

Abstract
Actin's functional complexity makes it a likely target of oxidative stress but also places it in a prime position to coordinate the response to oxidative stress. We have previously shown that the NADPH oxidoreductase Oye2p protects the actin cytoskeleton from oxidative stress. Here we demonstrate that the physiological consequence of actin oxidation is to accelerate cell death in yeast. Loss of Oye2p leads to reactive oxygen species accumulation, activation of the oxidative stress response, nuclear fragmentation and DNA degradation, and premature chronological aging of yeast cells. The oye2Δ phenotype can be completely suppressed by removing the potential for formation of the actin C285-C374 disulfide bond, the likely substrate of the Oye2p enzyme or by treating the cells with the clinically important reductant N-acetylcysteine. Because these two cysteines are coconserved in all actin isoforms, we theorize that we have uncovered a universal mechanism whereby actin helps to coordinate the cellular response to oxidative stress by both sensing and responding to oxidative load.
INTRODUCTION
The accumulation of oxygen-containing free radicals in the cell results in oxidative stress conditions that can cause apoptosis and cellular aging. The actin cytoskeleton is an early target of cellular oxidative stress (Dalle-Donne et al., 2001) and in certain disease conditions, the oxidative state of actin in the cell is very different from normal. For example, in sickle cell crisis, a major factor that contributes to the inflexibility of irreversibly sickled cells (ISCs) is the formation of an intracellular disulfide bond between C284 and C373 of β-actin (Shartava et al., 1995; Bencsath et al., 1996). We have previously shown that the yeast actin cytoskeleton is subject to the same form of oxidative damage as ISC actin. Our finding that an oxidoreductase called Oye2p (old yellow enzyme) regulates oxidation between C285 and C374 in Saccharomyces cerevisiae suggests that actin oxidation takes place in all eukaryotic cells and that the actin cytoskeleton is subject to redox regulation (Haarer and Amberg, 2004). That act1C285A- and act1C374A-bearing mutants are more resistant to oxidative stress than wild-type strains and that these alleles completely suppress the oxidative sensitivity of an old yellow enzyme (OYE) null strain proves that actin is a critical target for determining sensitivity to oxidative stress.
Actin's involvement in programmed cell death is gaining greater recognition in the apoptosis and cancer research fields (Rao and Li, 2004). Altering actin dynamics in lymphocytes by either stabilizing or destabilizing F-actin structures modulates apoptotic signaling upon withdrawal of growth factors (Posey and Bierer, 1999). In addition, during TNF-α apoptotic signaling, viral disruption at the β-actin locus dramatically reduces mitochondrial clustering and production of reactive oxygen species (ROS), indicating that actin participates in a programmed cell death program (Li et al., 2004). Furthermore, hyperstabilization of actin by addition of a drug called jasplakinolide (Jas) induced apoptosis in HL-60 cells, and blocking actin polymerization inhibited camptothesin-induced apoptosis, suggesting that the polymerization status of the actin cytoskeleton is crucial to apoptotic initiation and progression (Rao et al., 1999).
In recent years, the yeast model has been emerging as an important system to better understand the ancestrally conserved mechanisms of regulated cell death such as apoptosis, autophagy, and necrosis. The validity of the yeast model for studying programmed cell death is bolstered by the observations that yeast cells exhibit apoptotic markers in common with mammalian cells such as chromatin condensation, DNA fragmentation, phosphatidylserine exposure, and ROS accumulation (Madeo et al., 1999). In S. cerevisiae, decreased actin dynamics has been shown to increase the accumulation of ROS (Gourlay et al., 2004). Because the redox state of actin has been observed in some systems to be an important contributor to actin stability (Shartava et al., 1995, 1997; Haarer and Amberg, 2004), in the current study we asked whether the oxidation state of actin's cysteines 285 and 374 was able to regulate cell death in S. cerevisiae. Our data suggest that the oxidoreductase Oye2p plays a prosurvival role in the cell by protecting the actin cytoskeleton from oxidation-induced hyperstabilization. Our data support a model in which the actin cytoskeleton is a central signaling component that couples the accumulation of ROS to programmed cell death.
MATERIALS AND METHODS
Construction of Strains and Media Conditions
All yeast strains used in this study are of the S288C background. Yeast strains are listed in Table 1. Standard methods were used for growth, sporulation, transformation, and genetic analysis of yeast (Amberg et al., 2005).
Table 1.
Saccharomyces cerevisiae strains used in this study
| Name | Genotype | Source |
|---|---|---|
| DAY111x112 | MATa/MATα ura3-52/ura3-52 leu2Δ1/leu2Δ1 trp1Δ63/trp1Δ63 his3Δ200/his3Δ200 | D. Amberg |
| DAY127x128 | MATa/MATα ura3-52/ura3-52 leu2Δ1/leu2Δ1 trp1Δ63/trp1Δ63 his3Δ200/his3Δ200 oye2-Δ2::HIS3 /oye2-Δ2::HIS3 oye3-Δ2::TRP1/oye3-Δ2::TRP1 | Haarer and Amberg (2004) |
| DAY111 | MATaura3-52 leu2Δ1 trp1Δ63 his3Δ200 | Haarer and Amberg (2004) |
| DAY119 | MATaura3-52 leu2Δ1 trp1Δ63 his3Δ200 oye2-Δ2::HIS3 | Haarer and Amberg (2004) |
| DAY123 | MATaura3-52 leu2Δ1 trp1Δ63 his3Δ200 oye3-Δ2::TRP1 | Haarer and Amberg (2004) |
| DAY128 | MATaura3-52 leu2Δ1 trp1Δ63 his3Δ200 oye2-Δ2::HIS3 oye3-Δ2::TRP1 | Haarer and Amberg (2004) |
| MDY11 | MATaura3-52 leu2Δ1 trp1Δ63 his3Δ200 oye2-Δ2::HIS3 act1C374A::HIS3 | This study |
| MDY13 | MATaura3-52 leu2Δ1trp1Δ63 his3Δ200 oye2-Δ2::HIS3 act1C285A::HIS3 | This study |
| DAY173 | MATaura3-52 leu2Δ1 trp1Δ63 his3Δ200 oye2-Δ2::HIS3 oye3-Δ2::TRP1 act1C285A::HIS3 | Haarer and Amberg (2004) |
| DAY175 | MATaura3-52 leu2Δ1 trp1Δ63 his3Δ200 oye2-Δ2::HIS3 oye3-Δ2::TRP1 act1C374A::HIS3 | Haarer and Amberg (2004) |
| DAY169 | MATaura3-52 leu2ΔΔΔ200 act1C374A::HIS3 | Haarer and Amberg (2004) |
| DAY171 | MATaura3-52 leu2Δ1 trp1Δ63 his3Δ200 act1C285A::HIS3 | Haarer and Amberg (2004) |
| BHY313 | MATα his3Δ1 lys2Δ0 uraΔ0 act1-123::klURA3 mfa1Δ::PMFA1-sphis5+, can1Δ | B. Haarer |
Generation of rho0 Mutants
The method used to generate rho0 strains is as described (Goldring et al., 1970). In addition to streaking all colonies to YP + 3% glycerol to confirm lack of mitochondrial respiratory function, DAPI staining was performed as described to confirm the complete absence of mitochondrial DNA (Amberg et al., 2005).
Detection of Accumulation of ROS
Cultures were grown to logarithmic phase (about 3 × 107 cells/ml) and ROS were detected in vivo with the ROS-sensing dye dichlorodihydrofluorescein diacetate (H2DCFDA; Sigma, St. Louis, MO) as previously described (Madeo et al., 1999), except the cells were incubated for 3.5 h. Cells that appeared fluorescent in the FITC channel were scored as ROS positive. Cells (n = 500) were quantified for each experiment. The experiment was repeated at least three independent times for all strains, the number of cells that appeared fluorescent was averaged, and SDs were calculated.
Microscopy
The staining of the actin cytoskeleton and microscopy methods were performed as described (Amberg et al., 2005).
Localization of GFP-Yap1p
The URA3-marked, low copy CEN vector pGFP-Yap1p was obtained from the Moie-Rowley lab (Coleman et al., 1999). Transformed cells were grown in 3 ml SC-Ura medium until cells reach a density of ∼1 × 108 cells/ml. Cells were stained with 0.5 μg/ml DAPI for 15 min and observed for green fluorescent protein (GFP) fluorescence on a Zeiss Axioskop 2 MOT microscope (Thornwood, NY; Amberg et al., 2005). Two hundred cells of each strain were scored for nuclear GFP-Yap1p localization.
Detection of Chromatin Fragmentation
DAPI staining was used to detect changes in nuclear morphology. Cells were grown in YPD medium to logarithmic phase (at a density of 3 × 107 cells/ml) and stained as described (Amberg et al., 2005). Two hundred cells were quantified for the appearance of fragmented nuclei, and the experiment was repeated three independent times.
Detection of DNA Fragmentation
Cells were grown in YPD medium to logarithmic phase (3 × 107 cells/ml) and then fixed on a slide according to an immunofluorescence protocol (Amberg et al., 2005). Cells were then permeabilized with a solution of 0.1% Triton X-100 and 0.1% sodium citrate. After this, the In Situ Cell Death Detection Kit, Fluorescein (Susin et al., 1999) was used to label nicked DNA ends. Cells were then mounted in immunofluorescence mounting solution containing DAPI (Amberg et al., 2005) and visualized by fluorescence microscopy.
Actin Staining of N-acetyl-l-Cysteine–treated Cells
While cells were growing in YPD in log phase, N-acetyl-l-cysteine (NAC) was added to a final concentration of 31 mM, and the cells were incubated for an additional 5.5 h. Cells were fixed and stained with rhodamine phalloidin as described (Amberg et al., 2005).
Determination of Cell Viability
A fresh colony of each strain was inoculated into 5 ml of YPD in a well-aerated culture tube. For the length of the experiment, cultures were maintained on a rotating drum in a 30°C incubator. At the indicated time points, cells were gently sonicated with a Branson sonifier 450 (Branson Ultrasonics, Danbury, CT), and duplicate serial dilutions were made in YPD. The cells were diluted by a total factor of 106 of which 100 μl was plated onto YPD medium. Cell densities were obtained by making two 1:100 dilutions from each culture and then counting the cell numbers in duplicate in a hemacytometer. When a maximum cell density was obtained, this number was used as the cell density for all subsequent calculations of cell viability.
Cell viability was calculated as follows: Average colony forming units/maximum average cell density = % viability at each time point. Each experiment was repeated three times.
For the viability experiments that utilized NAC, 5-ml cultures of cells were grown to stationary phase and at day 2, and the cultures were split into separate tubes (control and experimental). Cells were plated at days 2, 5, 8, 11, 14, and 18. An aqueous solution of NAC was added to a final concentration of 31 mM to cultures at either day 4 or day 6. An equivalent volume of water was added to the control sample. The fraction of viable cells was calculated as described above.
Statistical Methods
The calculation of the arithmetic mean, x̄, was done as follows:
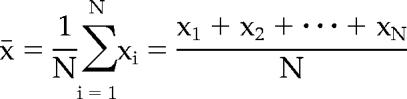 |
where xN represents each individual value and n is the total number of values.
The calculation of the SD, σ, was performed as follows:
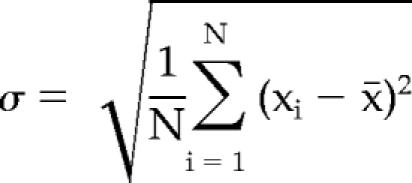 |
where σ is the SD, N is the total number of values, x̄ is the arithmetic mean, and xi is each individual value.
RESULTS
The Accumulation of ROS in Cells Lacking Oye2p Is Suppressed by act1C285A and act1C374A
ROS are central mediators of apoptosis in yeast and mammalian cells (Madeo et al., 1999; Chandra et al., 2000), being both necessary and sufficient to drive cells into programmed cell death (Madeo et al., 1999). We have previously shown that oye2Δ cells are sensitive to external sources of oxidative stress and that the recovery of their actin cytoskeleton and growth after oxidative stress is delayed in comparison to wild-type cells. Here we asked whether oye2Δ cells have elevated levels of ROS in vivo by quantifying the percentages of logarithmically growing cells that stain with the fluorescent, ROS indicator dye, H2DCFDA. Examples of ROS negative wild-type versus ROS-positive oye2Δ cells are shown in the inset of Figure 1. To confirm that the ROS-sensing dye, H2DCFDA, was specifically reacting with ROS in our cells, we showed that addition of the antioxidant NAC to the oye2Δ cells blocked H2DCFDA-induced fluorescence (our unpublished observations). As shown in Figure 1, there is an ∼30 and 20% increase in the number of H2DCFDA reactive cells in oye2Δ and oye3Δ cells, respectively, compared with wild-type cultures. In an oye2Δ strain, but not the oye3Δ strain, we were able to suppress increased ROS levels with either the act1C285A or the act1C374A alleles. These data suggest that Oye2p, but not Oye3p, is playing a specific role in protecting cells from an elevation of ROS that is in fact induced by the oxidation of the C285-C374 cysteine pair in actin. Consistent with these observations, our previous studies showed that Oye3p is unable to interact with actin and that oye3Δ cells have a normal actin cytoskeleton (Haarer and Amberg, 2004). The oye3Δ strain does show an elevation in ROS-positive cells that appears to be unrelated to actin oxidation. Consistent with these observations, the oye2Δ oye3Δ act1C374A and oye2Δ oye3Δ act1C285A strains have ROS levels equivalent to that of the oye3Δ strain. To examine the potential role of a direct interaction between actin and Oye2p, in regulating the in vivo accumulation of ROS, we examined a strain expressing a mutant form of actin (act1-123p) that we previously showed has a reduced affinity for Oye2p (Haarer and Amberg, 2004). As expected, the act1-123 strain showed a comparable percentage (27%) of ROS-positive cells to the oye2Δ strain.
Figure 1.

Actin oxidation in oyeΔ cells leads to ROS accumulation. Logarithmically growing cells were stained for 3.5 h with the ROS-sensing dye H2DCFDA, and the percentages of fluorescent cells was determined. □, rho+ cells; ▩, rho0 cells. The averages and SDs reported are for three to four experiments. Left inset, example of an ROS-negative wild-type cell; right inset, example of an ROS-positive oyeΔ cell.
Mitochondria have been accepted as the major source of ROS generation in the cell, specifically stemming from the electron transfer complexes of the respiratory chain (Fleury et al., 2002). To examine whether the mitochondrial respiratory chain is the source of oxygen-containing free radicals in our ROS-elevated mutants, we generated strains lacking mitochondrial DNA (rho0). These strains were tested for lack of growth on glycerol (a nonfermentable carbon source) and confirmed by DAPI staining to be devoid of mitochondrial DNA (data not shown) and are therefore lacking a functional mitchondrial respiratory chain. The loss of mitochondrial function was especially useful in identifying the major source of ROS generated in cells bearing the F-actin–stabilizing act1–159 allele (Gourlay et al., 2004). As shown in Figure 1, ROS-accumulating cells in oye2Δ and oye3Δ cultures were decreased by approximately half, indicating that half of the ROS in OYE-lacking mutants are generated from an origin other than the electron transport chain. We stained the actin cytoskeleton of rho0 oye2Δoye3Δ cells in order to examine if this level of oxidative stress was sufficient to cause oxidation-mediated stabilization of the actin cytoskeleton. We found that there was a partial suppression of the hyperstabilized actin phenotype of the OYE-lacking mutants (our unpublished observations) consistent with the observed decrease in ROS levels. As with our rho+ mutants, the number of rho0 oye2Δ cells with elevated ROS was suppressed to wild-type levels by both act1C285A and act1C374A. This suggests that the oxidative status of these cysteine residues of actin, and the consequent stabilization of the actin network, are key events in the mechanism governing ROS generation from both mitochondrial and nonmitochondrial sources.
Yap1p is a transcription factor that translocates to the nucleus upon exposure to oxidative stress (Coleman et al., 1999). It contains a carboxy-terminal cysteine-rich domain (c-CRD) that senses cytoplasmic oxidative stress and is necessary for cellular viability upon addition of external oxidants (Kuge et al., 1997; Wemmie et al., 1997). Furthermore, age-induced cell death has been shown to be delayed in Yap1p-overexpressing strains (Herker et al., 2004). As a second important indicator of oxidative stress, we found that in ∼32% of oye2Δ cells, GFP-Yap1p is localized to the nucleus in early stationary phase cells, whereas this nuclear localization pattern was observed in <1% of a comparable culture of wild-type cells (Figure 2). Importantly, in our double mutants oye2Δ act1C285A and oye2Δ act1C374A nuclear accumulation of Yap1p is suppressed to wild-type levels. These results indicate that in the absence of Oye2p, normal cellular levels of ROS lead to actin oxidation and a perception that the cell is under oxidative stress. This actin oxidation appears to trigger an elevation of ROS and in this way, actin appears to be both a sensor and an effector of ROS elevation.
Figure 2.

In the absence of Oye2p, actin oxidation activates the oxidative stress response. The oxidative stress responsive transcription factor Yap1p was expressed as a GFP fusion in various mutants and the percentages of cells with nuclear GFP-Yap1p was quantified by fluorescence microscopy. Nuclei were identified by DAPI staining. The results reported are for three experiments (n = 500 per experiment). Top insets, wild-type cell; bottom insets, oxidatively stressed oye2Δ cell.
Nuclear and DNA Fragmentation in OYE2-lacking Cells Is Suppressed by the act1C285A and act1C374A Alleles
To assess whether ROS accumulation in oye2Δ cells is associated with apoptotic markers, we tested for an increase in nuclear/chromatin fragmentation and DNA degradation. DAPI staining revealed that oye2Δ cells had an increase in the percentage of cells containing fragmented nuclei: ∼14% of oye2Δ cells showed nuclear fragmentation compared with <1% of wild-type cells (see Figure 3A). As expected, the positive control (wild-type cells treated for 200 min with 3 mM H2O2) also showed increased chromatin fragmentation (Madeo et al., 1999). As shown in Figure 3A, the fragmentation observed in oye2Δ cells was completely suppressed by the act1C285A and act1C374A alleles.
Figure 3.

In the absence of Oye2p, cells show nuclear fragmentation and DNA degradation. (A) DAPI staining was performed on logarithmically growing cells, and the percentages of cells displaying nuclear fragmentation for each strain is shown. The results reported are for three independent experiments (n = 200 per experiment). Examples of cells displaying normal nuclear morphology (wild type) versus fragmented nuclei (oye2Δ) are shown in the inset on the top right. (B) TUNEL staining was performed on logarithmically growing cells. Each panel displays a random field of cells from cultures of the indicated genotypes. Left, DIC staining; middle, TUNEL staining; right, DAPI staining. As a positive control, wildtype cells were treated with 3 U/ml Dnase I for 20 min at room temperature (wt+DNaseI). White arrow, nuclear staining; white arrowhead, mitochondrial staining.
In addition to DAPI staining, a TUNEL assay that is used to detect free 3′-OH ends of DNA (Gavrieli et al., 1992; Madeo et al., 1997) was used to examine whether the cellular DNA in the oye2Δ mutant was intact. DNase-I treatment of wild-type cells showed that the TUNEL assay accurately identified cells that exhibited an increase in free DNA ends (Figure 3B). We further showed that oye2Δ cells, relative to wild-type cells, displayed a large increase in the intensity of nuclear TUNEL staining (as a punctate dot; see white arrow) as well as mitochondrial TUNEL staining (see white arrowhead for an example). In contrast, the TUNEL staining of the oye2Δact1C285A and oye2Δact1C374A double mutants was nearly identical to wild type, indicating that DNA fragmentation in oye2Δ cells was also suppressed by the act1C285A and act1C374A alleles.
Chronological Aging of OYE-lacking Mutants Is Suppressed by act1C285A and act1C374A
It has previously been observed that aging yeast cells display an apoptotic phenotype, showing markers such as increased ROS accumulation, DNA degradation, extracellular phosphatidylserine exposure, and yeast caspase activation (Herker et al., 2004). To determine if the observed ROS elevation in oye-deficient cells is triggering cell death, we monitored the viability of our strains maintained in YPD over a period of 18 d. The survival of yeast strains varies dramatically in different media conditions. Wild-type cells that are grown in synthetic complete minimal medium lose viability after 10 d (our unpublished observations). In contrast, cells that are grown and maintained in stationary phase on rich media can maintain viability for weeks (Sinclair et al., 1998; Chen et al., 2005). We found that there was a rapid decline in the viability of our oye2Δ cells compared with wild-type cells (Figure 4). During 18 d at stationary phase, both the oye2Δ and oye2Δ oye3Δ cell viabilities dramatically decreased from day 2 to day 5, reaching <20% by day 18 compared with 70% viability observed for wild-type cells. Note that the loss of OYE3 appears to have little additive effect, suggesting that the loss of OYE2 alone is causing the observed lowering in cell viability. Importantly, the premature aging of the oye2Δ (and the oye2Δoye3Δ cells) was suppressed by both the act1C285A and act1C374A alleles. These data support our model that in the absence of Oye2p, oxidation of actin's cysteine residues, and the resulting elevation of ROS, leads to premature aging and cell death.
Figure 4.

In the absence of Oye2p, actin oxidation leads to premature aging and cell death. Strains were grown to and maintained in stationary phase for 18 d. On the indicated days, cells were plated and the percentages of viable cells were determined based on colony-forming units per total cells. The averages and SDs were calculated from three experiments, where n = 400, n being the number of cell bodies plated.
The Antioxidant NAC Suppresses the Actin Defects and Loss of Viability Observed in oye-lacking Mutants
NAC is an antioxidant that is emerging as an important therapeutic agent for the treatment of many conditions. Importantly, NAC is in stage II clinical trials as a treatment for sickle cell patients. It is believed to act by reducing the oxidation of thiol groups in the β -actin and spectrin of irreversibly sickled RBCs restoring plasiticity to the cortical cytoskeleton (Goodman, 2004; Gibson et al., 1998).
To ask if NAC could reverse the effects of actin oxidation in yeast, we examined the phenotype of oye2Δoye3Δ cells after treatment with NAC. As can be seen in Figure 5A, the characteristic actin organization defects of the OYE deficient strain (hyperstabilization of actin cables and patches) were suppressed by the addition of NAC. We next tested whether the accelerated death of the oye2Δ cells in stationary phase could be suppressed by the addition of 31 mM NAC (Figure 5B). The addition of NAC at day 4 increased the percentage of oye2Δ viable cells at day 5 from 78 to 92%. By day 8, addition of NAC at either day 4 or day 6 increased untreated oye2Δ viability from 63% to 82 and 86%, respectively. However, after day 8, the viability of both NAC-treated oye2Δ and wild-type cultures rapidly drops off perhaps as a consequence of NAC or NAC byproduct toxicity. In support of this observation, another group has reported that at concentrations of NAC equal to or greater than 10 mM, the ability of the compound to promote growth in yeast mutants lacking superoxide dismutase was less than that observed at lower concentrations (Zyracka et al., 2005). This indicates that at high concentrations, the antioxidant is toxic to yeast. However, in our viability experiments we have used a relatively high concentration of NAC because it most dramatically suppressed the hyperstable actin phenotype associated with the OYE-lacking mutant, whereas a lower concentration (6.2 mM) did not (our unpublished observations). Nonetheless, it appears that NAC can transiently protect the actin cytoskeleton of yeast from oxidative stress and from actin oxidation-induced cell death 8 d into stationary phase.
Figure 5.

N-acetyl-l-cysteine (NAC) suppresses the actin organization and accelerated cell death phenotype of oye2Δ cells. (A) NAC was added to logarithmically growing cultures at a final concentration of 31 mM for 5.5 h, after which the cells were fixed, stained with rhodamine phalloidin and visualized by fluorescence microscopy. (B) NAC was added at a final concentration of 31 mM to stationary cultures on days 4 or 6 (indicated by arrows). Cells were removed at the indicated time points, dilutions were plated, and percent viability was calculated based on colony-forming units per total cell density. The arrows with dotted lines show the increases in viability at day 5 for NAC addition at day 4 and at day 8 for NAC addition at days 4 and 6. (C) The box highlights a portion of Figure 4B, with only the oye2Δ samples highlighted at days 2, 5, and 8.
DISCUSSION
Actin is perhaps one of the most functionally diverse proteins in eukaryotic cells. Traditionally the actin cytoskeleton is thought of as playing structural and organizational roles as well as a being a direct participant in a wide range of motility processes. Recently actin has also been found to be involved in many nuclear processes as well including regulation of chromatin structure and transcription (Olave et al., 2002; Bettinger et al., 2004; Percipalle and Visa, 2006). This functional diversity makes the actin cytoskeleton an ideal candidate for integrating signaling between diverse cellular processes. Previous work has not extended actin's functions to include signaling but the results presented here show that a distinct change in the actin oxidation state activates an oxidative stress response that can culminate in programmed cell death during aging. Concerning the work presented here, it has become appreciated that the proper regulation of actin dynamics is an important determinant of cell survival; hyperstabilization of actin filaments leads to an apoptotic phenotype followed by programmed cell death in both yeast and mammalian cells (Rao et al., 1999; Gourlay et al., 2004)
We have furthered these findings by showing that cysteines 285 and 374 of actin are important physiological sensors of intracellular oxidative stress and are regulators of programmed cell death during chronological aging. Specifically, the act1C285A and act1C374A alleles are able to suppress the intracellular accumulation of ROS, Yap1p nuclear localization, nuclear fragmentation, DNA degradation, and accelerated cell death observed in oyeΔ cells. We have also shown that both the hyperstabilized actin phenotype and rapid decline of chronologically aging oye cultures is suppressed by treatment with the clinically relevant antioxidant NAC. NAC is becoming recognized as an important treatment for HIV infection (Kalebic et al., 1991; Roederer et al., 1991; Herzenberg et al., 1997), cancer (Morini et al., 1999), neurodegenerative diseases (Deigner et al., 2000), and other diseases and conditions. It is believed to function by protecting against cell death/as a promoter of cell survival (Mayer and Noble, 1994). Importantly, it is currently in phase II trials for the treatment for sickle cell crisis (Goodman, 2004). Its efficacy in treating sickle cell crisis is based on its ability to reduce C284-C373 oxidized actin and return proper plasticity to the red blood cell (RBC) cytoskeleton (Gibson et al., 1998). The results reported here further suggest that NAC may act by suppressing an actin-induced cell death pathway. Erythrocytes are capable of undergoing a form of apoptosis known as eryptosis. In patients with sickle cell anemia, erythrocytes have been shown to have a 6 d half-life, compared with the 60 d half-life of normal RBCs (Benjamin and Manning, 1986). It is also worth noting that sickle cells generate twice the amount of ROS as control RBCs, an observation similar to that of OYE-lacking mutants (Hebbel et al., 1982). Future studies of the mechanism of action of NAC in the treatment of sickle cell anemia and other diseases may find that the oxidative state of actin plays a large part in the determination of survival of mammalian cells and that a key target of NAC is oxidized actin.
If actin's cysteines 285 and 374 can become disulfide-bonded and ultimately cause cell death, then why have they been coconserved in all eukaryote actin isoforms? We theorize that it may be to aid in the destruction of unfit cells that have accumulated excessive ROS and intracellular oxidative damage. In support of this theory of adaptive aging, superoxide has been proposed to mediate an altruistic aging and death program that leads to the death of >90% of a yeast population to allow for the survival of fitter/healthier cells (Fabrizio et al., 2004).
To begin to understand the cellular mechanisms by which ROS are being generated in oye2Δ cells, we have used cells lacking a functional respiratory chain to show that mitochondrial ROS account for approximately half of the total ROS accumulation in oye2Δ cells. Many studies have shown that mitochondria and actin functionally interact. Regulation of actin dynamics is required for proper mitochondrial inheritance and for maintenance of mitochondrial morphology (Boldogh et al., 2001; Okamoto and Shaw, 2005). The actin-depolymerizing protein gelsolin has been shown to inhibit the activity of the voltage-dependent anion channel (VDAC), the major channel in the outer membrane of mitochondria that regulates superoxide release, thereby preventing apoptotic mitochondrial changes in mammalian cells (Kusano et al., 2000). Actin itself has been shown to regulate apoptotic changes in Neurospora crassa, where monomeric actin, not the filamentous form, appears to regulate this channel's activity (Xu et al., 2001). Yeast also contains the VDAC in their mitochondria (Forte et al., 1987). One can easily imagine that the release of ROS through pores in the mitochondrial membrane of yeast and mammalian cells may be an event that is regulated by the dynamic state of actin. In our model, actin would be further targeted by mitochondrial ROS, and this would lead to greater stabilization of actin and even more spilling of ROS from mitochondria in a positive feedback loop leading to cell death. However, half of the ROS generated in oyeΔ cells appears to derive from nonmitochondrial sources. We do not currently know the source of this pool of ROS, but it is tempting to speculate that oxidized actin, interacting with cellular redox systems, directly results in the production of ROS.
Our findings that both nuclear and mitochondrial DNA in OYE2-lacking cells are stained with TUNEL suggests that the apoptotic mechanism involves the breakdown of both nuclear and mitochondrial DNA. Although the TUNEL assay is primarily used to label fragmented nuclear DNA, we have shown that fragmented mitochondrial DNA may also be labeled by this method. We speculate that the accumulation of ROS in the Oye2p-lacking mutant leads to mitochondrial fragmentation and that the converse may also be true. Indeed, it has been noted in mammalian cells that ROS generated from mitochondria are able to attack mitochondrial DNA, causing large-scale mtDNA deletions (Hayakawa et al., 1992).
Although some of the molecular details remain to be elucidated, the work presented here is consistent with a model in which cellular oxidative load is measured via the oxidative formation of the C285-C374 disulfide bond of actin. Under normal levels of ROS, old yellow enzyme 2 is able to keep pace in reducing the disulfide but when the load becomes too high, or perhaps if Oye2p is inhibited or the NADPH levels required to keep Oye2p reduced become limiting, the resulting accumulation of oxidized actin results in stabilization of the actin cytoskeleton. This in turn triggers further elevation of ROS, in part via spilling from the mitochondria, leading to greater oxidation and stabilization of actin, thereby establishing a positive feedback loop that ultimately leads to cell death.
ACKNOWLEDGMENTS
We thank B. Haarer for helpful suggestions, P. Kane and Y. Huang for helpful discussions and ideas, and all members of the Amberg laboratory for helpful input, and to the Kane laboratory for use of reagents. Finally, we thank S. Moye-Rowley for the GFP-YAP1 vector and K. Ayscough and C. Gourlay for technical assistance. This research was supported by National Institutes of Health Grant GM-56189.
Abbreviations used:
- NAC
N-acetyl-l-cysteine
- OYE
old yellow enzyme
- ROS
reactive oxygen species.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E06-08-0718) on February 7, 2007.
REFERENCES
- Amberg D. C., Burke D. J., Strathern J. N. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2005. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. [Google Scholar]
- Bencsath F. A., Shartava A., Monteiro C. A., Goodman S. R. Identification of the disulfide-linked peptide in irreversibly sickled cell beta-actin. Biochemistry. 1996;35:4403–4408. doi: 10.1021/bi960063n. [DOI] [PubMed] [Google Scholar]
- Benjamin L. J., Manning J. M. Enhanced survival of sickle erythrocytes upon treatment with glyceraldehyde. Blood. 1986;67:544–546. [PubMed] [Google Scholar]
- Bettinger B. T., Gilbert D. M., Amberg D. C. Actin up in the nucleus. Nat. Rev. Mol. Cell Biol. 2004;5:410–415. doi: 10.1038/nrm1370. [DOI] [PubMed] [Google Scholar]
- Boldogh I. R., Yang H. C., Pon L. A. Mitochondrial inheritance in budding yeast. Traffic. 2001;2:368–374. doi: 10.1034/j.1600-0854.2001.002006368.x. [DOI] [PubMed] [Google Scholar]
- Chandra J., Samali A., Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 2000;29:323–333. doi: 10.1016/s0891-5849(00)00302-6. [DOI] [PubMed] [Google Scholar]
- Chen Q., Ding Q., Keller J. N. The stationary phase model of aging in yeast for the study of oxidative stress and age-related neurodegeneration. Biogerontology. 2005;6:1–13. doi: 10.1007/s10522-004-7379-6. [DOI] [PubMed] [Google Scholar]
- Coleman S. T., Epping E. A., Steggerda S. M., Moye-Rowley W. S. Yap1p activates gene transcription in an oxidant-specific fashion. Mol. Cell. Biol. 1999;19:8302–8313. doi: 10.1128/mcb.19.12.8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I., Rossi R., Milzani A., Di Simplicio P., Colombo R. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic. Biol. Med. 2001;31:1624–1632. doi: 10.1016/s0891-5849(01)00749-3. [DOI] [PubMed] [Google Scholar]
- Deigner H. P., Haberkorn U., Kinscherf R. Apoptosis modulators in the therapy of neurodegenerative diseases. Expert Opin. Investig. Drugs. 2000;9:747–764. doi: 10.1517/13543784.9.4.747. [DOI] [PubMed] [Google Scholar]
- Fabrizio P., Battistella L., Vardavas R., Gattazzo C., Liou L. L., Diaspro A., Dossen J. W., Gralla E. B., Longo V. D. Superoxide is a mediator of an altruistic aging program in Saccharomyces cerevisiae. J. Cell Biol. 2004;166:1055–1067. doi: 10.1083/jcb.200404002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury C., Mignotte B., Vayssiere J. L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131–141. doi: 10.1016/s0300-9084(02)01369-x. [DOI] [PubMed] [Google Scholar]
- Forte M., Adelsberger-Mangan D., Colombini M. Purification and characterization of the voltage-dependent anion channel from the outer mitochondrial membrane of yeast. J. Membr. Biol. 1987;99:65–72. doi: 10.1007/BF01870622. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y., Sherman Y., Ben-Sasson S. A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson X. A., Shartava A., McIntyre J., Monteiro C. A., Zhang Y., Shah A., Campbell N. F., Goodman S. R. The efficacy of reducing agents or antioxidants in blocking the formation of dense cells and irreversibly sickled cells in vitro. Blood. 1998;91:4373–4378. [PubMed] [Google Scholar]
- Goldring E. S., Grossman L. I., Krupnick D., Cryer D. R., Marmur J. The petite mutation in yeast. Loss of mitochondrial deoxyribonucleic acid during induction of petites with ethidium bromide. J. Mol. Biol. 1970;52:323–335. doi: 10.1016/0022-2836(70)90033-1. [DOI] [PubMed] [Google Scholar]
- Goodman S. R. The irreversibly sickled cell: a perspective. Cell Mol. Biol. (Noisy-le-grand) 2004;50:53–58. [PubMed] [Google Scholar]
- Gourlay C. W., Carpp L. N., Timpson P., Winder S. J., Ayscough K. R. A role for the actin cytoskeleton in cell death and aging in yeast. J. Cell Biol. 2004;164:803–809. doi: 10.1083/jcb.200310148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarer B. K., Amberg D. C. Old yellow enzyme protects the actin cytoskeleton from oxidative stress. Mol. Biol. Cell. 2004;15:4522–4531. doi: 10.1091/mbc.E04-06-0445. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hayakawa M., Hattori K., Sugiyama S., Ozawa T. Age-associated oxygen damage and mutations in mitochondrial DNA in human hearts. Biochem. Biophys. Res. Commun. 1992;189:979–985. doi: 10.1016/0006-291x(92)92300-m. [DOI] [PubMed] [Google Scholar]
- Hebbel R. P., Eaton J. W., Steinberg M. H., White J. G. Erythrocyte/endothelial interactions in the pathogenesis of sickle-cell disease: a “real logical” assessment. Blood Cells. 1982;8:163–173. [PubMed] [Google Scholar]
- Herker E., Jungwirth H., Lehmann K. A., Maldener C., Frohlich K. U., Wissing S., Buttner S., Fehr M., Sigrist S., Madeo F. Chronological aging leads to apoptosis in yeast. J. Cell Biol. 2004;164:501–507. doi: 10.1083/jcb.200310014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzenberg L. A., De Rosa S. C., Dubs J. G., Roederer M., Anderson M. T., Ela S. W., Deresinski S. C. Glutathione deficiency is associated with impaired survival in HIV disease. Proc. Natl. Acad. Sci. USA. 1997;94:1967–1972. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalebic T., Kinter A., Poli G., Anderson M. E., Meister A., Fauci A. S. Suppression of human immunodeficiency virus expression in chronically infected monocytic cells by glutathione, glutathione ester, and N-acetylcysteine. Proc. Natl. Acad. Sci. USA. 1991;88:986–990. doi: 10.1073/pnas.88.3.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuge S., Jones N., Nomoto A. Regulation of yAP-1 nuclear localization in response to oxidative stress. EMBO J. 1997;16:1710–1720. doi: 10.1093/emboj/16.7.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano H., Shimizu S., Koya R. C., Fujita H., Kamada S., Kuzumaki N., Tsujimoto Y. Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene. 2000;19:4807–4814. doi: 10.1038/sj.onc.1203868. [DOI] [PubMed] [Google Scholar]
- Li J., Li Q., Xie C., Zhou H., Wang Y., Zhang N., Shao H., Chan S. C., Peng X., Lin S. C., Han J. Beta-actin is required for mitochondria clustering and ROS generation in TNF-induced, caspase-independent cell death. J. Cell Sci. 2004;117:4673–4680. doi: 10.1242/jcs.01339. [DOI] [PubMed] [Google Scholar]
- Madeo F., Frohlich E., Frohlich K. U. A yeast mutant showing diagnostic markers of early and late apoptosis. J. Cell Biol. 1997;139:729–734. doi: 10.1083/jcb.139.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F., Frohlich E., Ligr M., Grey M., Sigrist S. J., Wolf D. H., Frohlich K. U. Oxygen stress: a regulator of apoptosis in yeast. J. Cell Biol. 1999;145:757–767. doi: 10.1083/jcb.145.4.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M., Noble M. N-acetyl-L-cysteine is a pluripotent protector against cell death and enhancer of trophic factor-mediated cell survival in vitro. Proc. Natl. Acad. Sci. USA. 1994;91:7496–7500. doi: 10.1073/pnas.91.16.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morini M., Cai T., Aluigi M. G., Noonan D. M., Masiello L., De Flora S., D'Agostini F., Albini A., Fassina G. The role of the thiol N-acetylcysteine in the prevention of tumor invasion and angiogenesis. Int. J. Biol. Markers. 1999;14:268–271. doi: 10.1177/172460089901400413. [DOI] [PubMed] [Google Scholar]
- Okamoto K., Shaw J. M. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- Olave I. A., Reck-Peterson S. L., Crabtree G. R. Nuclear actin and actin-related proteins in chromatin remodeling. Annu. Rev. Biochem. 2002;71:755–781. doi: 10.1146/annurev.biochem.71.110601.135507. [DOI] [PubMed] [Google Scholar]
- Percipalle P., Visa N. Molecular functions of nuclear actin in transcription. J. Cell Biol. 2006;172:967–971. doi: 10.1083/jcb.200512083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey S. C., Bierer B. E. Actin stabilization by jasplakinolide enhances apoptosis induced by cytokine deprivation. J. Biol. Chem. 1999;274:4259–4265. doi: 10.1074/jbc.274.7.4259. [DOI] [PubMed] [Google Scholar]
- Rao J., Li N. Microfilament actin remodeling as a potential target for cancer drug development. Curr. Cancer Drug Targets. 2004;4:345–354. doi: 10.2174/1568009043332998. [DOI] [PubMed] [Google Scholar]
- Rao J. Y., Jin Y. S., Zheng Q., Cheng J., Tai J., Hemstreet G. P., 3rd Alterations of the actin polymerization status as an apoptotic morphological effector in HL-60 cells. J. Cell Biochem. 1999;75:686–697. [PubMed] [Google Scholar]
- Roederer M., Raju P. A., Staal F. J., Herzenberg L. A. N-acetylcysteine inhibits latent HIV expression in chronically infected cells. AIDS Res. Hum. Retroviruses. 1991;7:563–567. doi: 10.1089/aid.1991.7.563. [DOI] [PubMed] [Google Scholar]
- Shartava A., Korn W., Shah A. K., Goodman S. R. Irreversibly sickled cell beta-actin: defective filament formation. Am. J. Hematol. 1997;55:97–103. doi: 10.1002/(sici)1096-8652(199706)55:2<97::aid-ajh8>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Shartava A., Monteiro C. A., Bencsath F. A., Schneider K., Chait B. T., Gussio R., Casoria-Scott L. A., Shah A. K., Heuerman C. A., Goodman S. R. A posttranslational modification of beta-actin contributes to the slow dissociation of the spectrin-protein 4.1-actin complex of irreversibly sickled cells. J. Cell Biol. 1995;128:805–818. doi: 10.1083/jcb.128.5.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair D., Mills K., Guarente L. Aging in Saccharomyces cerevisiae. Annu. Rev. Microbiol. 1998;52:533–560. doi: 10.1146/annurev.micro.52.1.533. [DOI] [PubMed] [Google Scholar]
- Susin S. A., et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Wemmie J. A., Steggerda S. M., Moye-Rowley W. S. The Saccharomyces cerevisiae AP-1 protein discriminates between oxidative stress elicited by the oxidants H2O2 and diamide. J. Biol. Chem. 1997;272:7908–7914. doi: 10.1074/jbc.272.12.7908. [DOI] [PubMed] [Google Scholar]
- Xu X., Forbes J. G., Colombini M. Actin modulates the gating of Neurospora crassa VDAC. J. Membr. Biol. 2001;180:73–81. doi: 10.1007/s002320010060. [DOI] [PubMed] [Google Scholar]
- Zyracka E., Zadrag R., Koziol S., Krzepilko A., Bartosz G., Bilinski T. Ascorbate abolishes auxotrophy caused by the lack of superoxide dismutase in Saccharomyces cerevisiae. Yeast can be a biosensor for antioxidants. J. Biotechnol. 2005;115:271–278. doi: 10.1016/j.jbiotec.2004.09.003. [DOI] [PubMed] [Google Scholar]