

Abstract
Corticotropin releasing hormone (CRH), a messenger of stress at the central level, is expressed in the epidermis where it operates within local equivalent of hypothalamo-pituitary axis. CRH inhibits NF-κB activity in human immortalized epidermal (PIG1) melanocytes. In melanocytes CRH stimulates pro-opiomelanocortin (POMC) mRNA and adrenocorticotropin (ACTH) peptide production. Knockdown of POMC levels by transfecting cells with antisense oligonucleotides blocks the effect of CRH on NF-κB signaling indicating that the above inhibition is indirect, e.g. through activation of POMC. We suggest that induction of POMC by CRH serves as a feedback mechanism to self-restrict inflammatory response in the skin.
Keywords: CRH, Melanocyte, NF-κB, POMC
1. Introduction
Corticotropin releasing hormone (CRH), a 41-amino acid long peptide [40] is a main trigger of the hypothalamo–pituitary–adrenal (HPA) axis [6,14]. Stress signals in the hypothalamus stimulate expression and release of CRH, resulting in pro-opiomelanocortin (POMC) expression and adrenocorticotropin (ACTH) release by the pituitary [6]. ACTH elicits cortisol release by adrenal cortex that is responsible for attenuation of the stress response at the central and peripheral level [6].
CRH is also produced in peripheral tissues, including the epidermis, where it can act as a regulatory element of local neuroendocrine interactions [28–30,35]. Nevertheless, the functional significance of CRH expression in peripheral tissues is still unresolved. CRH has been described to act as a growth factor [16,34], apoptosis regulator [3,7] and differentiation factor [8,9,22,46]. CRH also acts as pro-inflammatory factor, since it stimulates degranulation of mast cells and increases vascular permeability [38]. CRH also stimulates vascular endothelial growth factor release [4], IL-6 release by keratinocytes [43] and IL-1β release by monocytes [25]. CRH also has an anti-inflammatory activity since it inhibits release of IL-1β by keratinocytes [43], IL-1 and IL-6 by monocytes and mono-nuclear cells [11,25]. Thus, effects of CRH are dependent on the cell type and on the experimental conditions.
Production of inflammatory cytokines is regulated by several transcription factors including NF-κB [21]. CRH was shown to stimulate [47] as well as inhibit [15] NF-κB activity, indicating that its effects vary depending on the model used. Of note, effects of CRH on the stimulation of POMC levels have been linked to NF-κB signaling in pituitary AtT-20 cells [15].
Melanocytes regulate many epidermal processes. Most notably, their dendrites deliver melanin granules as well as a plethora of proteins, peptides and biogenic amines [31]. Each of these substances has the potential to regulate function of neighboring keratinocytes. Melanocytes also produce several inflammatory cytokines [17] such as IL-1β, IL-6, IL-8, TNF-α, GM-CSF, whose expression is regulated by NF-κB [39]. To this end, these cytokines participate in the inflammatory reaction in the epidermis. Primary human melanocytes also produce effectors of the HPA axis, i.e., ultraviolet B (UVB) treatment results in CRH expression and production [27], CRH treatment results in POMC expression and ACTH production, as wells as in cortisol and corticosterone release (this effect is ACTH-mediated) [33]. Based on the above we examined: (1) the effects of CRH on NF-κB in the melanocytes (since it is a transcription factor regulating production of inflammatory cytokines); (2) whether CRH-triggered downstream elements of HPA axis (e.g. POMC-derived ACTH or cortisol) are involved in the CRH mediated regulation of NF-κB signaling.
2. Materials and methods
2.1. Cell culture
The immortalized normal human epidermal melanocyte line PIG1, a gift of Dr. C. Le Poole, Loyola Medical Center, Maywood, IL [19], was maintained at 37 °C in medium 154 with Human Melanocyte Growth Supplement and antibiotics. For experiments PIG1 melanocytes were seeded at 10 000/cm2 cells in growth medium. After 24 h, the cells were washed with phosphate buffered saline (PBS) and refed with EpiLife medium with Epilife Defined Growth Supplement (EDGS) and antibiotics, which did not contain bovine pituitary extract (BPE). Melanocyte culture products were purchased from Cascade Biologics Inc., Portland, OR. After another 24 h, the medium was changed to serum free Ham’s F10 medium (Cellgro, Mediatech Inc., Herndon, VA). CRH (Sigma, St. Louis, MO) was added at the time of addition of Ham’s F10 medium and 12 h later. The cells were incubated for total of 24 h.
2.2. NF-κB assays
Melanocytes were transfected using transfection reagents (sc-29528 and sc-36868, Santa Cruz Biotechnology Inc., Santa Cruz, CA) in EpiLife medium with EDGS with firefly luciferase reporter gene plasmid pNF-κB-Luc containing 4× NF-κB cis elements (5′-GGGGACTTTCC-3′) and with phRL-TK (it expressed Renilla luciferase and served as normalization control; Promega, Madison, WI). pNF-κB-Luc and pP1-Luc (control without κB sites, empty vector) plasmids were constructed as described previously [30,45]. At 24 h after transfection, the cells cultures were incubated in serum-free medium with or without CRH for 24 h. Then, cells were lysed and luciferase and Renilla luciferase signals were recorded after sequential addition of Luciferase Assay Reagent II and Stop-Glo Reagent (Promega, Madison, WI) using TD-20/20 luminometer (Turner Designs, Sunnyvale, CA). After subtraction of background, specific signal was normalized to the Renilla signal. Obtained values were divided by mean of control (cells transfected with NF-κB construct and incubated in Ham’s F10 medium without CRH). In some assays antalarmin (Sigma, St. Louis, MO) was used. NF-κB binding activity was estimated with p65 activity ELISA [26,36]. Assay was performed according to manufacturer’s protocol (TransAM NF-κB p65 transcription factor assay, Active Motif, Carlsbad, CA). In brief, cells were lysed and total cell lysate was added to wells coated with oligonucleotide probe containing κB-binding sites. NF-κB dimers were detected with anti-p65 antibody and secondary antibody linked to horseradish peroxidase. NF-κB binding activity of cell extracts was normalized to total protein content (quantified with BCA reagent, Pierce Biotechnology Inc., Rockford, IL) of cell extracts.
2.3. Western blot analysis of IκB-α and β
Total cell extracts were prepared in RIPA buffer with Sigma protease inhibitor cocktail (1:100) and clarified by centrifugation (10 000 × g, 20 min). Cytosolic extracts were prepared according tothe methoddescribedby Lavagno etal. [18]. Inbrief,cellswere detached, washedwith ice-cold PBS,and centrifugedat 1000 × g for 5 min at 4 °C. The cell pellet was resuspended in 300 μl of lysis buffer (10 mM HEPES, pH 7.9, 1.5 mM magnesium chloride, 10 mM potassium chloride, 0.5 mM phenylmethylsulfonyl fluoride, and 0.5 mM dithiothreitol) and incubated on ice for 15 min. At the end of this incubation, 20 μl of 10% Igepal p-630 was added and, after centrifugation at 13 000 × g for 1 min at 4 °C, cytosolic extracts were obtained. Nuclei extracts were prepared as previously described [41]. Cell lysates (20 μg) were separated on 12% SDS-PAGE gels and transferred to polyviny-lidene fluoride membranes. After blocking with Tris buffered saline, 0.05% Triton X-100 and 5% milk, the membranes were incubated with primary rabbit anti-human IκB-α (sc-371, 1:200) or IκB-β (sc-9130, 1:200) antibodies, followed by incubation with horseradish peroxidase conjugated goat anti-rabbit IgG (1:5000) (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Membranes were stripped and re-probed with anti-actin antibody (sc-1615, 1:200). The proteins were visualized with Supersignal West Pico Chemiluminescent Substrate (Pierce Biotechnology Inc., Rockford, IL). The chemiluminescent signal was acquired on a Fluor-S MultiImager and analyzed with Quantity One software (Bio-Rad Laboratories, Hercules, CA).
2.4. Immunolocalization of p65 and IκB-α (confocal laser microscopy)
Cells were seeded in 8-well Lab-Tek II chamber slides (Nalge Nunc Inc., Naperville, IL). Cells pre-incubated in Epilife with EDGS for 24 h, were then stimulated with 100 nM CRH in Ham’s F10 medium for 24 h and then fixed with 4% paraformaldehyde in PBS for 10 min. The cells were permeabilized with 1:1 methanol/acetone for 5 min and blocked with 1% bovine serum albumin (BSA; in PBS) for 30 min. The cells were incubated consecutively with rabbit primary anti-human p65 antibody (sc-372, 1:200) or rabbit primary anti-human IκB-α antibody (sc-371, 1:200) (Santa Cruz Biotechnology Inc., Santa Cruz, CA) for 1 h, anti-rabbit streptavidin conjugate for 1 h and fluorescein isothiocyanate (FITC)-avidin conjugate (Vector Laboratories Inc., Burlingame, CA) for 30 min in buffer containing 1% BSA in PBS. The slides were extensively washed with PBS between staining, and then fixed Vectashield mounting medium with (for IκB-α) or without (for p65) propidium iodide (Vector Laboratories Inc., Burlingame, CA). Slides not incubated with primary antibody were used as background controls. Slides were observed with laser scanning confocal fluorescent microscope (LSM 510, Carl Zeiss GmbH, Jena, Germany).
2.5. Real-time PCR
Total RNA was isolated using Trizol, treated with DNAse I and reverse transcribed with SuperScript First-Strand Synthesis System (Invitrogen Life Technologies, Carlsbad, CA). The following primers were used—POMC: forward, CTA CGG CGG TTT CAT GAC CT; reverse, CCC TCA CTC GCC CTT CTT G (amplicon 100 bp); 18S rRNA: forward, TTC GGA ACT GAG GCC ATG AT; reverse, TTT CGC TCT GGT CCG TCT TG (101 bp). Reactions were performed using Sybr Green PCR Master Mix (ABI, Foster City, CA), and the data collected on an ABI Prism 7700 and analyzed using the Sequence Detector 1.9.1 (ABI, Foster City, CA). POMC mRNA amounts were calculated relative to 18S rRNA by a comparative CT method.
2.6. Enzyme linked immunosorbent assays (ELISAs) for ACTH, cortisol and corticosterone
To measure ACTH extracts were prepared by lysing the cells in PBS with 0.5% Triton X-100 and Sigma protease inhibitor cocktail (1:100) for 30 min on ice, followed by centrifugation at 14 000 × g for 20 min. The assay was performed according to manufacturer’s protocol (Alpco Diagnostics, Windham, NH). Amounts of cortisol or corticosterone in supernatants were determined by ELISA (R&D Systems Inc., Minneapolis, MN). ACTH concentrations in cell extracts and steroids in the supernatants were normalized to total protein content (quantified with BCA reagent, Pierce Biotechnology Inc., Rockford, IL) of cell extracts.
2.7. Knockdown of POMC gene
The cells were transfected with phosphorothioated antisense oligonucleotide complementary to the first 25 bp of POMC exon 3 (synthesized by IDT, Coralville, IA) according to the method of [10]. Final concentration of oligonucleotides was 5 μM. Cells were transfected at the time of media change to EpiLife with EDGS using transfection reagents (sc-29528 and sc-36868) from Santa Cruz Biotechnology Inc., Santa Cruz, CA) according to manufacturer’s protocols. In the presence of POMC antisense oligonucleotide ACTH levels in cellular lysates decreased ~2-fold, as measured by ELISA (not shown). A scrambled oligonucleotide, sequence as described by [10], was used for control. Transfection media were changed after 24 h and cells treated as above.
2.8. Statistical analysis
Data was analyzed by Student’s t-tests using Prism 4.00 (GraphPad Software, San Diego), and presented as mean ± S.E.M. (n = 3 or 4).
3. Results and discussion
3.1. CRH inhibits activation of NF-κB in PIG1 melanocytes
PIG1 melanocytes were transfected with a luciferase construct driven by κB sites (pNF-κB-Luc) or a construct without κB sites in the growth medium without BPE. After 24 h medium was changed to serum free Ham’s F10 medium with or without 100 nM CRH. The following day, the cells were lysed and luciferase activity measured. NF-κB activity in cells incubated in the Ham’s F10 serum free medium for 24 h was 3.2-fold higher than in the cells incubated in the medium containing growth factors (Fig. 1). CRH inhibited NF-κB dependent transcriptional activity by 72 ± 10% (Fig. 1A) as compared to cells incubated in serum free medium without addition of the peptide. Addition of antalarmin (10 μM), a specific non-peptide antagonist of CRH receptor type 1 [15], 1 h prior to CRH treatment abrogated the effects of CRH on NF-κB-dependent transcription, which confirmed the specificity of the action of the hormone (Fig. 1A). Treatment of transfected cells with media, CRH and antalarmin did not affect transcriptional activities of construct that did not contain κB sites in the promoter (Fig. 1A). The activation of subunits p65 of NF-κB was also demonstrated by electrophoretic mobility shift assays for NF-κB (not shown).
Fig. 1.
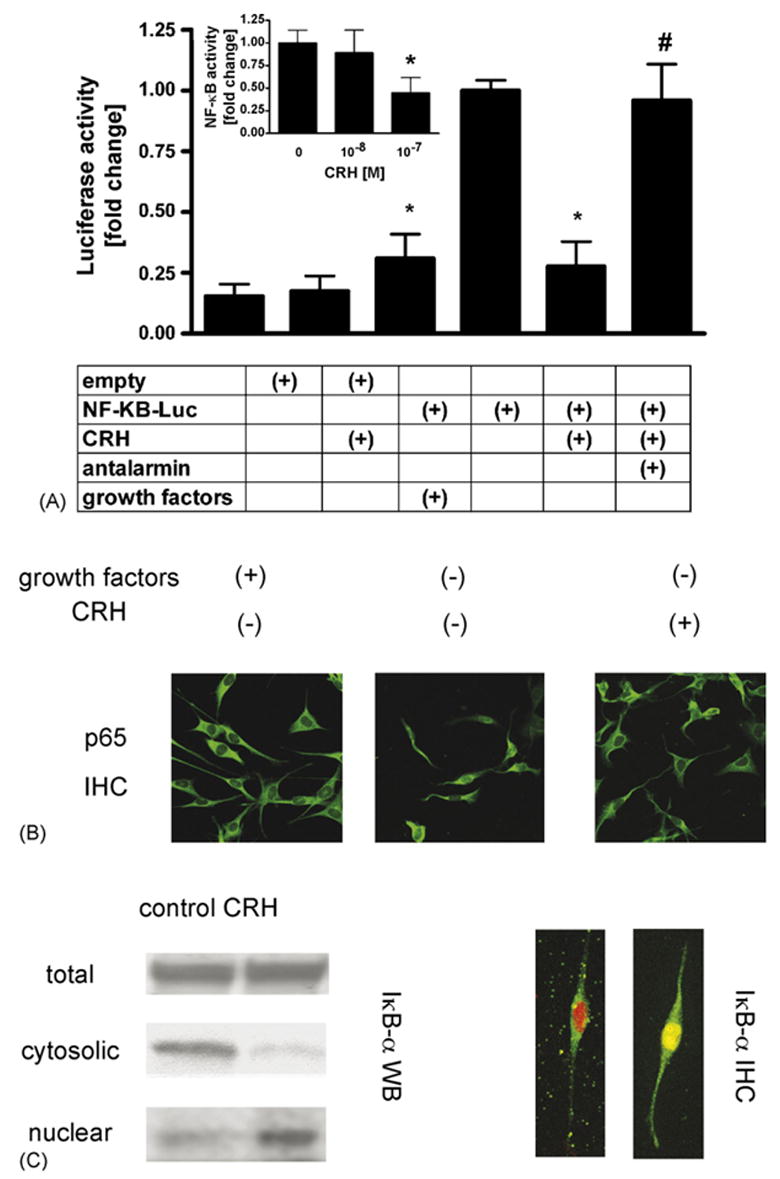
CRH decreases NF-κB activity in PIG1 melanocytes and induces translocation of IκB-α from the cytoplasm to the nucleus. (A) Cells were transfected with κB-driven or empty luciferase reporter constructs, pre-incubated in Epilife medium with EDGS (containing growth factors) for 24 h, then medium was changed to Epilife with EDGS or serum free Ham’s F10 and 100 nM CRH was added for 24 h. Antalarmin (10 μM) was added 1 h before medium change. Then cells were lysed and NF-κB transcriptional activity was measured. Inset: effect of increasing doses of CRH on NF-κB activity. Data is presented as mean ± S.E.M. (n = 3; inset: n = 4) in comparison to the cells incubated in medium without growth factors and CRH. *P < 0.05 control vs. CRH. #P < 0.05 CRH vs. CRH and antalarmin. There is no statistical difference between non-treated and treated empty vector controls. (B) Cells were fixed and stained with anti-p65 antibody followed by secondary antibody conjugated to FITC (green). The p65 was distributed in the cytoplasm in the cells incubated in the medium supplemented with serum; incubation of cells in serum free medium resulted in nuclear localization of p65. Incubation of cells with CRH resulted in decreased nuclear localization of p65 and increased cytoplasmic localization. Results are representative of two separate experiments. (C) Cells were pre-incubated in Epilife medium with EDGS for 24 h and then incubated in Ham’s F10 medium without growth factors for 24 h with or without 100 nM CRH. Upper panel: total, cytosolic and nuclear fractions were prepared as described in Section 2. Presence of IκB-α in the lysates separated by SDS-PAGE was estimated with Western blot. Results represent two separate experiments. Lower panel: translocation of IκB-α protein to the nucleus was assessed by confocal microscopy. IκB-α was stained with antibodies conjugated to FITC (green) and propidium iodide was used to stain the nucleus (red). In the absence of CRH IκB-α is localized in the cytoplasm. In the presence of CRH IκB-α was co-localized with nuclear stain (yellow). Result is representative of 4 separate treatments per condition. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
In general, NF-κB resides in the cytoplasm. Upon activation by various stimuli NF-κB translocates into the nucleus to regulate gene transcription. To further characterize the effect of CRH on activation of NF-κB in melanocytes, we have also analyzed subcellular distribution of p65 protein by confocal fluorescent microscopy. After pre-incubation for 24 h in the medium without BPE, the cells were stimulated with or without 100 nMCRH for 24 h in serum freeHam’s F10 medium.Then, the cells were fixed and stained with anti-p65 antibody followed by secondary antibody conjugated to FITC. In the cells grown in the growth factors-containing medium, p65 was localized in the cytoplasm.In growth factor-free medium, clear translocation of NF-κB into the nucleus was evident, consistent with NF-κB activation. In contrast, CRH addition to growth factor-free medium blocks nuclear translocation of NF-κB (Fig. 1B).
In immortalized human epidermal neonatal PIG1 melanocytes, the inhibitory effect of CRH on NF-κB pathway is mediated by pro-opiomelanocortin. CRH inhibits NF-κB in corticotrophs, neurons and HaCaT cells [15,20,44]. POMC derived peptides are potent anti-inflammatory agents that inhibit NF-κB in leukocytes, keratinocytes, melanocytes and melanoma cells [12,13,24,42]. In corticotroph AtT-20 cells, induction of POMC by CRH decreases NF-κB activity [15]. Decrease of NF-κB activity by secondary POMC-dependent mechanism in the melanocyte is thus consistent with the mechanism observed in the cell classically targeted by CRH at the systemic level [6] and inhibition of NF-κB by POMC-derived peptides in skin cells [13].
3.2. Decrease of NF-κB activity depends on IκB-α translocation from the cytoplasm to the nucleus simultaneously with NF-κB
In the canonical pathway of NF-κB activation, after binding of ligand to its receptor, several types of kinases are activated that then converge into the IκB kinase 1/IκB kinase 2/NF-κB essential modulator complex. Subsequent phosphorylation and ubiquitination of IκB proteins leads to the release of NF-κB complexes, uncovering of NLS sequences on them and their translocation to the nucleus to affect transcription of relevant genes [21]. Inhibition of NF-κB signaling may result from increased synthesis of IκB, decreased degradation of IκB or translocation of IκB/NF-κB complex to the nucleus [21,37]. We have analyzed the effect of CRH on the levels of IκB-α in cellular extracts as well as cytoplasmic and nuclear fractions of melanocytes. Extracts were prepared from cells treated as above. As presented in Fig. 1C (left panel), CRH neither affected the cellular levels of IκB-α nor IκB-β (not shown). However, addition of CRH results in decreased IκB-α levels in the cytoplasm with a subsequent increase in the nucleus (Fig. 1C, left panel). Confocal microscopy of the cells incubated with or without CRH confirms results obtained with cell fractionation and Western blot. In control cells IκB-α is expressed predominantly in the cytoplasm. CRH treatment of cells results in a marked increase of IκB-α in the nucleus (Fig. 1C, right panel).
The above data indicate that inhibitory effect of CRH results from a CRH-mediated increase of IκB-α in the nucleus following classical pathway described in the literature [1,2,5,24,37]. Thus, IκB-α maintains NF-κB in unstimulated cells in an inactive form. Upon cell stimulation, IκB-α is rapidly degraded and NF-κB translocates into the nucleus. NF-κB stimulates in turn transcription of IκB-α gene. Newly synthesized IκB-α inhibits NF-κB signaling by enhanced maintenance of NF-κB in the cytoplasm. IκB-α also translocates into the nucleus, associates with NF-κB subunits and blocks their binding and transcriptional activity [2,37]. The latter mechanism functions to terminate response to typical NF-κB-inducing stimuli such as IL-1 or TNF-α [1,5]. Of note, nitric oxide, ACTH and α-melanocyte stimulating hormone (α-MSH) all are reported to inhibit NF-κB activity by the above pathway [24,37]. Since we have observed upon CRH treatment an increased presence of IκB-α in the nucleus and decreased presence of p65 in the nucleus as compared to the cytoplasm, we expect that IκB-α displace NF-κB from its gene promoters as in the models described above.
3.3. CRH increases POMC expression and ACTH immunoreactivity in PIG1 melanocytes
CRH stimulates POMC expression, ACTH production, and cortisol and corticosterone release by normal human neonatal epidermal melanocytes [33] and these effects on steroid production are dependent on POMC production. We therefore analyzed POMC expression, ACTH production, and cortisol and corticosterone release by PIG1 melanocytes. To determine the level of POMC mRNA, serum deprived PIG1 melanocytes were incubated in the presence or absence of CRH. Then RNA was extracted and mRNA measured by real-time RT-PCR. As shown in Fig. 2A, CRH (100 nM) stimulated POMC expression by ~100-fold.
Fig. 2.
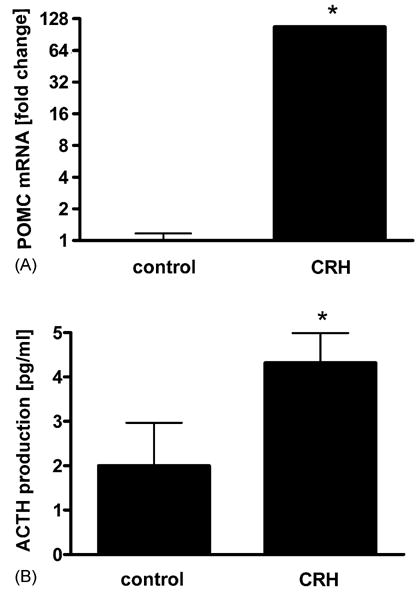
CRH stimulates POMC and ACTH in PIG1 melanocytes. Cells were pre-incubated in Epilife medium with EDGS for 24 h, and then incubated with or without 100 nM CRH in the Ham’s F10 medium without growth factors for 24 h. Then POMC expression was examined with real time RT-PCR. (A) Cells were incubated with or without 100 nM CRH in the same conditions as above and ACTH immunoreactivity in the cell lysates was estimated with ELISA. (B) The data is presented as mean ± S.E.M. (n = 3) in comparison to cells not treated with CRH. *P < 0.05 control vs. CRH.
To determine the level of ACTH, cells were incubated in serum free medium in the presence or absence of CRH, lysed with buffer consisting of PBS and Triton X-100 and ACTH immunoreactivity in the lysates was measured with ELISA. ACTH immunoreactivity in cellular lysates increased 212 ± 33% (Fig. 2B). Steroid release into the media (collected from cells treated with CRH) was also measured by ELISA. In contrast to primary neonatal melanocytes [33], immortalized PIG1 melanocytes did not produce cortisol or corticosterone. This suggests that POMC-derived factor but not glucocorticoid is responsible for inhibition of NF-κB activity.
Induction of POMC expression follows cAMP/PKA activation after binding of CRH to CRH receptor [23]. We have documented that cAMP/protein kinase A (PKA) is stimulated by CRH in the normal neonatal and immortalized (PIG1) melanocytes [34]. This suggests that intracellular pathway leading from CRH receptor to POMC gene activation in the melanocytes is similar to the one described in corticotrophs.
3.4. Effect of CRH on NF-κB activity is diminished in the presence of POMC gene silencer
Having shown that CRH dramatically increases POMC expression, we determined the role of POMC on the observed effects of CRH on NF-κB signaling. To inhibit POMC expression, we used POMC antisense oligonucleotides. In separate (control) experiment we demonstrated that the presence of POMC antisense oligonucleotide decreased by ~2-fold ACTH levels in cellular lysates, indicating specificity of the pathway tested (see Section 2).
After transfection with POMC antisense or scrambled oligonucleotides, cells were incubated with or without CRH in serum free medium, then lysed and the binding of p65 to κB sites was determined by ELISA. In the CRH-treated cells transfected with the POMC antisense oligonucleotide, binding of p65 to κB sites increased 4.6-fold as compared to the CRH-treated untransfected cells (Fig. 3). In contrast, transfection with the scrambled oligonucleotide did not affect DNA binding activity in CRH-treated cells, confirming the specificity of the assay (Fig. 3). In separate experiments we measured NF-κB transcriptional activity (Fig. 3, inset). As shown in the inset to Fig. 3, in the presence of POMC antisense oligonucleotide NF-κB-dependent transcriptional activity increased by ~11-fold in comparison to non-transfected cells, as measured by reporter gene assay. Thus, measurements of both NF-κB DNA binding activity and of its transcriptional activity demonstrate that CRH-induced POMC has an inhibitory effect on NF-κB pathway in melanocytes.
Fig. 3.
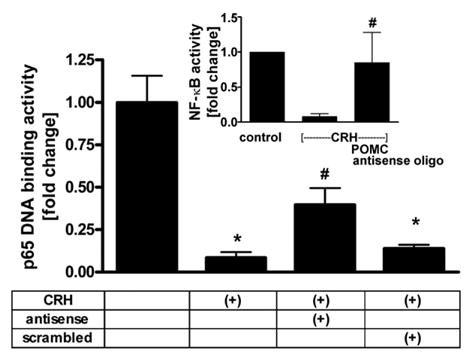
Knockdown of POMC expression results in the decrease of inhibitory effect of CRH on NF-κB signaling. Cells were transfected with POMC antisense or scrambled oligonucleotides, pre-incubated in Epilife medium with EDGS for 24 h and then incubated in Ham’s F10 medium without growth factors for 24 h without or with 100 nM CRH. NF-κB DNA binding activity was estimated by p65 activity ELISA. Inset: cells were transfected with NF-κB construct and POMC antisense or scrambled oligonucleotide, pre-incubated in Epilife medium with EDGS for 24 h and then incubated in Ham’s F10 medium without growth factors for 24 h with or without 100 nM CRH. NF-κB transcriptional activity was estimated by the luciferase signal. The data is presented as mean ± S.E.M. (n = 3) in comparison to the cells not treated with CRH (=100%). *P < 0.05 control vs. CRH, #P < 0.05 cells treated with CRH and transfected with POMC antisense oligonucleotides vs. transfected with scrambled antisense oligonucleotides.
Mediation of effect of CRH on cellular phenotype by POMC-derived peptides was observed in corticotrophs [15] and melanocytes [33]. In other cell types, such as keratinocytes, CRH did not stimulate POMC [32]. CRH acted there directly stimulating rather than inhibiting inflammatory response [45].
4. Concluding remark
The data and information reported above indicate that in human epidermal melanocytes CRH diminishes NF-κB activation through CRH-R1 mediated stimulation of the melanocyte’s POMC system. This is in contrast to normal epidermal keratinocytes where CRH acts as a pro-inflammatory agent via activation of CRH-R1 [45]. Thus, the same molecule (CRH) acting on the same receptor (CRH-R1) can induce opposite effects (anti- versus pro-inflammatory) in the same tissue (epidermis) in a cell type specific fashion through overlapping but distinct mechanisms. Since melanocytes are able to produce several inflammatory cytokines whose expression is under control of NF-κB, one can expect that effect of CRH-POMC on its activity will diminish production and release of aforementioned cytokines. This might represent termination of inflammatory response and return of the tissue to homeostasis. Accordingly, we suggest that the phenomenon described above represent a novel mechanism of self-restriction of inflammation imposed by cells of neural crest origin (melanocytes) at the local level (skin). In this context human epidermis serves as an extremely fertile ground for studies on the role of CRH and POMC systems in the integration of the neuroendocrine signals with skin immune system.
Acknowledgments
The work was supported by National Institutes of Health grants CA73753 (LMP), AR047079 and AR052190 from NIAMS (AS), Johnson and Johnson Skin Research Training Grant (BZ; AS mentor) and William J. Cunliffe Scientific Award to AS. Confocal microscopy was performed on the equipment obtained through Shared Instrumentation Grant from National Center for Research Purposes at the National Institutes of Health (S10 RR13725-01). Real-time RT-PCR was performed on ABI Prism 7770 in the Molecular Resource Center at the University of Tennessee, Memphis. We thank Dr. C. Le Poole for immortalized normal human epidermal melanocyte line PIG1. Technical assistance of Dr. M. Zmijewski is greatly appreciated.
References
- 1.Arenzana-Seisdedos F, Thompson J, Rodriguez MS, Bachelerie F, Thomas D, Hay RT. Inducible nuclear expression of newly synthesized I kappa B alpha negatively regulates DNA-binding and transcriptional activities of NF-kappa B. Mol Cell Biol. 1995;15:2689–96. doi: 10.1128/mcb.15.5.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arenzana-Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay RT, Virelizier JL, et al. Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J Cell Sci. 1997;110:369–78. doi: 10.1242/jcs.110.3.369. [DOI] [PubMed] [Google Scholar]
- 3.Bayatti N, Zschocke J, Behl C. Brain region-specific neuroprotective action and signaling of corticotropin-releasing hormone in primary neurons. Endocrinology. 2003;144:4051–60. doi: 10.1210/en.2003-0168. [DOI] [PubMed] [Google Scholar]
- 4.Cao J, Cetrulo CL, Theoharides TC. Corticotropin releasing hormone induces vascular endothelial growth factor release from human mast cells via the cAMP/protein kinase A/p38 mitogen-activated protein kinase pathway. Mol Pharmacol. 2006;69:998–1006. doi: 10.1124/mol.105.019539. [DOI] [PubMed] [Google Scholar]
- 5.Chen LF, Greene WC. Shaping the nuclear action of NF-kappa B. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 6.Chrousos GP. The hypothalamic–pituitary–adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–63. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- 7.Dermitzaki E, Tsatsanis C, Gravanis A, Margioris AN. Corticotropin releasing hormone induces Fas ligand production and apoptosis in PC12 cells via activation of p38 mitogen-activated protein kinase. J Biol Chem. 2002;277:12280–7. doi: 10.1074/jbc.M111236200. [DOI] [PubMed] [Google Scholar]
- 8.Emanuel RL, Torday JS, Asokananthan N, Sunday ME. Direct effects of corticotropin releasing hormone and thyrotropin-releasing hormone on fetal lung explants. Peptides. 2000;21:1819–29. doi: 10.1016/s0196-9781(00)00343-0. [DOI] [PubMed] [Google Scholar]
- 9.Ferrari A, Petraglia F, Gurpide E. Corticotropin releasing factor decidualizes human endometrial stromal cells in vitro. Interaction with progestin. J Steroid Biochem Mol Biol. 1995;54:251–5. doi: 10.1016/0960-0760(95)00142-m. [DOI] [PubMed] [Google Scholar]
- 10.Frankel B, Longo SL, Rodziewicz GS, Hodge CJ., Jr Antisense oligonucleotide-induced inhibition of adrenocorticotropic hormone release from cultured human corticotrophs. J Neurosurg. 1999;91:261–7. doi: 10.3171/jns.1999.91.2.0261. [DOI] [PubMed] [Google Scholar]
- 11.Hagan P, Poole S, Bristow AF. Immunosuppressive activity of corticotrophin releasing factor. Inhibition of interleukin-1 and interleukin-6 production by human mononuclear cells. Biochem J. 1992;281:251–4. doi: 10.1042/bj2810251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haycock JW, Wagner M, Morandini R, Ghanem G, Rennie IG, Mac Neil S. Alpha-melanocyte-stimulating hormone inhibits NF-kappa B activation in human melanocytes and melanoma cells. J Invest Dermatol. 1999;113:560–6. doi: 10.1046/j.1523-1747.1999.00739.x. [DOI] [PubMed] [Google Scholar]
- 13.Haycock JW, Rowe SJ, Cartledge S, Wyatt A, Ghanem G, Morandini R, et al. Alpha-melanocyte-stimulating hormone reduces impact of proinflammatory cytokine and peroxide-generated oxidative stress on keratinocyte and melanoma cell lines. J Biol Chem. 2000;275:15629–36. doi: 10.1074/jbc.275.21.15629. [DOI] [PubMed] [Google Scholar]
- 14.Hillhouse EW, Grammatopoulos DK. The molecular mechanisms underlying the regulation of the biological activity of corticotropin releasing hormone receptors: implications for physiology and pathophysiology. Endocr Rev. 2006 doi: 10.1210/er.2005-0034. [DOI] [PubMed] [Google Scholar]
- 15.Karalis KP, Venihaki M, Zhao J, van Vlerken LE, Chandras C. NF-kappa B participates in the corticotropin releasing hormone-induced regulation of the pituitary proopiomelanocortin gene. J Biol Chem. 2004;279:10837–40. doi: 10.1074/jbc.M313063200. [DOI] [PubMed] [Google Scholar]
- 16.Korbonits M, Morris DG, Nanzer A, Kola B, Grossman AB. Role of regulatory factors in pituitary tumour formation. Front Horm Res. 2004;32:63–95. doi: 10.1159/000079038. [DOI] [PubMed] [Google Scholar]
- 17.Kruger-Krasagakes S, Krasagakis K, Garbe C, Diamantstein T. Production of cytokines by human melanoma cells and melanocytes. Recent Results Cancer Res. 1995;139:155–68. doi: 10.1007/978-3-642-78771-3_11. [DOI] [PubMed] [Google Scholar]
- 18.Lavagno L, Gunella G, Bardelli C, Spina S, Fresu LG, Viano I, et al. Anti-inflammatory drugs and tumor necrosis factor-alpha production from monocytes: role of transcription factor NF-kappa B and implication for rheumatoid arthritis therapy. Eur J Pharmacol. 2004;501:199–208. doi: 10.1016/j.ejphar.2004.07.101. [DOI] [PubMed] [Google Scholar]
- 19.Le Poole IC, van den Berg FM, van den Wijngaard RM, Galloway DA, van Amstel PJ, Buffing AA, et al. Generation of a human melanocyte cell line by introduction of HPV16 E6 and E7 genes. In Vitro Cell Develop Biol Anim. 1997;33:42–9. doi: 10.1007/s11626-997-0021-6. [DOI] [PubMed] [Google Scholar]
- 20.Lezoualc’h F, Engert S, Berning B, Behl C. Corticotropin releasing hormone-mediated neuroprotection against oxidative stress is associated with the increased release of non-amyloidogenic amyloid beta precursor protein and with the suppression of nuclear factor-kappa B. Mol Endocrinol. 2000;14:147–59. doi: 10.1210/mend.14.1.0403. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Verma IM. NF-kappa B regulation in the immune system. Nat Rev Immunol. 2002;2:725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 22.Linton EA, Woodman JR, Asboth G, Glynn BP, Plested CP, Bernal AL. Corticotrophin releasing hormone: its potential for a role in human myometrium. Exp Physiol. 2001;86:273–81. doi: 10.1113/eph8602183. [DOI] [PubMed] [Google Scholar]
- 23.Lundblad JR, Roberts JL. Regulation of proopiomelanocortin gene expression in pituitary. Endocr Rev. 1988;9:135–58. doi: 10.1210/edrv-9-1-135. [DOI] [PubMed] [Google Scholar]
- 24.Moustafa M, Szabo M, Ghanem GE, Morandini R, Kemp EH, MacNeil S, et al. Inhibition of tumor necrosis factor-alpha stimulated NF-kappa B/p65 in human keratinocytes by alpha-melanocyte stimulating hormone and adrenocorticotropic hormone peptides. J Invest Dermatol. 2002;119:1244–53. doi: 10.1046/j.1523-1747.2002.19602.x. [DOI] [PubMed] [Google Scholar]
- 25.Paez Pereda M, Sauer J, Perez Castro C, Finkielman S, Stalla GK, Holsboer F, et al. Corticotropin releasing hormone differentially modulates the interleukin-1 system according to the level of monocyte activation by endotoxin. Endocrinology. 1995;136:5504–10. doi: 10.1210/endo.136.12.7588301. [DOI] [PubMed] [Google Scholar]
- 26.Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, et al. Development of a sensitive multi-well colorimetric assay for active NF-kappa B. Nucl Acids Res. 2001;29:E21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slominski A, Baker J, Ermak G, Chakraborty A, Pawelek J. Ultraviolet B stimulates production of corticotropin releasing factor (CRF) by human melanocytes. FEBS Lett. 1996;399:175–6. doi: 10.1016/s0014-5793(96)01315-4. [DOI] [PubMed] [Google Scholar]
- 28.Slominski A, Wortsman J, Luger T, Paus R, Solomon S. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol Rev. 2000;80:979–1020. doi: 10.1152/physrev.2000.80.3.979. [DOI] [PubMed] [Google Scholar]
- 29.Slominski A, Wortsman J, Pisarchik A, Zbytek B, Linton EA, Mazurkiewicz JE, et al. Cutaneous expression of corticotropin releasing hormone (CRH), urocortin, and CRH receptors. FASEB J. 2001;15:1678–93. doi: 10.1096/fj.00-0850rev. [DOI] [PubMed] [Google Scholar]
- 30.Slominski A, Pisarchik A, Tobin DJ, Mazurkiewicz JE, Wortsman J. Differential expression of a cutaneous corticotropin-releasing hormone system. Endocrinology. 2004;145:941–50. doi: 10.1210/en.2003-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84:1155–228. doi: 10.1152/physrev.00044.2003. [DOI] [PubMed] [Google Scholar]
- 32.Slominski A, Zbytek B, Semak I, Sweatman T, Wortsman J. CRH stimulates POMC activity and corticosterone production in dermal fibroblasts. J Neuroimmunol. 2005;162:97–102. doi: 10.1016/j.jneuroim.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 33.Slominski A, Zbytek B, Szczesniewski A, Semak I, Kaminski J, Sweatman T, et al. CRH stimulation of corticosteroids production in melanocytes is mediated by ACTH. Am J Physiol Endocrinol Metabol. 2005;288:E701–6. doi: 10.1152/ajpendo.00519.2004. [DOI] [PubMed] [Google Scholar]
- 34.Slominski A, Zbytek B, Pisarchik A, Zmijewski RM, Wortsman J. CRH functions as a growth factor/cytokine in the skin. J Cell Physiol. 2006;206:780–91. doi: 10.1002/jcp.20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slominski A, Zbytek B, Zmijewski M, Slominski RM, Kauser S, Wortsman J, et al. Corticotropin releasing hormone and the skin. Front Biosci. 2006;11:2230–48. doi: 10.2741/1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sorokina EM, Merlo JJ, Jr, Tsygankov AY. Molecular mechanisms of the effect of herpesvirus saimiri protein StpC on the signaling pathway leading to NF-kappa B activation. J Biol Chem. 2004;279:13469–77. doi: 10.1074/jbc.M305250200. [DOI] [PubMed] [Google Scholar]
- 37.Spiecker M, Peng HB, Liao JK. Inhibition of endothelial vascular cell adhesion molecule-1 expression by nitric oxide involves the induction and nuclear translocation of I kappa B alpha. J Biol Chem. 1997;272:30969–74. doi: 10.1074/jbc.272.49.30969. [DOI] [PubMed] [Google Scholar]
- 38.Theoharides TC, Donelan JM, Papadopoulou N, Cao J, Kempuraj D, Conti P. Mast cells as targets of corticotropin-releasing factor and related peptides. Trends Pharmacol Sci. 2004;25:563–8. doi: 10.1016/j.tips.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 39.Thomson AW, Lotze MT. The cytokine handbook. 4. London, San Diego: Elsevier Science Ltd; 2003. [Google Scholar]
- 40.Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–7. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- 41.Yang CH, Murti A, Pfeffer SR, Basu L, Kim JG, Pfeffer LM. IFN-alpha/beta promotes cell survival by activating NF-kappa B. PNAS. 2000;97:13631–6. doi: 10.1073/pnas.250477397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon SW, Goh SH, Chun JS, Cho EW, Lee MK, Kim KL, et al. Alpha-melanocyte-stimulating hormone inhibits lipopolysaccharide-induced tumor necrosis factor-alpha production in leukocytes by modulating protein kinase A, p38 kinase, and nuclear factor kappa B signaling pathways. J Biol Chem. 2003;278:32914–20. doi: 10.1074/jbc.M302444200. [DOI] [PubMed] [Google Scholar]
- 43.Zbytek B, Mysliwski A, Slominski A, Wortsman J, Wei ET, Mysliwska J. Corticotropin releasing hormone affects cytokine production in human HaCaT keratinocytes. Life Sci. 2002;70:1013–21. doi: 10.1016/s0024-3205(01)01476-x. [DOI] [PubMed] [Google Scholar]
- 44.Zbytek B, Pfeffer LM, Slominski AT. Corticotropin releasing hormone inhibits nuclear factor-kappa B pathway in human HaCaT keratinocytes. J Invest Dermatol. 2003;121:1496–9. doi: 10.1111/j.1523-1747.2003.12612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zbytek B, Pfeffer LM, Slominski AT. Corticotropin releasing hormone stimulates NF-kappa B in human epidermal keratinocytes. J Endocrinol. 2004;181:R1–7. doi: 10.1677/joe.0.181r001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zbytek B, Slominski AT. Corticotropin releasing hormone induces keratinocyte differentiation in the adult human epidermis. J Cell Physiol. 2005;203:118–26. doi: 10.1002/jcp.20209. [DOI] [PubMed] [Google Scholar]
- 47.Zhao J, Karalis KP. Regulation of nuclear factor-kappa B by corticotropin releasing hormone in mouse thymocytes. Mol Endocrinol. 2002;16:2561–70. doi: 10.1210/me.2001-0334. [DOI] [PubMed] [Google Scholar]