

Abstract
To regulate mammalian water homeostasis, arginine-vasopressin (AVP) induces phosphorylation and thereby redistribution of renal aquaporin-2 (AQP2) water channels from vesicles to the apical membrane. Vice versa, AVP (or forskolin) removal and hormones activating PKC cause AQP2 internalization, but the mechanism is unknown. Here, we show that a fraction of AQP2 is modified with two to three ubiquitin moieties in vitro and in vivo. Mutagenesis revealed that AQP2 is ubiquitinated with one K63-linked chain at K270 only. In Madin–Darby canine kidney cells, AQP2 ubiquitination occurs preferentially when present in the apical membrane, is transiently increased with forskolin removal or PKC activation, and precedes its internalization. Internalization kinetics assays with wild type (wt) and ubiquitination-deficient (K270R) AQP2 revealed that ubiquitination enhances AQP2 endocytosis. Electron microscopy showed that a translational fusion of AQP2 with ubiquitin (AQP2-Ub) localized particularly to internal vesicles of multivesicular bodies (MVBs), whereas AQP2-K270R largely localized to the apical membrane, early endosomes, and the limiting membrane of MVBs. Consistent with this distribution pattern, lysosomal degradation was extensive for AQP2-Ub, low for AQP2-K270R, and intermediate for wt-AQP2. Our data show that short-chain ubiquitination is involved in the regulated endocytosis, MVB sorting, and degradation of AQP2 and may be the mechanism used by AVP removal and PKC-activating hormones to reduce renal water reabsorption. Moreover, because several other channels are also (short-chain) ubiquitinated, our data suggest that ubiquitination may be a general mediator for the regulated endocytosis and degradation of channels in higher eukaryotes.
Keywords: homeostasis, internalization, signal transduction, sorting, transmembrane protein
Aquaporin (AQP) water channels are important for rapid and selective osmotic water transport across cell membranes. Most AQPs have a constitutive open pore, and regulation of transmembrane water transport is thus controlled by channel insertion into and retrieval from the cell surface (1–3). AQP2 is one of the best-characterized AQPs and confers water permeability to the kidney-collecting duct to serve body water homeostasis. This process is of pathophysiological importance, because inadequate cell-surface expression of AQP2 results in nephrogenic diabetes insipidus (4, 5), whereas increased cell surface expression and excessive water reabsorption is observed in congestive heart failure, preecclampsia, and the syndrome of inappropriate release of the hormone arginine-vasopressin (AVP) (6).
Hypernatremia and hypovolemia induce the release of AVP, which regulates the cell-surface expression of AQP2. AVP occupation of its renal type 2 receptors initiates a signaling cascade resulting in phosphorylation of AQP2 at S256. Phosphorylation of AQP2 is necessary for the fusion of AQP2-containing vesicles with the apical membrane, where AQP2 facilitates water reabsorption (2, 7–11). Moreover, AVP also regulates water reabsorption by arresting endocytosis, as demonstrated by comparison of isolated tubule perfusion and mathematical modeling (12) and by the large increase of coated pits in the apical membrane of vasopressin-stimulated isolated tubules (13). After correction of the water balance, AVP levels decrease, and AQP2 is internalized (14, 15). Moreover, several hormones induce AQP2 internalization through activation of PKC, thereby counteracting AVP (16). At least two hormones (prostaglandin E2 and dopamine) cause AQP2 internalization independent of S256 dephosphorylation (17, 18). Similarly, in Madin–Darby canine kidney (MDCK) cells, activation of PKC by the phorbol ester 12-tetradecanoylphorbol-13-acetate (TPA) causes the internalization of AQP2 without affecting its phosphorylation at S256 or inducing any other phosphorylation of AQP2 (9). Thus, although AQP2 insertion into and retrieval from the apical membrane are equally important processes in maintaining body water homeostasis, the mechanism of AQP2 internalization is poorly understood.
Monoubiquitination and K63-linked short-chain ubiquitination regulate endocytosis and degradation of receptors, channels, and transporters in yeast (19). In higher eukaryotes, this type of ubiquitination clearly enhances lysosomal degradation, but its role in endocytosis is still controversial (20–22). Here, we show that removal of AVP (mimicked by forskolin washout) and PKC activation cause K63-linked ubiquitination of AQP2, which enhances endocytosis, sorting to intraendosomal vesicles and degradation of AQP2.
Results
AQP2 Is Ubiquitinated in Vitro and in Vivo.
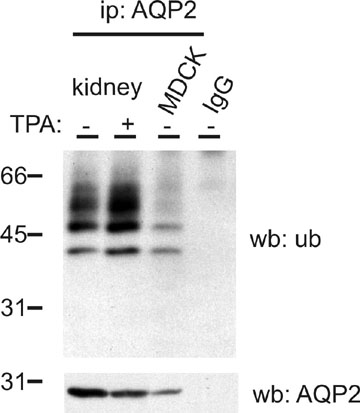
To analyze whether AQP2 is ubiquitinated, it was immunoprecipitated from solubilized goat kidney medulla membranes and MDCK-AQP2 (23) cells. Subsequent immunoblotting for ubiquitin revealed ubiquitinated AQP2 bands of mainly 43, 50, and 57 kDa (Fig. 1A). Furthermore, the smeary higher-molecular-weight forms (between ≈50 and 60 kDa) disappeared after endoglycosidase-F treatment (not shown), indicating that these bands are both ubiquitinated and glycosylated. Incubation of goat membrane lysates with protein A beads only or loading of AQP2 antibodies did not yield any signal. Together, these data indicate that AQP2 is modified with a limited number of ubiquitin molecules, both in vitro and in vivo.
Fig. 1.

Ubiquitination of AQP2 occurs at the apical plasma membrane. (A) AQP2 was immunoprecipitated (ip: AQP2) from solubilized goat kidney inner medulla membranes (kidney) or MDCK-AQP2 cells (MDCK). Solubilized kidney membranes were also subjected to precipitation with protein A-coupled beads without antibodies (prot.A). Throughout this study, nonequivalents (5% and 95%) of the (immuno)precipitates were immunoblotted (ib) for AQP2 or ubiquitin (ub), respectively. AQP2 antibodies (IgG) serve as negative control. (B) MDCK cells expressing wt-AQP2 (wt), AQP2-S256A (SA) or AQP2-S256D (SD) were left untreated (−) or pretreated (±) with forskolin (forsk) for 45 min. Subsequently, AQP2 was immunoprecipitated and immunoblotted for ubiquitin or AQP2. (C) MDCK cells expressing wt-AQP2 were treated with forskolin for 45 min, surface-biotinylated, treated with MesNa to strip surface biotin where indicated (+), and lysed. AQP2 was immunoprecipitated, released from the beads, and a part (10%) of the sample was used for direct immunoblotting (total), whereas the remainder (90%) was first subjected to streptavidin pulldowns to recover biotinylated AQP2 (apical) and subsequently immunoblotted.
Short-Chain Ubiquitination of AQP2 Preferentially Occurs at the Plasma Membrane.
If AQP2 ubiquitination plays a role in regulation of its apical internalization, the most likely site of ubiquitination is the apical plasma membrane. As in vivo, stimulation of MDCK cells with AVP or forskolin induces the translocation of AQP2 to the apical membrane (23). Therefore, we immunoprecipitated AQP2 from MDCK cells pretreated with forskolin or not. Immunoblotting and signal quantification revealed that the relative amount of ubiquitinated AQP2 was 2.2 ± 0.4 times higher after forskolin stimulation (Fig. 1B; n = 3; P = 0.014). Moreover, we analyzed the ubiquitination of AQP2-S256A and AQP2-S256D, which mimic nonphosphorylated and phosphorylated AQP2, and which show a forskolin-independent steady-state localization in vesicles or the apical membrane, respectively (7–9). The level of AQP2-S256A ubiquitination was similar to that of unstimulated wild-type (wt)-AQP2 (0.8 ± 0.1 times that of unstimulated wt-AQP2, n = 3, P = 0.23), whereas that of AQP2-S256D was 1.4 ± 0.1 times higher than that of unstimulated wt-AQP2 (Fig. 1B; n = 3, P = 0.022).
To directly measure whether plasma membrane AQP2 is preferentially ubiquitinated, forskolin-stimulated MDCK-AQP2 cells were subjected to cell surface biotinylation. After AQP2 immunoprecipitation, 10% was taken out, whereas the remaining 90% of AQP2 was enriched for plasma membrane AQP2 by streptavidin pulldowns. Subsequent immunoblotting (Fig. 1C Left) and relative quantification revealed that 2.1 ± 0.5 times more AQP2 was ubiquitinated in the apical membrane than in the total pool (n = 4; P = 0.041). To ensure that detected AQP2 was not from biotinylation of intracellular AQP2 or nonspecific binding of AQP2 to the streptavidin beads, the experiment was now done with biotin stripped from the plasma membrane directly after biotinylation. Immunoblotting of this streptavidin pulldown showed no signal for AQP2 or ubiquitin (Fig. 1C Right). These data indicate that AQP2 is preferentially ubiquitinated when present in the apical plasma membrane.
Ubiquitination of AQP2 Precedes Its Internalization.
Removal of forskolin after 45 min and PKC activation with TPA in the presence of forskolin induce the internalization of AQP2 in MDCK cells (9). To investigate whether this correlates with an increase in ubiquitination, we used these treatments to compare the time kinetics of AQP2 ubiquitination and internalization. With constant levels of immunoprecipitated AQP2 (Fig. 2 Bottom), forskolin washout for 5 min revealed increased levels of AQP2 ubiquitination (2.0 ± 0.2 times that of forskolin treated, n = 3; P = 0.004), whereas a washout for 15 and 45 min resulted in the gradual decrease in AQP2 ubiquitination to prewashout levels (Fig. 2 Top). Addition of TPA for 5 min to forskolin-treated cells also resulted in an increase in AQP2 ubiquitination (4.8 ± 0.3 times that of forskolin treated, n = 3; P < 0.01), whereas 15- and 45-min TPA treatment again showed a gradual decrease in AQP2 ubiquitination. Biochemical internalization assays using biotinylation and 2-sodium mercaptoethanesulfonic acid (MesNa) revealed that at 15 and 45 min after forskolin washout or TPA addition, but not at 5 min, increased amounts of internalized AQP2 were observed (Fig. 2 Middle). Because ubiquitination peaks at 5 min, these data indicate that AQP2 ubiquitination precedes its internalization. It needs to be noted that, because the fraction of ubiquitinated AQP2 is small, internalized AQP2 was visualized by using AQP2 antibodies. Using kidney tissue, we show that TPA also increases AQP2 ubiquitination ex vivo (Fig. 6, which is published as supporting information on the PNAS web site).
Fig. 2.

Ubiquitination of AQP2 occurs before its internalization. MDCK cells expressing wt-AQP2 were pretreated with forskolin for 45 min (0). Subsequently, forskolin was washed out (forsk wash out) or the phorbol ester TPA was added (forsk + TPA) for the indicated times. Cells were lysed, and AQP2 was immunoprecipitated and immunoblotted for ubiquitin or AQP2. In parallel, forskolin-treated MDCK-AQP2 cells were surface-biotinylated in the cold, followed by an incubation at 37°C without forskolin (forsk wash out) or with forskolin and TPA (forsk + TPA) for the indicated times. Then, surface-bound biotin was removed with MesNa, and the cells were lysed. Biotinylated proteins were recovered by using immobilized streptavidin and immunoblotted for AQP2 (internalized AQP2).
AQP2 Is Ubiquitinated with One K63-Linked Chain at K270.
Because AQP2 has three putative attachment sites for ubiquitin (cytosolic lysines) at positions 228, 238, and 270, AQP2 mutants having one, two, or all three lysines replaced by arginines were expressed in MDCK cells. Ubiquitination analyses revealed that all mutants having the K270R replacement were no longer ubiquitinated, whereas all other mutants showed the same pattern and intensity of ubiquitination as wt-AQP2 (Fig. 3A). These data reveal that only K270 is a substrate for ubiquitination in AQP2. Because ubiquitin fused to receptor tyrosine kinases provided information on the role of ubiquitin in their regulation (24), we expressed a similar fusion with AQP2 (AQP2-Ub). Because K270 is the penultimate amino acid of AQP2, we designed AQP2-Ub as a translational fusion of AQP2-K270R lacking A271 and coupled to ubiquitin-K48R-ΔG75/76. The K48R exchange and deletion of the C-terminal glycines (G75/76) were introduced to prevent polyubiquitination (24). In MDCK cells, AQP2-Ub migrates at ≈36 kDa (Fig. 3B Left), which is consistent with the addition of one ubiquitin moiety (7 kDa) to AQP2 (29 kDa). When immunoblotted for ubiquitin, additional bands of ≈43, ≈50, and ≈57 kDa are detected, consistent with AQP2 modified with two, three, and four ubiquitin moieties, respectively (Fig. 3B Center). Ubiquitinated wt-AQP2 migrates similarly and with similar relative intensities of their ≈43- and ≈50-kDa bands, indicating that the main ubiquitinated forms of wt-AQP2 contain two and three ubiquitin moieties, respectively. Ubiquitin immunoblots with increased levels of ubiquitination, as realized with a 5-min TPA treatment, also revealed the monoubiquitinated wt-AQP2 band (Fig. 3B Right, arrowhead). Moreover, even after TPA treatment, AQP2-K270R was not ubiquitinated (Fig. 3B).
Fig. 3.

AQP2 is ubiquitinated at K270 with one short K63 linked chain. (A) Lysates of nontransfected MDCK cells (−) or of those expressing wt-AQP2 (wt) or its indicated mutants were subjected to AQP2 immunoprecipitation. Immunoprecipitates were immunoblotted for ubiquitin or AQP2. (B) MDCK cells stably expressing wt-AQP2 or its mutants were not treated or treated (+) with TPA. AQP2 immunoprecipitates from these cells were immunoblotted for ubiquitin or AQP2. The open arrowhead indicates AQP2 modified with one ubiquitin molecule. Signals from immunoglobulins (IgG) used in the immunoprecipitations are indicated.
Strikingly, AQP2-Ub was still modified with several ubiquitin molecules, despite the lack of K270 in AQP2, and of K48 and G75/76 in ubiquitin. Of the seven lysines in ubiquitin, ubiquitination of K63 is involved in protein sorting (22). Therefore, we expressed a mutant in which this lysine was replaced by an arginine in AQP2-Ub (AQP2-Ub-K63R) in MDCK cells. Indeed, AQP2-Ub-K63R was detected only as a 36-kDa protein (Fig. 3B Center). Together, these data show that AQP2 is ubiquitinated at K270 with one K63-linked chain of mainly two to three ubiquitin moieties.
Ubiquitination of AQP2 Increases Its Endocytosis.
To determine the steady-state localization of AQP2 mutants, MDCK cells were fixed and subjected to immunocytochemistry. Confocal analysis revealed that wt-AQP2, AQP2-K270R, and AQP2-Ub are present in intracellular compartments (Fig. 4A). After forskolin stimulation, both wt-AQP2 and AQP2-K270R were redistributed to the apical membrane, whereas the steady-state localization of AQP2-Ub did not change (Fig. 4A).
Fig. 4.

Ubiquitination of AQP2 increases its endocytosis. (A) MDCK cells expressing wt-AQP2, AQP2-K270R or AQP2-Ub were left untreated (−) or treated with forskolin, fixed, and subjected to immunocytochemistry using AQP2 antibodies. Confocal images were obtained in the XY and XZ planes. (B) MDCK cells expressing wt-AQP2 or AQP2-K270R were treated with forskolin, followed by TPA addition (forsk. + TPA) for the indicated times. Then, cells were fixed and analyzed as under A. (C) Internalization of wt-AQP2 or AQP2-K270R after forskolin treatment followed by TPA addition (forsk. + TPA) was determined as in Fig. 3 for the indicated times. Semiquantification of AQP2 (n = 3) was done as described in Materials and Methods. Filled and open data points represent wt-AQP2 and AQP2-K270R, respectively.
To further analyze the effect of ubiquitination on AQP2 internalization, MDCK-AQP2 and MDCK-AQP2-K270R cells were treated with forskolin, followed by TPA addition for several time points, fixed, and subjected to immunocytochemistry. Confocal analysis clearly revealed internalized AQP2 at 15 and 45 min of TPA treatment (Fig. 4B), consistent with the kinetics of AQP2 internalization as biochemically determined (Fig. 2A). Interestingly, internalization of AQP2-K270R was not observed at these time points. Forskolin washout showed similar effects as found for the TPA treatment (Fig. 7A, which is published as supporting information on the PNAS web site). Biochemical internalization assays using biotinylation and MesNa confirmed that with 45 min of TPA treatment, the internalized fraction of wt-AQP2 was larger than that of AQP2-K270R (Fig. 7B).
To establish whether the enhanced internalization of ubiquitinated AQP2 is due to increased endocytosis, the kinetics of AQP2 internalization was determined by using the internalization assay at 5, 10, 20, and 30 min of TPA addition. Densitometric analyses of the AQP2 immunoblot signals revealed that wt-AQP2 was internalized faster than AQP2-K270R (Fig. 4C). Forskolin washout showed similar effects as TPA treatment (Fig. 7C). These data indicate that ubiquitination of AQP2 enhances its endocytosis.
Ubiquitinated AQP2 Is Targeted for Degradation in the Multivesicular Bodies (MVBs)/Lysosome Pathway.
Immunogold labeling for AQP2 on ultrathin cryosections prepared from MDCK cells revealed endosomal expression of wt-AQP2, AQP2-K270R, and AQP2-Ub, albeit with distinct distributions (Fig. 5A). Labeled endosomes were classified as early (type I), late (type II), and late endosomes/lysosomes (type III), according to their luminal content (25–27). Lysosomes were not labeled, presumably because of epitope degradation. Quantification of the labeling revealed that the majority of AQP2-Ub-positive endosomes were of the late endosomal/lysosomal type, whereas no substantial staining was found in early endosomes (Fig. 5 and Table 1). In contrast, only 40% of the AQP2-K270R positive endosomes were of the late endosome/lysosome type, and early endosomes were now often found positive. In addition, the relative labeling of the distinct endosomal subdomains was also significantly different (Table 1). The vast majority of endosomal AQP2-Ub was associated with internal vesicles, whereas 45% of endosome-associated AQP2-K270R resided on the limiting membrane. wt-AQP2 showed an inter- and intraendosomal distribution intermediate to that of AQP2-K270R and AQP2-Ub (Table 1). To determine whether the differential localizations were reflected in their levels of degradation, MDCK cells expressing wt-AQP2 or AQP2-K270R were treated with cycloheximide to block translation. Subsequent treatments with TPA revealed that wt-AQP2 was more rapidly degraded than AQP2-K270R, which could partially be prevented by treatment with the lysosome inhibitor chloroquine (Fig. 5B). Furthermore, the low abundance of the AQP2-Ub protein increased markedly after treatment with chloroquine, indicating that AQP2-Ub is strongly targeted for lysosomal degradation (Fig. 5C). Together, these data indicate that ubiquitination of AQP2 induces its transport to late endosomes/lysosomes, incorporation into the intraendosomal vesicles and subsequent lysosomal degradation or excretion in exosomes.
Fig. 5.

Ubiquitination of AQP2 increases its lysosomal degradation. (A) Cryosections of fixed MDCK cells expressing AQP2-K270R (1, 2) or AQP2-Ub (3) were immunolabeled for AQP2 (10-nm gold particles). Endosomes are divided into three types: I = early endosome, II = late endosome, and III = late endosome/lysosome (1). Overview of the apical part of an MDCK cell showing all three types of endosomes. The arrows point to labeled dense vesicles that mediate either endocytosis or recycling (2). Detail of an early endosome with AQP2-K270R label at the limiting membrane (3). Detail of late endosomes/lysosomes with the majority of label associated with internal membranes. (Scale bars, 200 nm.) (B) Protein synthesis in MDCK-AQP2 or MDCK-AQP2-K270R was inhibited (cycloheximide), and TPA was added in the absence or presence of chloroquine for the indicated times. Subsequently, cells were lysed and immunoblotted for AQP2. (C) MDCK cells expressing wt-AQP2 or AQP2-Ub were treated with chloroquine for the indicated times. Subsequently, cells were lysed and immunoblotted for AQP2.
Table 1.
Differential endosomal distribution of wt-AQP2, AQP2-K270R, and AQP2-Ub
| Endosome | Distribution of endosomal AQP2, % | |||
|---|---|---|---|---|
| Type I | Type II | Type III | Limiting membrane | |
| wt-AQP2 | 10 | 40 | 50 | 20 |
| AQP2-K270R | 30 | 30 | 40 | 45 |
| AQP2-Ub | 0 | 27 | 73 | 5 |
Quantifications were performed on ultrathin cryosections of MDCK cells labeled for AQP2. For each counting, 30 endosomes were analyzed. Endosomes are divided into three types: I, early endosome; II, late endosome; III, late endosome/lysosome.
Discussion
Short-Chain Ubiquitination of AQP2 Induces Its Endocytosis.
In higher eukaryotes, cargo ubiquitination is involved in the endocytic sorting of receptors and channels, but the means by which internalization is accomplished (increased endocytosis, decreased exocytosis, degradation, etc.) has remained controversial (20–22, 24). A complicating factor in delineating the role of cargo ubiquitination in its endocytosis is that cargo ubiquitination itself may be dispensable but requires an intact ubiquitin conjugation system. For example, after ligand binding, G protein-coupled receptors (GPCRs) are endocytosed through the interaction with the ancillary protein β-arrestin. Although monoubiquitination of β-arrestin is essential in endocytosis, (poly)-ubiquitination of the GPCRs themselves affects only their degradation (28). Here, we show that K63-linked short-chain ubiquitination of AQP2 at K270 is exploited for the regulated endocytosis of this water channel, because AQP2 ubiquitination occurs particularly in the plasma membrane, is induced by TPA and forskolin removal, and precedes and enhances AQP2 internalization, and because the ubiquitin-deficient AQP2-K270R is delayed in its endocytosis. As such, our data unveil that higher eukaryotes also use short-chain ubiquitination of cargo to mediate endocytosis.
That AQP2-K270R is still internalized, albeit with a slower rate, indicates that AQP2 ubiquitination is not the only signal for endocytosis. Interestingly, Sigismund et al. (29) showed that a low dose of epithelial growth factor (EGF) causes internalization of nonubiquitinated wt EGF-receptor mainly through the clathrin pathway, whereas a high dose of EGF resulted in receptor ubiquitination and internalization through a clathrin-independent pathway (29), suggesting that ubiquitination directs the cargo protein from the constitutive to the regulated endocytic pathway. Our data show that AQP2 also may be endocytosed through a constitutive and regulated pathway, because TPA stimulation and forskolin removal mediate the ubiquitination-enhanced (regulated) endocytosis, whereas the constitutive endocytosis of AQP2-K270R occurs independent of its ubiquitination. However, it remains to be established whether these pathways indeed differ in the mechanism by which endocytosis is accomplished.
Ubiquitination Targets AQP2 for Degradation.
In the endocytic sorting pathway, monoubiquitinated cargo proteins are clustered by the endosomal sorting complexes required for transport and delivered to vesicles that invaginate into the lumen of MVBs, eventually resulting in lysosomal degradation (30–32). Non- or deubiquitinated proteins might exit this pathway in an early stage or will remain in the MVB limiting membrane, from where they may recycle. Our data indicate that AQP2 follows a similar pathway. First, the interendosomal distributions of AQP2-K270R, wt-AQP2, and AQP2-Ub correlate with their relative levels of ubiquitination. Second, AQP2-Ub is mainly found in internal vesicles of MVBs, whereas AQP2-K270R is observed in the limiting membranes. Third, the TPA-induced ubiquitination of wt-AQP2 increases its degradation, which could partially be inhibited by the lysosome inhibitor chloroquine. Fourth, chloroquine strongly reduces the degradation of AQP2-Ub. It needs to be noted that our experiments do not reveal whether AQP2-Ub is sorted to MVBs by the plasma membrane or directly from the Golgi network.
Ubiquitination-Mediated Endocytosis of AQP2, the Other Side of the Coin.
Mammalian water homeostasis is regulated by the insertion into and removal of AQP2 from the apical plasma membrane (2, 33, 34). Although the insertion of AQP2 into the apical plasma membrane depends on the AVP-induced phosphorylation of AQP2 at S256 (7–9), AQP2 internalization does not involve its dephosphorylation (17, 18), and therefore the underlying mechanism has remained elusive. Our present study unveils this step in AQP2 regulation in that a transient short-chain ubiquitination of AQP2 induces its endocytosis and MVB targeting, after either AVP withdrawal or activation of PKC.
We propose the following model for AQP2 regulation in renal principal cells (Fig. 8, which is published as supporting information on the PNAS web site): whereas AVP increases AQP2 phosphorylation resulting in increased steady-state expression of AQP2 in the apical membrane of renal principal cells, AVP removal and AVP-counteracting hormones activating PKC reverse this location by inducing its ubiquitination, endocytosis, and sorting to intraendosomal vesicles of MVBs. From these vesicles, AQP2 is targeted for degradation in lysosomes or shed as exosomes from principal cells into the urine. As shown for the vacuolar hydrolase carboxypeptidase S and the epithelial growth factor receptor (22, 30), the extent of AQP2 degradation by MVBs likely depends on its ubiquitination state en route to MVBs, which is modulated by the activity of (de-)ubiquitinating enzymes. When ubiquitin-conjugated at the limiting membrane of MVBs, AQP2 is likely targeted to internal vesicles and degraded (or shed), whereas when deubiquitinated, it is available for a next AVP-induced insertion into the plasma membrane.
Our hypothesis is supported by several studies. In vivo, AQP2 is increasingly ubiquitinated with TPA, is localized in recycling vesicles and MVBs (2), and colocalizes with Hrs-2 (35), which mediates endocytic sorting of ubiquitinated cargo (36). Moreover, the amount of urinary AQP2 in exosomes is increased with elevated AVP levels (37, 38). In vivo and in vitro, AQP2 is a stable protein with a half-life of 6–12 h (39), and several rounds of AVP addition and removal revealed that AQP2 can recycle at least three times in transfected LLC-PK1 cells (40).
It remains to be established whether AQP2 ubiquitination and the dynamic interplay of (de)-ubiquitinating enzymes play a role in several disease states with aberrant localization/degradation of AQP2 (i.e., acquired nephrogenic diabetes insipidus and syndrome of inappropriate release of AVP), whether and which AVP-counteracting hormones also induce AQP2 internalization by ubiquitination, and which (de)-ubiquitinating and ubiquitin-interacting proteins regulate AQP2 sorting and degradation.
Short-Chain Ubiquitination, a General Mechanism to Regulate the Endocytosis of Channels in Higher Eukaryotes.
Although short-chain ubiquitination and monoubiquitination mediate endocytosis of yeast transporters and channels (19, 41), our data show this pathway is also used for the hormone-regulated endocytosis of AQP2 in renal cells. Besides AQP2, we also found that AQP3 and AQP5 are short-chain-ubiquitinated (data not shown). Moreover, ubiquitination was reported to coincide with the increased degradation of the CLC5 chloride channel, ENaC, and the ROMK potassium channel (42–44). Although the type (mono- or polyubiquitination) and a definitive role for ubiquitination in the endocytosis of these proteins still have to be unraveled, it suggests that ubiquitination might also be a general mechanism to endocytose channels in higher eukaryotes.
Materials and Methods
Constructs, Cell Culture, Treatments, and Stable Transfection.
MDCK-AQP2 and -AQP2-S256A/D have been described (9). The fusion construct encoding AQP2-Ub was obtained by PCR on ubiquitin (a gift from D. Bohmann, European Molecular Biology Laboratory, Heidelberg, Germany) using the reverse primer 5′-ccatcgattaaccacctctcagacgcagg-3′ (ClaI underlined) and the forward primer 5′-ggggtaccagaatgcagatcttcgtgaaaaccc-3′ [AQP2 coding sequence in bold and the recognition site for KpnI (endogenous in AQP2 cDNA) is underlined]. The PCR fragment was subcloned into the KpnI and ClaI restriction sites of pCB6-AQP2. MDCK cell culture and transfection, immunocytochemistry, and collection of images were performed as described (10). A minimum of three cell lines expressing the same protein were analyzed. Cells were treated with forskolin (10−5 M), TPA (10−7 M), cycloheximide (0.05 mM), and/or chloroquine (0.1 mM) in culture medium.
Immunoprecipitation from Kidney Inner Medulla and MDCK Cells.
Goat kidney inner medulla slices were treated with or without TPA in DMEM at 37°C for 15 min. Subsequently, the tissue was homogenized in 20 ml of 300 mM sucrose/25 mM imidazole/1 mM EDTA/20 mM N-ethylmaleimide (NEM). Membranes were isolated by centrifugation of the precleared homogenate at 200,000 × g and solubilized in 2% SDS [in 150 mM NaCl/25 mM Hepes (pH 7.4)/20 mM NEM] at 37°C for 30 min. These lysates were diluted 20× with 1% Triton X-100/150 mM NaCl/25 mM Hepes (pH 7.4)/20 mM NEM. The supernatant was precleared from immunoglobulins by three sequential incubations with 100 μl of immobilized protein A. MDCK cells were grown to confluence, treated as described, and lysed [1% Triton X-100/150 mM NaCl/25 mM Hepes (pH 7.4)/20 mM NEM]. Both the kidney and MDCK lysates were subjected to AQP2 immunoprecipitation.
Cell Surface Biotinylation and Biochemical Internalization Assays.
MDCK cells were grown to confluence, treated, placed on ice, and washed with ice-cold PBS-CM (1 mM CaCl2/0.1 mM MgCl2). Surface proteins were biotinylated twice for 20 min with ice-cold 1.5 mg/ml Sulfo-NHS-SS-biotin (Pierce, Rockford, IL) in 10 mM triethanolamine (pH 8.9)/2 mM CaCl2/125 mM NaCl. After biotin removal, excess biotin was quenched with ice-cold 50 mM NH4Cl in PBS-CM. For biotinylation experiments, cells were lysed in 1 ml of lysis buffer [1% Triton X-100/150 mM NaCl/5 mM EDTA/50 mM Tris (pH 7.5)] for 30 min at room temperature. For internalization experiments, surface proteins were permitted to become internalized by warming the cells to 37°C. Next, the cells were washed with ice-cold PBS-CM and treated 3× 20 min with ice-cold 100 mM MesNa in 100 mM NaCl/1 mM EDTA/50 mM Tris (pH 8.6)/0.2%BSA. The cell-impermeable MesNa removes cell-surface-bound biotin, while internalized biotinylated proteins are protected. Excess MesNa was quenched with ice-cold 120 mM iodoacetic acid in PBS-CM, and cells were lysed as with the biotinylation. Subsequently, biotinylated proteins were pulled down from with immobilized streptavidin, followed by extensive washing and immunoblotting. For quantification of internalization, signals were determined by densitometry and comparison with a dilution series of AQP2. Then, internalization of AQP2 was determined as the percentage of biotinylated AQP2 after stripping compared with that of initially biotinylated AQP2, corrected for the stripping control (0 min internalization with stripping).
Immunoblotting and Densitometric Quantification.
PAGE and blotting, blocking, antibody incubation, and chemiluminescence of the membranes were performed as described (8). Ubiquitin (Zymed, San Francisco, CA) antibodies were diluted to a concentration of 1:250, followed by 1:10,000-diluted biotinylated anti-mouse IgGs and 1:8,000-diluted streptavidin-peroxidase. Representative figures are shown. To quantify protein signals from the immunoblot, we calculated the amount of protein (in arbitrary units) from a 2-fold dilution series of the same protein that was parallel immunoblotted. The amount of ubiquitinated AQP2 was normalized to the amount of 29-kDa AQP2. Only signals in the linear range of the immunoblot were used. P values between two variables was calculated by using Student's t test and between multiple variables by using ANOVA.
Immunoelectron Microscopy.
MDCK cells were fixed in 2% freshly prepared formaldehyde plus 0.2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, for 2 h at room temperature. Subsequent processing, cryosectioning (50–60 nm), and labeling with antibodies and protein A gold was done as described (45). Quantitative analysis of AQP2 distribution over distinct types of endosomes as well as over the internal and limiting endosomal membranes was done as follows. Sections were screened at random for a positively labeled endosome. Each positive endosome was classified as type I (with zero to five internal vesicles), type II (with more then five internal vesicles), or type III (with internal vesicles as well as dense material). These morphological criteria were previously linked to kinetic and molecular characterizations of endosomes and represent a general classification into early endosomes, late endosomes, and late endosomes/lysosomes, respectively (25–27). For each endosome, the number of gold particles was counted and ascribed to the limiting membrane or internal vesicles. Per cell type, 30 endosomes were analyzed. The relative distribution of AQP2 in the limiting membrane (in percent) was calculated from the total of all gold particles in the limiting membrane of the 30 analyzed endosomes and the sum of all gold particles in the limiting membrane and internal vesicles of those endosomes. In total 55, 63, and 69 gold particles were counted for wt-AQP2, AQP2-K270R, and AQP2-Ub, respectively.
Supplementary Material
Acknowledgments
We thank Dr. O. Staub for helpful discussions, M. van Peski and R. Scriwanek for the preparation of the EM figures, and M. van Beest for suggestions. This study was supported by grants from The Netherlands Organization for Scientific Research (to E.-J.K.) (NWO Grant 916.36.122) and from the Dutch Kidney Foundation (C03.2060, to P.M.T.D.).
Abbreviations
- AQP
aquaporin
- AQP2
AQP-2
- wt
wild type
- AVP
arginine-vasopressin
- TPA
12-tetradecanoylphorbol-13-acetate
- MDCK
Madin–Darby canine kidney
- MVB
multivesicular body
- AQP2-Ub
fusion of AQP2 and ubiquitin
- MesNa
2-sodium mercaptoethanesulfonic acid.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
References
- 1.Sidhaye V, Hoffert JD, King LS. J Biol Chem. 2005;280:3590–3596. doi: 10.1074/jbc.M411038200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nielsen S, Digiovanni SR, Christensen EI, Knepper MA, Harris HW. Proc Natl Acad Sci USA. 1993;90:11663–11667. doi: 10.1073/pnas.90.24.11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marinelli RA, Pham L, Agre P, LaRusso NF. J Biol Chem. 1997;272:12984–12988. doi: 10.1074/jbc.272.20.12984. [DOI] [PubMed] [Google Scholar]
- 4.Deen PMT, Verdijk MAJ, Knoers N. V. A. M., Wieringa B, Monnens LAH, van Os CH, van Oost BA. Science. 1994;264:92–95. doi: 10.1126/science.8140421. [DOI] [PubMed] [Google Scholar]
- 5.Marples D, Christensen S, Christensen EI, Ottosen PD, Nielsen S. J Clin Invest. 1995;95:1838–1845. doi: 10.1172/JCI117863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frokiaer J, Marples D, Knepper MA, Nielsen S. Am J Med Sci. 1998;316:291–299. doi: 10.1097/00000441-199811000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Katsura T, Gustafson CE, Ausiello DA, Brown D. Am J Physiol. 1997;41:F816–F822. [PubMed] [Google Scholar]
- 8.Kamsteeg EJ, Heijnen I, van Os CH, Deen PMT. J Cell Biol. 2000;151:919–930. doi: 10.1083/jcb.151.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Balkom BWM, Savelkoul PJ, markovich D, Hofman E, Nielsen S, van der Sluijs P, Deen PMT. J Biol Chem. 2002;277:41473–41479. doi: 10.1074/jbc.M207525200. [DOI] [PubMed] [Google Scholar]
- 10.De Mattia F, Savelkoul PJ, Kamsteeg EJ, Konings IB, van der SP, Mallmann R, Oksche A, Deen PM. J Am Soc Nephrol. 2005;16:2872–2880. doi: 10.1681/ASN.2005010104. [DOI] [PubMed] [Google Scholar]
- 11.McDill BW, Li SZ, Kovach PA, Ding L, Chen F. Proc Natl Acad Sci USA. 2006;103:6952–6957. doi: 10.1073/pnas.0602087103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knepper MA, Nielsen S. Am J Physiol. 1993;265:F214–F224. doi: 10.1152/ajprenal.1993.265.2.F214. [DOI] [PubMed] [Google Scholar]
- 13.Nielsen S, Muller J, Knepper MA. Am J Physiol. 1993;265:F225–F238. doi: 10.1152/ajprenal.1993.265.2.F225. [DOI] [PubMed] [Google Scholar]
- 14.Katsura T, Verbavatz JM, Farinas J, Ma T, Ausiello DA, Verkman AS, Brown D. Proc Natl Acad Sci USA. 1995;92:7212–7216. doi: 10.1073/pnas.92.16.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nielsen S, Chou CL, Marples D, Christensen EI, Kishore BK, Knepper MA. Proc Natl Acad Sci USA. 1995;92:1013–1017. doi: 10.1073/pnas.92.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deen PMT, Van Balkom BWM, Kamsteeg EJ. J Cell Biol. 2000;79:523–530. doi: 10.1078/0171-9335-00075. Eur. [DOI] [PubMed] [Google Scholar]
- 17.Zelenina M, Christensen BM, Palmer J, Nairn AC, Nielsen S, Aperia A. Am J Physiol. 2000;278:F388–F394. doi: 10.1152/ajprenal.2000.278.3.F388. [DOI] [PubMed] [Google Scholar]
- 18.Nejsum LN, Zelenina M, Aperia A, Frokiaer J, Nielsen S. Am J Physiol Renal Physiol. 2005;288:F930–F938. doi: 10.1152/ajprenal.00291.2004. [DOI] [PubMed] [Google Scholar]
- 19.Galan JM, Haguenauer-Tsapis R. EMBO J. 1997;16:5847–5854. doi: 10.1093/emboj/16.19.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, Beguinot L, Geiger B, Yarden Y. Genes Dev. 1998;12:3663–3674. doi: 10.1101/gad.12.23.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haglund K, Dikic I. EMBO J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A. Mol Cell. 2006;21:737–748. doi: 10.1016/j.molcel.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 23.Deen PMT, Rijss JPL, Mulders SM, Errington RJ, van Baal J, van Os CH. J Am Soc Nephrol. 1997;8:1493–1501. doi: 10.1681/ASN.V8101493. [DOI] [PubMed] [Google Scholar]
- 24.Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Nat Cell Biol. 2003;5:461–466. doi: 10.1038/ncb983. [DOI] [PubMed] [Google Scholar]
- 25.Kleijmeer MJ, Morkowski S, Griffith JM, Rudensky AY, Geuze HJ. J Cell Biol. 1997;139:639–649. doi: 10.1083/jcb.139.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sachse M, Urbe S, Oorschot V, Strous GJ, Klumperman J. Mol Biol Cell. 2002;13:1313–1328. doi: 10.1091/mbc.01-10-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sachse M, Ramm G, Strous G, Klumperman J. Histochem Cell Biol. 2002;117:91–104. doi: 10.1007/s00418-001-0348-0. [DOI] [PubMed] [Google Scholar]
- 28.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 29.Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, Di Fiore PP, Polo S. Proc Natl Acad Sci USA. 2005;102:2760–2765. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katzmann DJ, Babst M, Emr SD. Cell. 2001;106:145–155. doi: 10.1016/s0092-8674(01)00434-2. [DOI] [PubMed] [Google Scholar]
- 31.Katzmann DJ, Odorizzi G, Emr SD. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- 32.Glickman MH, Ciechanover A. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 33.Wade JB, Stetson DL, Lewis SA. Ann NY Acad Sci. 1981;372:106–117. doi: 10.1111/j.1749-6632.1981.tb15464.x. [DOI] [PubMed] [Google Scholar]
- 34.Fushimi K, Uchida S, Hara Y, Hirata Y, Marumo F, Sasaki S. Nature. 1993;361:549–552. doi: 10.1038/361549a0. [DOI] [PubMed] [Google Scholar]
- 35.Shukla A, Hager H, Corydon TJ, Bean AJ, Dahl R, Vajda Z, Li H, Hoffmann HJ, Nielsen S. Am J Physiol. 2001;281:F546–F556. doi: 10.1152/ajprenal.2001.281.3.F546. [DOI] [PubMed] [Google Scholar]
- 36.Polo S, Sigismund S, Faretta M, Guidi M, Capua MR, Bossi G, Chen H, De Camilli P, Di Fiore PP. Nature. 2002;416:451–455. doi: 10.1038/416451a. [DOI] [PubMed] [Google Scholar]
- 37.Kanno K, Sasaki S, Hirata Y, Ishikawa S, Fushimi K, Nakanishi S, Bichet DG, Marumo F. N Engl J Med. 1995;332:1540–1545. doi: 10.1056/NEJM199506083322303. [DOI] [PubMed] [Google Scholar]
- 38.Pisitkun T, Shen RF, Knepper MA. Proc Natl Acad Sci USA. 2004;101:13368–13373. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hendriks G, Koudijs M., van Balkom BW, Oorschot V, Klumperman J, Deen PMT, van der Sluijs P. J Biol Chem. 2004;279:2975–2983. doi: 10.1074/jbc.M310767200. [DOI] [PubMed] [Google Scholar]
- 40.Katsura T, Ausiello DA, Brown D. Am J Physiol. 1996;39:F548–F553. doi: 10.1152/ajprenal.1996.270.3.F548. [DOI] [PubMed] [Google Scholar]
- 41.Hicke L. Cell. 2001;106:527–530. doi: 10.1016/s0092-8674(01)00485-8. [DOI] [PubMed] [Google Scholar]
- 42.Hryciw DH, Ekberg J, Lee A, Lensink IL, Kumar S, Guggino WB, Cook DI, Pollock CA, Poronnik P. J Biol Chem. 2004;279:54996–55007. doi: 10.1074/jbc.M411491200. [DOI] [PubMed] [Google Scholar]
- 43.Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D. EMBO J. 1997;16:6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin DH, Sterling H, Wang Z, Babilonia E, Yang B, Dong K, Hebert SC, Giebisch G, Wang WH. Proc Natl Acad Sci USA. 2005;102:4306–4311. doi: 10.1073/pnas.0409767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE. J Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}