or repressed overlapped for the different hypertrophic stimuli. (C) Bar graph indicates the fold change in expression during both TAB- and CnA-induced hypertrophy compared with the baseline of the miRNAs of interest. (D) Northern blot analysis of miRNAs in WT and CnA Tg hearts. U6 RNA was detected as a loading control.
Northern blot analysis of cardiac RNA from WT and CnA Tg animals confirmed an increased expression of miR-21, miR-23, miR-24, miR-125b, miR-195, miR-199a, and miR-214, and decreased expression of miR-29c, miR-93, miR-150, and miR-181b (Fig. 1D and Table 3, which is published as supporting information on the PNAS web site). We were unable to detect a transcript for miR-217 by Northern blot analysis, and sequence identity between the a and b isoforms of miR-23 and miR-27, and miR-133a-1, miR-133a-2, and miR-133b prohibited us from specifically determining differences in their expression (Table 4, which is published as supporting information on the PNAS web site). Collectively, these data indicate that distinct miRNAs are regulated during cardiac hypertrophy, suggesting the possibility that they might function as modulators of this process.
miRNA Expression in Human Heart Failure.
Ventricular hypertrophy develops in response to numerous forms of cardiac stress and often leads to heart failure in humans (1). Northern blot analysis of the hypertrophy-regulated miRNAs in idiopathic end-stage failing human hearts showed increased expression of miR-24, miR-125b, miR-195, miR-199a, and miR-214, whereas the expression for miR-23 appeared to be variable within the nonfailing and failing groups (Fig. 2). We found no change in expression of miR-21, miR-27, miR-29c, miR-93, miR-150, and miR-181b (data not shown). Thus, the altered pattern of miRNA expression in the failing human heart overlapped that of the hypertrophic mouse heart, suggesting that these miRNAs represent a molecular signature of adverse cardiac remodeling.
Fig. 2.

miRNA expression in human heart failure. Northern blot analysis of miRNAs in four normal (Left) and six failing (Right) human hearts. The average fold change of each miRNA in the failing samples is shown.
Overexpression of miRNAs Evokes Morphological Changes in Cardiomyocytes.
To examine their effect on cardiomyocytes, we overexpressed single miRNAs by using adenoviruses created by inserting rat genomic fragments encoding the miRNAs of interest and 200–400 bp of 5′ and 3′ flanking sequence into an RNA polymerase II-driven adenoviral vector. Mature miRNAs are formed by nuclear cleavage of pri-miRNAs into a ≈70-nt pre-miRNA (the so-called stem-loop), followed by cytoplasmic processing into the 18- to 22-nt miRNA. Northern blot analysis in myocytes cultured in serum-free media or infected at a multiplicity of infection (MOI) of 5, 50, or 100 showed efficient overexpression of the mature miRNAs, with the exception of miR-21 (Fig. 3A). Intriguingly, the ratio between pre-miRNA and mature miRNA and the level of overexpression differed for all miRNAs tested. Because our expression strategy was comparable for all miRNAs and every virus generated a pre-miRNA, this finding suggests that the processing of pre-miRNAs into mature miRNAs may be miRNA-specific.
Fig. 3.

Overexpression of miRNAs in primary cardiomyocytes. (A) Primary rat cardiomyocytes in serum-free medium (SF) were infected with adenoviruses expressing individual miRNAs at the indicated MOI. miRNAs were detected by Northern blot analysis on 10 μg of RNA. Positions of the pre-miRNAs and mature miRNAs are indicated. (B) Actinin staining of cardiomyocytes shows that adenoviral overexpression of several miRNAs induces morphological changes characteristic of hypertrophy. Cells were infected with pAd β-gal as a negative control. As a positive control, cells were stimulated with phenylephrine (PE), a potent inducer of hypertrophy.
Actinin staining revealed that myocytes exposed to miRNA overexpression underwent dramatic morphological changes (Fig. 3B and Table 1). miR-23a, miR-23b, miR-24, miR-195, and miR-214, all of which were up-regulated during cardiac hypertrophy, appeared to be capable of inducing hypertrophic growth in vitro. Hypertrophy in response to these miRNAs was comparable to that evoked by the adrenergic agonist phenylephrine, among the most potent hypertrophic stimuli known. Overexpression of miR-199a resulted in an especially pronounced morphological response in which cardiomyocytes became elongated, reminiscent of a phenotype of eccentric cardiac hypertrophy that results from serial assembly of sarcomeres, associated with dilated cardiomyopathy. The morphological responses of cardiomyocytes to overexpression of these miRNAs do not represent a general response to miRNA overexpression because miR-150 and miR-181b, which were down-regulated during hypertrophy, caused a reduction in cardiomyocyte cell size. Overexpression of miR-125b and miR-133a had no effect on cardiomyocyte morphology (data not shown).
Table 1.
Morphological effects of miRNA overexpression on cardiomyocytes
| miRNA type | Phenotypic effect |
|---|---|
| Up-regulated | |
| miR-23a | Myocyte hypertrophy |
| miR-23b | Myocyte hypertrophy |
| miR-24 | Myocyte hypertrophy |
| miR-125b | No effect |
| miR-195 | Myocyte hypertrophy |
| miR-199a | Elongated, lined-up myocytes |
| miR-214 | Myocyte hypertrophy |
| Down-regulated | |
| miR-93 | Small clusters of cells |
| miR-133a | No effect |
| miR-150 | Small, rounded up myocytes |
| miR-181b | Small, aggregated myocytes |
Cardiac Overexpression of miR-195 Is Sufficient to Drive Cardiac Hypertrophy.
We overexpressed miR-24, miR-195, and miR-214 specifically in the heart under the control of the α-myosin heavy chain (MHC) promoter. We were unable to obtain F1 offspring for miR-24, suggesting that cardiac overexpression of this miRNA causes embryonic lethality. Variable survival rates were observed for different miR-195 Tg lines (Table 5, which is published as supporting information on the PNAS web site). Because all offspring of miR-195 Tg line 3 died in the first 2 weeks after birth because of heart failure (Fig. 4A), we focused on a Tg line (line 1) for miR-195, which was viable. Northern blot analysis showed miR-195 to be expressed at levels ≈25-fold above normal in Tg line 1 (Fig. 4B). Overexpression of miR-195 initally induced cardiac growth with disorganization of cardiomyocytes, which progressed to a dilated phenotype by 6 weeks of age. Although there were some fibrotic lesions, more striking was the dramatic increase in size of individual myocytes in miR-195 Tg mice compared with normal mice (Fig. 4C).
Fig. 4.

Cardiac-specific overexpression of miR-195 is sufficient to drive cardiomyopathy. (A) H&E-stained sections on hearts of WT and miR-195 Tg, line 3, indicate that overexpression of miR-195 induces death 2 weeks after birth because of cardiac dilation. (Scale bar: 2 mm.) (B) Northern blot analysis on hearts from WT and miR-195 line 3, miR-195 line 1, and miR–214 Tg line 1, confirming a 29-, 26-, and 15-fold cardiac-specific miRNA overexpression, respectively. (C) H&E-stained sections show that overexpression of miR-195 induces cardiac growth at 2 weeks of age, which within 6 weeks progresses to a dilated phenotype, whereas cardiac overexpression of miR-214 has no phenotypic effect. The bottom two panels show a high magnification of the H&E section, indicating severe myocyte disorganization in the miR-195 Tg animals compared with WT. (Scale bars: Top and Middle, 2 mm; Bottom, 20 μm.) (D) H&E-stained sections show that overexpression of miR-214 has no morphological effect on the heart.
Echocardiography on 6-week-old animals showed that miR-195 Tg mice displayed thinning of the left ventricular walls (anterior wall in systole and posterior wall in systole), an increase in left ventricular diameter (left ventricular internal diameter in diastole and systole) and a deterioration in cardiac function, as indicated by decreased fractional shortening (Fig. 5A). Ratios of heart weight to body weight were also dramatically increased in miR-195 Tg animals compared with WT littermates, indicating that overexpression of miR-195 was sufficient to stimulate cardiac growth (Fig. 5B). Real-time PCR analysis on cardiac tissue from miR-195 Tg animals compared with their WT littermates revealed dramatic up-regulation of the hypertrophic markers atrial natriuretic factor, b-type natriuretic protein, and β-MHC in response to cardiac overexpression of miR-195 (Fig. 5C).
Fig. 5.

Overexpression of miR-195 induces cardiac dysfunction caused by cardiac growth. (A) Echocardiographic analyses indicate miR-195 Tg mice show left ventricle dilation and wall thinning, resulting in a decreased fractional shortening compared with WT littermates. Representative m-mode images are shown. (B) The ratio of heart weight to body weight in response to cardiac-specific overexpression of miR-195. (C) Real-time PCR analysis shows an up-regulation of hypertrophic genes in miR-195 Tg animals compared with WT animals (n = 3; mean ± SEM; ∗P < 0.05).
In contrast to the dramatic effects of miR-195 on cardiac structure, function, and gene expression, cardiac overexpression of miR-214 at levels comparable with those of miR-195 had no phenotypic effect (Fig. 4 B and D and data not shown). Thus, the cardiac remodeling induced in the miR-195 Tg animals is specifically caused by the functional effects of this miRNA rather than a general nonspecific effect resulting from miRNA overexpression. These results indicate that increased expression of miR-195 induces hypertrophic signaling, leading to cardiac failure.
Discussion
miRNAs play essential roles in modulating basic physiologic processes including proliferation, differentiation, and apoptosis. The results of this study reveal a collection of miRNAs that are regulated during cardiac hypertrophy and heart failure and appear to play an active role in pathological cardiac remodeling.
A Signature of miRNAs Associated with Cardiac Hypertrophy and Failure.
Adverse cardiac remodeling in response to acute and chronic stress signaling is typically thought to result from the activation and repression of genes encoding proteins that regulate cardiac contractility and structure. Our results point to an additional layer of regulation of cardiac growth mediated by stress-responsive miRNAs. Constitutive CnA signaling and TAB resulted in the up- and down-regulation of common sets of miRNAs, suggesting that these miRNAs represent a genetic signature of the cardiac stress response.
Although most mammalian miRNAs are encoded by separate genes with their own promoters, some miRNAs are also generated by processing of intron sequences of protein-coding genes. A significant number of miRNAs are also expressed in clusters in which two or three miRNAs are generated from a common pri-mRNA. This combined genomic organization enables miRNAs to function in a coordinate manner and suggests that such clustered miRNAs might constitute a regulatory network to target specific mRNAs. Previously, miR-23a, miR-27a, and miR-24-2, which were up-regulated during cardiac hypertrophy, were shown to be generated by processing of a 2.2-kb RNA precursor (19). It will be interesting to determine whether the promoter region of this miRNA cluster or the genes encoding other stress-inducible miRs contain functional NFAT or MEF2 response elements, which have been shown to mediate gene activation in response to CnA signaling and pressure overload (13, 20).
The differential accumulation of pre-miRNA versus mature miRNAs for each of the miRNAs we examined suggests that pre-miRNA processing is regulated in an miRNA-specific manner. Differential processing of pre-miRNAs into mature miRNAs was recently shown to contribute to tissue and developmental-specific miRNA expression in mammals (21). Because our data also indicate that a posttranscriptional mechanism determines the processing efficiency of individual miRNAs, we envision there to be a system that regulates temporal and tissue-specific expression of mature miRNAs in the heart.
Several of the miRNAs we found to be up-regulated during cardiac hypertrophy have been implicated in differentiation and apoptosis in other cell types. Among these, miR-21, has been reported to possess antiapoptotic functions (22) and correlate with the proliferation index of human cancers (23). Additionally, miR-23 and miR-125b are up-regulated during cell differentiation and are critical for cell proliferation (24–26). The up-regulation of these specific miRNAs during cell differentiation and proliferation might imply that they fulfill a functional role in cell division or, in the case of cardiomyocytes, cell growth.
Pathological Cardiac Remodeling in Response to Stress-Inducible miRNAs.
Overexpression of individual stress-inducible miRNAs in primary cardiomyocytes in vitro evoked hypertrophic growth and sarcomere assembly, and cardiac overexpression of miR-195 in vivo was sufficient to drive cardiac hypertrophy, which rapidly transitioned to heart failure. The ability of miR-195 to promote cardiac growth contrasts with that of miR-1, a muscle-specific miRNA that inhibits cardiac growth by suppressing the expression of the basic helix–loop–helix protein Hand2 (27). miR-1 is highly expressed in the adult heart, but miR-195 is apparently capable of overriding its inhibitory influence on cardiac growth.
The ability of individual miRNAs to modulate cardiac phenotypes suggests that regulated expression of miRNAs is a cause rather than simply a consequence of cardiac remodeling. However, it should be pointed out that the levels of miRNA expression achieved in these overexpression assays exceed those of the endogenous miRNAs. Thus, we feel that cardiac phenotypes resulting from regulated expression of endogenous miRNAs most likely reflect the combined actions of multiple miRNAs rather than the action of single miRNAs. Although individual miRNAs were expressed at supraphysiological levels in our overexpression assays, it is unlikely that their effects on cardiac remodeling are nonspecific because overexpression of miR-214 and others (data not shown) does not evoke an adverse cardiac remodeling response.
It is especially interesting that overexpression in cardiomyocytes of the miRNAs that were down-regulated during hypertrophy caused an apparent reduction in cell size, an effect opposite that evoked by the up-regulated miRs. One interpretation of these results is that these miRNAs normally function to suppress growth and are therefore down-regulated to enhance hypertrophy. Among the miRNAs that were down-regulated during cardiac hypertrophy, miR-133 has been reported to repress serum response factor (SRF) (28), a positive regulator of cardiac growth and heart failure (29). Thus, down-regulation of miR-133 during cardiac hypertrophy would be predicted to result in an increase in SRF expression, which could contribute to the adverse remodeling response. It is also notable that miR-133a is encoded by the same pre-miRNA as miR-1, an inhibitor of cardiac growth (28). It will be interesting to determine whether forced overexpression of miR-133 and other miRNAs that are down-regulated during cardiac hypertrophy suppress cardiac growth in vivo.
Future Questions.
In summary, our data suggest that dysregulation of miRNA expression contributes to heart disease. An important aim for the future will be to identify the mRNA targets of the miRNAs responsible for adverse cardiac remodeling. Typically, miRNAs inhibit expression of target mRNAs either by perfect base-pairing, which results in mRNA degradation, or imperfect base-pairing, which results in translational inhibition. As miRNAs often have numerous target mRNAs, the effects of individual miRNAs on cardiac growth and function may reflect altered expression of the products of multiple mRNAs. Further analysis of the functions of the regulatory miRNAs described in this study during adverse cardiac remodeling, and during heart development, promises to provide insights into heart disease and potential therapeutic targets. Given that most of the miRNAs we found to be regulated in response to stress signaling in the heart are also expressed in other tissues, it will be of interest to determine whether they represent conserved downstream effectors of stress response pathways in other cell types.
Materials and Methods
miRNA Microarrays.
Low-molecular-weight RNA was isolated from cardiac tissue by using the mirVana miRNA Isolation Kit (Ambion, Austin, TX). RNA from three animals in each group was pooled and used for miRNA expression analysis by using the miRMAX microarray (30). A total of 186 miRNAs examined showed comparable relative expression at baseline (Fig. 6). Differentially regulated miRNAs were defined as those with either <0.5- or >1.5-fold changes in expression for both arrays compared with the baseline expression levels from sham or WT mouse hearts. All animal protocols were approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee.
RNA Analysis.
Cardiac tissue samples of left ventricles of anonymous humans diagnosed as having nonfailing or failing hearts were obtained from Myogen, Inc. (Westminster, CO). The Institutional Review Board at the University of Texas Southwestern Medical Center in accordance with the Department of Health and Human Services considers this work to meet the criteria of exempt review. Equal loading was confirmed by staining Northern blot gels with ethidium bromide. Total RNA was isolated from mouse and human cardiac tissue samples or isolated myocytes by using TRIzol reagent (Gibco/BRL, Carlsbad, CA). Northern blots were performed according to standard procedures. Expression of a subset of hypertrophic markers was analyzed by quantitative real-time PCR. Details can be found in Supporting Text, which is published as supporting information on the PNAS web site.
Cloning and Expression of miRNAs.
Specific miRNA-expressing adenoviruses were generated by PCR from rat genomic DNA fragments (for PCR primers and detailed information see Table 6, which is published as supporting information on the PNAS web site).
Primary Cardiomyocyte Cultures, Infections, and Immunocytochemistry.
Neonatal rat ventricular myocytes were isolated from 1- to 2-day-old Sprague–Dawley rats, infected, and stained as described (31). See Supporting Text for the method used.
Generation of Tg Mice.
A mouse genomic fragment flanking the miRNA of interest was subcloned into a cardiac-specific expression plasmid containing the α-MHC and human growth hormone (GH) poly(A)+ signal (32) (for PCR primers, see Table 7, which is published as supporting information on the PNAS web site). Genomic DNA was isolated from mouse tail biopsies and analyzed by PCR using primers specific for the human GH poly(A)+ signal.
Histological Analysis.
Histological analysis was performed by using standard procedures (33).
Transthoracic Echocardiography.
Cardiac function and heart dimensions were evaluated by echocardiography as described (34). See Supporting Text for more details.
Supplementary Material
Acknowledgments
We thank Dr. Joseph Hill (University of Texas Southwestern Medical Center) for providing cardiac tissue from animals that received TAB; Erik Bush (Myogen, Inc.) for the human cardiac samples; Drs. Rhonda Bassel-Duby and Dan Garry for comments on the manuscript; and Cheryl Nolen for excellent technical assistance. This work was supported by grants from the National Institutes of Health, the Donald W. Reynolds Cardiovascular Clinical Research Center, and the Robert A. Welch Foundation (to E.N.O.).
Abbreviations
- miRNA
microRNA
- TAB
thoracic aortic banding
- CnA
calcineurin A
- Tg
transgenic
- MHC
myosin heavy chain
- MOI
multiplicity of infection.
Footnotes
The authors declare no conflict of interest.
References
- 1.Arad M, Seidman JG, Seidman CE. Hum Mol Genet. 2002;11:2499–2506. doi: 10.1093/hmg/11.20.2499. [DOI] [PubMed] [Google Scholar]
- 2.Dorn GW, 2nd, Robbins J, Sugden PH. Circ Res. 2003;92:1171–1175. doi: 10.1161/01.RES.0000077012.11088.BC. [DOI] [PubMed] [Google Scholar]
- 3.Chien KR. Cell. 1999;98:555–558. doi: 10.1016/s0092-8674(00)80043-4. [DOI] [PubMed] [Google Scholar]
- 4.Frey N, Olson EN. Annu Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 5.Bartel DP. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.Moss EG, Tang L. Dev Biol. 2003;258:432–442. doi: 10.1016/s0012-1606(03)00126-x. [DOI] [PubMed] [Google Scholar]
- 7.Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Muller P, et al. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- 8.Esquela-Kerscher A, Slack FJ. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 9.Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM. Proc Natl Acad Sci USA. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammond SM. Curr Opin Genet Dev. 2006;16:4–9. doi: 10.1016/j.gde.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Ambros V. Cell. 2003;113:673–676. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 12.Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- 13.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barad O, Meiri E, Avniel A, Aharonov R, Barzilai A, Bentwich I, Einav U, Gilad S, Hurban P, Karov Y, et al. Genome Res. 2004;14:2486–2494. doi: 10.1101/gr.2845604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shingara J, Keiger K, Shelton J, Laosinchai-Wolf W, Powers P, Conrad R, Brown D, Labourier E. RNA. 2005;11:1461–1470. doi: 10.1261/rna.2610405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Babak T, Zhang W, Morris Q, Blencowe BJ, Hughes TR. RNA. 2004;10:1813–1819. doi: 10.1261/rna.7119904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu CG, Calin GA, Meloon B, Gamliel N, Sevignani C, Ferracin M, Dumitru CD, Shimizu M, Zupo S, Dono M, et al. Proc Natl Acad Sci USA. 2004;101:9740–9744. doi: 10.1073/pnas.0403293101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obernosterer G, Leuschner PJ, Alenius M, Martinez J. RNA. 2006;12:1161–1167. doi: 10.1261/rna.2322506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan JA, Krichevsky AM, Kosik KS. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 23.Roldo C, Missiaglia E, Hagan JP, Falconi M, Capelli P, Bersani S, Calin GA, Volinia S, Liu CG, Scarpa A, Croce CM. J Clin Oncol. 2006;24:4677–4684. doi: 10.1200/JCO.2005.05.5194. [DOI] [PubMed] [Google Scholar]
- 24.Kawasaki H, Taira K. Nucleic Acids Symp Ser. 2003;3:243–244. doi: 10.1093/nass/3.1.243. [DOI] [PubMed] [Google Scholar]
- 25.Lee YS, Kim HK, Chung S, Kim KS, Dutta A. J Biol Chem. 2005;280:16635–16641. doi: 10.1074/jbc.M412247200. [DOI] [PubMed] [Google Scholar]
- 26.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, et al. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 27.Zhao Y, Samal E, Srivastava D. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 28.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. Nat Genet. 2006;38:228–233. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parlakian A, Charvet C, Escoubet B, Mericskay M, Molkentin JD, Gary-Bobo G, De Windt LJ, Ludosky MA, Paulin D, Daegelen D, et al. Circulation. 2005;112:2930–2939. doi: 10.1161/CIRCULATIONAHA.105.533778. [DOI] [PubMed] [Google Scholar]
- 30.Goff LA, Yang M, Bowers J, Getts RC, Padgett RW, Hart RP. RNA. 2005;2:e9–e16. doi: 10.4161/rna.2.3.2059. [DOI] [PubMed] [Google Scholar]
- 31.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. J Clin Invest. 2006;116:1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gulick J, Subramaniam A, Neumann J, Robbins J. J Biol Chem. 1991;266:9180–9185. [PubMed] [Google Scholar]
- 33.Shelton JM, Lee MH, Richardson JA, Patel SB. J Lipid Res. 2000;41:532–537. [PubMed] [Google Scholar]
- 34.Harrison BC, Kim MS, van Rooij E, Plato CF, Papst PJ, Vega RB, McAnally JA, Richardson JA, Bassel-Duby R, Olson EN, McKinsey TA. Mol Cell Biol. 2006;26:3875–3888. doi: 10.1128/MCB.26.10.3875-3888.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
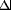{kind=link}