

Abstract
Gem is a protein of the Ras superfamily that plays a role in regulating voltage-gated Ca2+ channels and cytoskeletal reorganization. We now report that GTP-bound Gem interacts with the membrane–cytoskeleton linker protein Ezrin in its active state, and that Gem binds to active Ezrin in cells. The coexpression of Gem and Ezrin induces cell elongation accompanied by the disappearance of actin stress fibers and collapse of most focal adhesions. The same morphological effect is elicited when cells expressing Gem alone are stimulated with serum and requires the expression of ERM proteins. We show that endogenous Gem down-regulates the level of active RhoA and actin stress fibers. The effects of Gem downstream of Rho, i.e., ERM phosphorylation as well as disappearance of actin stress fibers and most focal adhesions, require the Rho-GAP partner of Gem, Gmip, a protein that is enriched in membranes under conditions in which Gem induced cell elongation. Our results suggest that Gem binds active Ezrin at the plasma membrane–cytoskeleton interface and acts via the Rho-GAP protein Gmip to down-regulate the processes dependent on the Rho pathway.
INTRODUCTION
Gem is an atypical protein of the Ras superfamily whose expression is induced by mitogenic stimulations in several cell types (Maguire et al., 1994; Vanhove et al., 1997; Iyer et al., 1999). Together with Rad (Reynet and Kahn, 1993), Rem (Finlin and Andres, 1997), and Rem2 (Finlin et al., 2000), it forms a subfamily of proteins, often termed RGK (for Rad and Kir/Gem), that share several striking characteristics. First, these proteins present significant variations, compared with other Ras family proteins, in the regions involved in binding the phosphate moieties of guanine nucleotides; in essence, the conserved threonine residue of the switch 1 region (often referred to as the effector region) that coordinates the magnesium ion complexing the β and γ phosphates of GTP is absent, and the DXXG region of switch 2 is profoundly modified to DXWE. The consequences of these variations on the biochemical and structural characteristics of Gem were recently described (Opatowsky et al., 2006; Splingard et al., 2007). Second, Gem presents a 72-amino acid N-terminal extension compared with Ras, to which no function has yet been ascribed. Third, it presents a C-terminal extension that does not contain consensus lipid modification motifs, but appears necessary for the localization of Gem to the plasma membrane (Maguire et al., 1994); this region, which is shared with other RGK proteins, carries a functional Ca2+/Calmodulin-binding domain (Fischer et al., 1996; Moyers et al., 1997), and binding of Calmodulin maintains Gem in the cytosol (Beguin et al., 2001; Beguin et al., 2005).
RGK proteins have been shown to carry two distinct functions, reorganization of the actin cytoskeleton and regulation of Ca2+ channels. Rad, Gem, Rem, and Rem2 inhibit the activity of L-type Ca2+ channels by directly interacting with their β subunits at the plasma membrane (Beguin et al., 2001; Finlin et al., 2003). In the case of Gem, this interaction only occurs with the GTP-bound form of the protein and is inhibited by its interaction with Calmodulin as well as with 14-3-3, which sequester Gem away from the plasma membrane (Beguin et al., 2001, 2005; Ward et al., 2004). In this way, Gem was shown to down-regulate Ca2+-dependent hormone exocytosis in neuroendocrine cells (Beguin et al., 2001), and Rem2 attenuates insulin secretion in pancreatic beta cells (Finlin et al., 2005). A growing body of evidence points to a role of RGK proteins in cytoskeletal organization. For instance, Ges, the human orthologue of murine Rem, promotes cytoskeletal changes as well as sprouting of endothelial cells (Pan et al., 2000). Several reports have shown that the overexpression of Gem affects cell morphology in connection with the cytoskeleton and have suggested possible functional links. Gem was reported to interact with microtubules through the kinesin-like protein KIF9 (Piddini et al., 2001). Moreover, the overexpression of Gem in neuroblastoma cells was reported to promote neurite outgrowth and cell flattening through inhibition of the Rho–ROCK pathway (Ward et al., 2002); this activity is regulated by the phosphorylation of Gem on 2 serine residues of the C-terminal extension by yet unidentified kinases (Ward et al., 2004).
To gain further insight into the function(s) of Gem and their underlying molecular mechanisms, we searched for its cellular partners using yeast two-hybrid screens. We previously showed that Gem interacts with a novel Rho-GAP protein, Gmip, that specifically down-regulates the activity of Rho (Aresta et al., 2002). In this article, we report that Gem also interacts with Ezrin, a member of the Ezrin/Radixin/Moesin family. These proteins function as membrane- cytoskeletal linkers and contribute to the formation of specialized structures of the plasma membrane (see Mangeat et al., 1999; Tsukita and Yonemura, 1999; Bretscher et al., 2002 for reviews). ERM proteins carry two conserved domains: an N-terminal region that binds the cytoplasmic domain of integral membrane proteins either directly or through an adaptor protein and a C-terminal domain that contains the F-actin–binding site. These domains, respectively called N- and C-ERMAD, interact with each other with high affinity, resulting in dormant molecules (either in the form of closed monomers or as head to tail oligomers) that are able to bind neither transmembrane proteins nor F-actin. Activation of Ezrin is thought to proceed via a two-step mechanism initially involving its interaction with PIP2 at the plasma membrane (Barret et al., 2000; Hamada et al., 2000; Fievet et al., 2004), which somehow results in opening the molecule or disrupting oligomers, hence freeing the N- and C-ERMADs for their respective interactions with membrane proteins and actin filaments. The active state of ERM proteins is then stabilized by phosphorylation of a conserved C-terminal threonine (T567 in the case of Ezrin), which blocks the interaction between the N- and C-ERMADs; several protein kinases such as the Rho effector ROCK (Matsui et al., 1998, though this point has been challenged by Matsui et al., [1999]), the θ (Pietromonaco et al., 1998) and α (Ng et al., 2001) isoforms of PKC, or the Nck-interacting kinase NIK (Baumgartner et al., 2006) have been implicated in this mechanism. Here, we show that the active form of Gem is able to interact with activated Ezrin via its N-ERMAD and that both proteins associate in vivo. Together, Gem and Ezrin induce morphological changes in HeLa and NIH 3T3 cells consisting in an elongated cell morphology, disappearance of actin stress fibers, collapse of focal adhesions, and a concomitant enhancement of the actin cortical network. We further show that Gem exerts these activities by down-regulating the Rho pathway via the RhoGAP protein Gmip. Our results show that Gem associates with Ezrin at the plasma membrane and acts via Gmip to regulate the organization of the actin cytoskeleton.
MATERIALS AND METHODS
Plasmids, Small Interfering RNAs, Recombinant Proteins, and Antibodies
pRc/CMV-FLAG-Gem and pGBTGem were previously described (Aresta et al., 2002). The S89A and S89N mutations were introduced into the Gem coding sequence by overlapping PCR. The GemΔCt mutant deleted from its 72 C-terminal residues (amino acids 1-265), and Gem-core mutant missing both its N- and C-terminal extensions (amino acids 73-265) were generated by PCR. All mutants were subcloned into pGBT10 for yeast two-hybrid assays and into the mammalian expression vector pRK5; their integrity was verified by sequencing. Full-length recombinant Gem was produced in BL21 Escherichia coli fused to the C-terminus of Maltose Binding Protein (pMal-C2 vector, New England Biolabs, Beverly, MA), purified on amylose-containing resin according to the manufacturer's instructions, and used to produce antiserum in rabbits (Eurogentec, Seraing, Belgium). Anti-Gem antibodies were affinity-purified on nitrocellulose strips loaded with Gem-(His)6 protein, produced from recombinant E. coli transformed with the pET31b vector (Novagen, Madison, WI) containing the Gem coding sequence, as described (Beranger et al., 1991). Gem-core protein (residues 73—265) was produced as a glutathione S-transferase (GST)-fusion protein in E. coli from a modified pGEX expression vector containing a tobacco etch virus (TEV) protease cleavage site (Sheffield et al., 1999); the fusion protein was purified on glutathione-Sepharose beads, and Gem-core was cleaved from GST with recombinant TEV (Splingard et al., 2007). Expression vectors (pRK5) for Gem, Rad, Rem, and Rem2 with an N-terminal HA tag were generated by PCR and verified by sequencing. Mammalian expression vectors for Ezrin (wild-type, T567A, T567D, and PIP2 mutants), or the N-ERMAD (residues 1-309), all tagged with a C-terminal VSV-G epitope (pCB6-Ezrin-VSVG; Barret et al., 2000; Gautreau et al., 2000), and rabbit polyclonal antibodies directed against Ezrin (Algrain et al., 1993) or ERM proteins phosphorylated on Threonine 567 (P-ERM) were generous gifts of Dr. Monique Arpin (Institut Curie, Paris, France). Alternatively, P-ERM antibodies were purchased from Cell Signaling Technology (Beverly, MA). GST fused to the N-terminal (GST-EzN residues 1-309) or C-terminal (GST-EzC residues 280-586) domains of Ezrin were produced in bacteria and purified on glutathione-Sepharose beads as described (Andreoli et al., 1994); EzC was cleaved from GST with thrombin. Protein concentrations were measured using a colorimetric assay (Bradford reagent, Bio-Rad, Richmond, CA). Antibodies reacting with all three ERM proteins were from Santa Cruz Biotechnology (C19; Santa Cruz, CA). Antibodies specifically reacting with Moesin and Radixin were a generous gift from Dr. M. Martin and C. Roy (Montpellier, France), those against Merlin were from Dr. A McClatchey (Boston, MA), and P5D4 anti-VSVG antibodies were from Dr. B Goud. 12CA5 anti-HA antibodies were from Roche (Basel, Switzerland), and monoclonal anti-Vinculin and anti-α-adaptin were from Sigma (St. Louis, MO) and BD Bioscience (San Jose, CA), respectively. To knock down all three ERM proteins in HeLa cells, a mixture of three small interfering RNAs (SiRNAs), each directed against one of these proteins and synthesized by Eurogentec, was used according to Pust et al. (2005). SiRNAs m1 and m2 directed against mouse Gem, 5′ GAACGAATGGCTCCATGAC 3′ and 5′ TGACCACTGCATGCAGGTC 3′, were synthesized by Dharmacon (Boulder, CO), whereas those directed against human Gem, siGem1 5′ AAGTTCATCGAGACCTCTGCA 3′ and siGem2 5′ AAGAATATGGCCTTCAAGCTC 3′, as well as those directed against human Gmip, 5′ GCGAUUUCUUCAGGAGCUC 3′ and 5′ CGUGACCCUUGAGAUGCUG 3′, were from Proligo (Paris, France). SiRNAs were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Anti-Gmip antibodies were previously described (Aresta et al., 2002).
Two-Hybrid Screen
Total RNA was prepared from human placenta using the RNeasy kit (Qiagen, Chatsworth, CA); Poly A+ RNA was purified on oligo-dT Cellulose columns (Amersham Pharmacia Biosciences, Piscataway, NJ) and reverse-transcribed with M-MuLV reverse transcriptase (Amersham Timesaver cDNA kit) using fully degenerated nonamers as primers. Fragments shorter than 400 base pairs were eliminated by size exclusion chromatography through a CHROMA SPIN+TE-400 gel filtration column (Clontech, Palo Alto, CA), annealed to Sfi1 linkers, and cloned into pGADT3T7, a derivative of pGADGH containing T3 and T7 promoters on each side of the polylinker and asymmetrical Sfi1 cloning sites in the center of the polylinker. DNA was electroporated into competent DH5α E. coli. The library had an estimated complexity of 3 × 106 independent clones; it was transformed into competent Y187 Saccharomyces cerevisiae and screened by mating with HF7c yeast containing pGBTGem as described (Fromont-Racine et al., 1997). Yeast cultures and two-hybrid assays were performed according to standard methods (Kaiser et al., 1994).
In Vitro Interactions
Purified recombinant Gem-core protein was stripped of nucleotide with 10 mM EDTA or loaded with 100 μM GDP or GTPγS and incubated with glutathione-Sepharose beads covered with GST or GST-EzN fusion protein for 3 h at 4°C in 20 mM HEPES buffer, pH 7.5, containing 0.1 M NaCl, 1 mM DTT, 10 mM MgCl2, and 0.1% Triton X-100. In some cases, GST-EzN–covered beads had been preincubated with an excess of purified EzC (the relative amounts of both proteins were estimated by scanning their respective bands on Coomassie Blue–stained SDS gels; not shown). Material remaining bound to the beads after a 3-h incubation and extensive washing was analyzed by Western blotting.
Immunoprecipitations
HeLa cells, grown in DMEM containing 10% fetal calf serum, were transfected with plasmids using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions. In some cases, they were transfected 24 h later with SiRNAs using Oligofectamine (Invitrogen). After a further 24-h incubation, the cells were washed twice with ice-cold PBS and lysed in buffer A (20 mM Tris, pH 7.5, 1 mM EGTA, 100 mM NaCl, 1% Triton X-100, 5% glycerol, and 1 mM DTT) containing protease inhibitors (complete without EDTA, Roche) as well as phosphatase inhibitors (1 mM sodium orthovanadate, 10 mM β-glycero-phosphate, 5 mM sodium pyrophosphate, and 50 mM sodium fluoride), and debris were removed by a 5-min centrifugation at 13,000 × g. Extracts were normalized for protein content and immunoprecipitated for 2 h with affinity-purified anti-Gem antibodies or FLAG-M2 antibodies (Sigma) coupled to protein G-Sepharose. Beads were washed five times in buffer A and boiled in SDS-PAGE sample buffer, and immunoprecipitated proteins were visualized by Western blotting.
Fractions enriched in active Ezrin were prepared by a differential detergent solubilization procedure (Granes et al., 2000). Cells were lysed as above with buffer A containing only 0.2% Triton X-100; the lysate was centrifuged for 30 min at 45,000 rpm in a Beckman TLA45 rotor (Fullerton, CA), and the supernatant represented the 0.2% Triton X-100 extract. The pellet was resuspended in buffer A (containing 1% Triton X-100), incubated for 15 min on ice, and centrifuged again as above; this second supernatant represented the 1% Triton X-100 extract. Fractions were analyzed by immunoprecipitation as above. To assess the relative cellular content in phosphorylated ERM, FAK, and PYK2, cells were directly lysed in SDS gel sample buffer containing phosphatase inhibitors as above supplemented with 1 μM each okadaic acid and calyculin A. Western blots were probed with antibodies directed against total ERM, FAK, and PYK2 (BD Transduction Laboratories, Lexington, KY), as well as phosphoERM, phosphoFAK (pTyr397, Sigma), and phosphoPYK2 (pTyr579/580, Sigma). When indicated, the relative levels of Ezrin and phospho-ERM were quantified by exposing the blots to a Fuji CCD camera (Tokyo, Japan) using the Image Gauge software.
Immunofluorescence
HeLa or NIH3T3 cells were plated on glass coverslips, transfected with 0.2 μg of pRK5-Gem and pCB6-Ezrin expression vectors as described above, or 100 nM SiRNAs using Lipofectamine 2000, and incubated for 16 h in the absence of serum. They were stimulated for 2 h with 10% fetal calf serum when indicated and processed for immunofluorescence as previously described (Beranger et al., 1991). Briefly, cells were fixed with 4% paraformaldehyde for 20 min at room temperature, quenched for 20 min with 50 mM ammonium chloride, and permeabilized with 0.1% Triton X-100 in PBS containing 0.2% BSA (PBS-BSA) for 7 min. Fixed cells were incubated with rabbit affinity-purified anti-Gem (1:50) and either monoclonal anti-VSVG (P5D4, ascites fluid 1:100), monoclonal anti-vinculin (1:1000, Sigma-Aldrich), ALEXA-488-phalloidin or ALEXA-350-phalloidin (1:250, Molecular Probes, Eugene, OR) diluted in PBS-BSA for 1 h. Secondary antibodies conjugated to ALEXA-488 or Cy3 (Molecular Probes) were used at a 1:1000 dilution. Slides were mounted in Mowiol; images were either captured with a Leica TCS-2 confocal laser-scanning microscope (Wetzlar, Germany), or with a Leica DMRA microscope using the 63× lens equipped with a Princeton Instruments Micromax camera using the Metaview software (Roper Scientific, Tucson, AZ). Green and red images were combined using Metamorph (Roper Scientific), and figures were mounted using Adobe Photoshop 7 (San Jose, CA) with adjustments for luminosity and contrast.
Morphological Characterization of HeLa Cells Ectopically Expressing Gem
HeLa cells grown on glass coverslips were transfected as above with expression vectors encoding Gem (untagged normal and mutant proteins) and Ezrin, or SiRNAs directed against ERM proteins together with Gem expression vector. After 48 h in the absence of serum, they were fixed and stained as above for the presence of Gem and Ezrin. The maximal length (L) and width (l) of cells expressing both Gem and Ezrin were measured using the Metamorph software; cell elongation was calculated as L/l. At least 100 cells expressing Gem, Ezrin, or both Gem and Ezrin were scored in each experiment. Alternatively, HeLa cells transfected solely with expression vectors encoding normal or mutant Gem proteins were cultured for 48 h in the absence of serum and stimulated for 2 h with 10% fetal bovine serum before fixation and staining with anti-Gem antibodies. The elongation of cells expressing Gem was measured as above.
Fractionation
HeLa cells transfected and treated as above were washed twice with PBS, once with 20 mM HEPES buffer, pH 7.4, containing 10 mM NaCl, and incubated for 10 min on ice in the same buffer containing protease and phosphatase inhibitors as described above in the text. Cells were harvested and lysed using a Dounce homogenizer; lysis was assessed by light microscopy and staining with trypan blue. Large debris and nuclei were eliminated by a 5-min centrifugation at 1000 × g; cytosolic and membrane fractions were obtained by ultracentrifugation for 30 min at 50,000 × g at 4°C and subjected to Western blotting (50 μg protein from each cytosol and membranes).
RNA Preparation and RT PCR
Total cellular RNA was isolated from barely confluent 85-mm-diameter dishes of NIH 3T3 cells using TRIzol (Invitrogen), and cDNA was synthesized using 4 μg RNA with “Ready-to-Go You prime First Strand Beads” (Amersham Pharmacia Biosciences). Specific transcripts were amplified with the following primers (Proligo) for Gem: forward 5′ CAACCTCCGAAACCGCCACTC 3′ and reverse 5′ CACCTGTCGCACAATGCCCTC 3′) and GAPDH: forward 5′ CCTCAACTACATGGTCTACA 3′ and reverse 5′ TTCTCGTGGTTCACACCCAT 3′, using AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA). The products were resolved by electrophoresis in 0.8% agarose gels and viewed by ethidium bromide staining.
Rho, Rac, and Cdc42 Activity Assays
Rho-GTP levels were measured by a pulldown assay using GST fused to the Rho-binding domain of Rhotekin and monoclonal anti-RhoA antibodies (Santa Cruz) as described (Ren and Schwartz, 2000); respective levels of Rho-GTP and total Rho were quantified using a STORM phosphorimager (GE Healthcare, Waukesha, WI). Rac-GTP and Cdc42-GTP levels were similarly measured using GST fused to the CRIB domain of PAK and GST fused to the Cdc42-binding domain of WASP, respectively (Sander et al., 1999); they were detected using monoclonal anti-Rac1 and Cdc42 antibodies (BD Biosciences), respectively.
Adhesion Assay
Twenty-four–well plates (Costar, Cambridge, MA), were coated overnight at 4°C with 10 μg/ml fibronectin when indicated, washed with PBS, and saturated with 1% BSA for 2 h at 37°C. HeLa cells overexpressing wild-type or S89A Gem were detached with Trypsin/EDTA, washed, recovered in serum-free medium, and plated (50,000 cells/well) in triplicate wells for 30 min. Nonadherent cells were removed by washing, and adhering cells were stained with crystal violet (0.1% in 20% methanol) for 30 min at room temperature. Cells were lysed, and adhesion was quantified by measuring absorbance at 405 nm using an ELISA reader (Bio-Rad Laboratories, Hercules, CA).
RESULTS
Gem Interacts with Ezrin in the Yeast Two-Hybrid System and In Vitro
To gain insight into the cellular functions of Gem, we used the yeast two-hybrid system to search for new cellular partners. To this end, we screened a random-primed cDNA library from human placenta with the full-length Gem protein fused to the C-terminus of the GAL4 DNA-binding domain as a bait. Several positive clones represented a fragment of the ROCK I kinase, an interaction that had previously been described (Ward et al., 2002). In addition, we found three clones containing the N-terminal domain of the membrane-cytoskeletal linker protein Ezrin; this domain, known as the N-ERMAD (Mangeat et al., 1999; Tsukita and Yonemura, 1999; Bretscher et al., 2002), is involved in the interaction of Ezrin with membrane-anchor proteins. This interaction is specific of Gem, because the N-ERMAD of Ezrin did not interact with the closely related protein Rem (data not shown). We used deletion mutants of Gem to show that binding to the Ezrin N-ERMAD requires the presence of neither the N- nor the C-terminal extensions of Gem and that its Ras-homologous core (Gem-core) was sufficient for the interaction (Table 1). Furthermore, the Ezrin N-ERMAD no longer interacted with a Gem mutant in which Ser 89, the residue equivalent to the highly conserved Ser 17 in Ras, is mutated to Ala or Asn; in the case of most Ras-related proteins, these mutants exhibit a reduced affinity for nucleotides, especially for GTP, and represent an inactive, or dominant-negative, form of the protein. The fact that Ezrin only interacts with the active form of Gem was directly shown in vitro, using purified recombinant proteins. Indeed, purified Gem-core interacted with the N-ERMAD of Ezrin when bound to GTPγS, but not in its GDP-bound form or after having been stripped of nucleotide by treatment with EDTA (Figure 1A). Conversely, this interaction was prevented when an excess of purified Ezrin C-ERMAD had previously been bound to the N-ERMAD (Figure 1B), a complex that mimics the inactive dormant form of Ezrin. Hence Gem directly interacts Ezrin in vitro, and this interaction requires both proteins to be in their active state.
Table 1.
Interaction of Gem with its partners in the yeast two-hybrid assay
| AD fusion | DBD fusion |
||||
|---|---|---|---|---|---|
| pGBT10 | Gem | ΔC | Core | S89N/A | |
| neoGNJC | − | − | − | − | − |
| Ezrin | − | ++ | ++ | ++ | − |
| ROCK I | − | ++ | ++ | ++ | − |
| Gmip | − | ++ | ++ | ++ | − |
The following fragments of Gem were used as baits: Gem (full length 1-296), Gem-core (core, 73-265), and GemΔC (ΔC, 1-265). S89N/A designates Gem carrying the S89N or S89A substitutions, which exhibited identical results. Preys were Ezrin (residues 1-309), ROCKI (residues 769-1117), and Gmip (1-721). pGBT10 and NeoGNJC, respectively, were used as empty vector controls. HF7c yeast were co-transformed with the combination of bait and prey plasmids indicated in the table; the expression of Gem and its mutants was checked in all double transformants. Interactions were scored with identical results for growth on selective medium lacking histidine and β-galactosidase activity.
Figure 1.

In vitro interaction of Gem with Ezrin. (A) The Ezrin N-ERMAD interacts with Gem-GTP. Purified Gem-core protein, 6 pmol, were loaded with GDP (D) or GTPγS (T) or were depleted from nucleotide by incubation with EDTA (E) and then incubated with GST alone or fused to the N-ERMAD of Ezrin. Top, Ponceau stain of the blot; bottom, presence of Gem associated with the beads revealed by Western blotting. (B) Binding of Gem-GTP to the N-ERMAD is inhibited by binding of the C-ERMAD. Purified N-ERMAD fused to GST on glutathione-Sepharose beads was preincubated with a 2× or 6× molar excess of purified C-ERMAD. After washing of excess free C-ERMAD, the N-ERMAD/C-ERMAD complex (visualized on the top Ponceau stain), was incubated with GTPγS-bound Gem-core, and the amount of Gem bound to the beads was revealed by Western blotting.
In Vivo Interaction between Gem and Ezrin
Using several independent methods, we show that Gem and Ezrin indeed interact in cells. Both proteins were coimmunoprecipitated from lysates of HeLa cells ectopically expressing FLAG-tagged Gem and VSV-G–tagged full-length Ezrin or its N-ERMAD (Figure 2A). Using antibodies that specifically react with Gem, but not with other proteins of the RGK family in immunoprecipitation and Western blotting (Figure 2C), we also show that endogenous Gem and Ezrin could be coimmunoprecipitated from HeLa (Figure 2B) and NIH 3T3 cells (not shown), two cell lines that constitutively express Gem (Hatzoglou and de Gunzburg, unpublished observations). Remarkably, Gem exhibited some selectivity in its interaction with ERM proteins as endogenous Gem coimmunoprecipitated with Ezrin and Moesin, but not with Radixin or with the more distantly related protein Merlin (Figure 2D). Because Ezrin, Moesin, and Radixin share 85% sequence identity between their N-ERMADs (Mangeat et al., 1999), we sought to use this property to identify the residues of Ezrin involved in its interaction with Gem. Only four residues that are common to the Ezrin and Moesin N-ERMADs are different in Radixin, and two among them (Ser 144 and Val 174 in Ezrin) are in regions that undergo structural changes during ERM activation (Smith et al., 2003). However, mutation of these two residues of Ezrin into the corresponding residues of Radixin failed to affect the interaction between Gem and Ezrin (data not shown).
Figure 2.

Gem interacts with Ezrin in vivo. (A) Coimmunoprecipitation of ectopically expressed Gem and Ezrin. HeLa cells were cotransfected with pRC/CMV-FLAG-Gem, and empty vector (−), pCB6-EzN-VSVG (EzN) or pCB6-Ezrin-VSVG (Ez). Forty-eight hours later, cell lysates were subjected to immunoprecipitation with anti-FLAG antibodies, followed by immunoblotting with anti-VSVG to detect Ezrin, or anti-Gem antibodies. (B) Association of endogenous Gem with Ezrin. HeLa cell lysates were immunoprecipitated either with anti-Gem polyclonal antibody or with an nonrelated antibody (NR) as control. Coprecipitated Ezrin was detected using polyclonal anti-Ezrin antibodies. (C) Specificity of Gem antibodies. HeLa cells were transfected with constructs encoding HA-tagged Gem, Rad, Rem, and Rem2. Lysates were either immunoblotted with HA (top) or affinity-purified Gem antibodies (middle); alternatively, they were immunoprecipitated with Gem antibodies followed by blotting with HA antibodies. (D) Specificity of the interaction of Gem with ERM proteins. Hela cell lysates were immunoprecipitated with Gem antibodies. Proteins present in the lysates or coimmunopricipitated with Gem were detected by Western blotting with antibodies specifically reacting with Ezrin, Radixin, Moesin, or Merlin.
Because Gem associates selectively with the active form of Ezrin in vitro, we proceeded to investigate whether such was also the case in cells and to establish whether complexes comprising Gem bound to active Ezrin were actually found in vivo. To this end, we used a differential detergent solubilization procedure whereby cells are sequentially extracted with 0.2 and 1% Triton X-100 (Figure 3), yielding two fractions that had previously been shown to be enriched in unphosphorylated and phosphorylated ERMs, respectively (Granes et al., 2000); ERM proteins phosphorylated in their C-terminal region at the position equivalent to Thr 567 in Ezrin were detected using phospho-specific antibodies. As shown in Figure 3, HeLa cells stimulated for 15 min with serum contained a significant level of activated endogenous ERM proteins, the bulk of which was not solubilized by 0.2% Triton and was selectively recovered in the 1% Triton extract. In contrast, unphosphorylated Ezrin was predominantly found in the 0.2% Triton fraction. Upon immunoprecipitation, equivalent amounts of Gem were recovered from both fractions. Gem-associated Ezrin in the low-detergent fraction was essentially unphosphorylated. This either suggests that Gem is able to interact with inactive dormant Ezrin in this fraction, a conclusion in contradiction with the results from in vitro interaction experiments (Figure 1); alternatively, it may indicate that the Ezrin molecules interacting with Gem represent a form of the protein where the N-ERMAD is not bound to the C-ERMAD, therefore presenting the hallmark of active Ezrin, but remain unphosphorylated in that particular compartment. In contrast, Gem-associated Ezrin recovered from the 1% Triton fraction was enriched in the phosphorylated form, showing that in this compartment at least, Gem indeed interacts with the active form of Ezrin.
Figure 3.
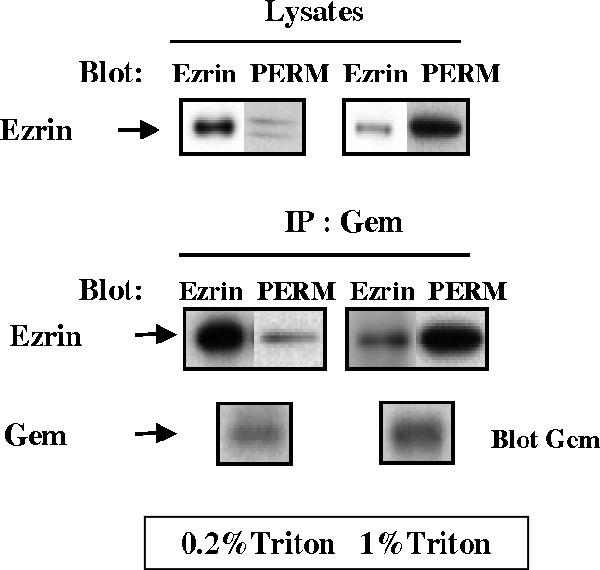
Gem associates with active Ezrin in vivo. Serum-starved HeLa cells were stimulated for 15 min with serum and sequentially extracted with 0.2 and 1% Triton X-100 as described in Materials and Methods. Extracts were immunoprecipitated with anti-Gem antibodies, and proteins were detected by Western blotting with anti-Gem or anti P-ERM antibodies and reblotted with anti-Ezrin antibodies.
The Coexpression of Gem with Ezrin Induces Cell Elongation
Previous studies had showed that the overexpression of Gem in various cells in the presence of serum leads to profound morphological alterations (Piddini et al., 2001; Ward et al., 2002). However, the expression of Gem in serum-starved HeLa (Figure 4a) or NIH 3T3 (not shown) cells did not affect their overall morphology; such was also the case for the expression of Ezrin alone (Figure 4b). Both individually expressed proteins exhibited a diffuse cytoplasmic staining, as well as some labeling of the plasma membrane and its protrusions. In contrast, the coexpression of Gem together with Ezrin resulted in a considerable elongation of serum-starved HeLa and NIH 3T3 cells, as well as a significant colocalization of both proteins at the plasma membrane (see Figure 4, d–f and j–o). This striking morphological effect was quantified for HeLa cells as depicted in Figure 5A; the effect was consistent across the population of cells ectopically expressing both proteins and represented a fourfold enhancement of their length/width ratio compared with control cells. We sought to characterize the dependence of this phenomenon on the activities of Gem and Ezrin. Qualitative (Figure 4) as well as quantitative (Figure 5A) results show that cell elongation required full-length and active Gem. Though the central core domain of Gem was sufficient to interact with Ezrin (Table 1 and Figure 1), it was unable to cooperate with it to induce cell elongation; in fact, deletion of the C-terminal extension of Gem, reported to be necessary for its membrane localization (Maguire et al., 1994), was sufficient to abrogate the morphological effect. Furthermore, the S89A (Figure 4c) or S89N (not shown) mutants of Gem, that partially localize to the plasma membrane similarly to the wild-type protein, equally failed to cause any morphological effect in cooperation with Ezrin. Reciprocally, cell elongation required a fully activatable Ezrin molecule: neither a mutant incapable of interacting with the plasma membrane because of its incapacity to bind the lipid PIP2 (Barret et al., 2000), nor a mutant in which the activated state could not be stabilized because of the substitution of Thr 567 by Ala, were able to cooperate with Gem for cell elongation (Figure 5B). Furthermore, expression of the constitutively active T567D Ezrin mutant, in the absence of ectopically expressed Gem, was not sufficient to induce the morphological effect. Hence our results show that in the absence of serum, the overexpression of Gem together with Ezrin causes cell elongation, and this effect requires the cooperation of full-length and intact Gem and Ezrin molecules.
Figure 4.

Morphological changes induced by the expression of Gem and Ezrin. Top, HeLa cells were transfected with expression vectors for Gem (a), GemS89A (c), or VSVG-tagged Ezrin (b) alone, or Gem and Ezrin together (d–f, wild-type Gem; g–i, GemS89A). Bottom, NIH 3T3 cells were cotransfected with vectors encoding Gem and VSVG-tagged Ezrin (j–l). Images in the bottom row (m–o) depict enlargements of the images from j–l, respectively. Sixteen hours after transfection, cells were fixed and stained with anti-Gem (a, c, d, g, j, and m) or anti-VSVG (b, e, h, k, and n) antibodies; merged green and red images are shown in f, i, l, and o. Bar, 20 μm.
Figure 5.

Quantification of cell elongation induced by Gem and Ezrin. (A) HeLa cells were transfected with empty vector (mock) or expression vectors encoding Gem, GemΔC, Gem-core, or GemS89A either alone or together with Ezrin. (B) HeLa cells were transfected with empty vector (mock) or expression vectors encoding Ezrin, the constitutively active T567D mutant (T/D), or the inactive PIP2 or T567A (T/A) mutants, either alone or in combination with Gem. Cells were incubated for 48 h in the absence of serum and processed for immunofluorescence to detect the expression of Gem and Ezrin; the elongation of transfected cells was calculated as described in Materials and Methods. The data represent the average of three independent experiments. (C) HeLa cells were transfected with expression vectors for Gem and its mutants as above. They were stimulated with 10% serum for 2 h, processed for immunofluorescence, and analyzed as above. The data represent the average of three separate experiments. (D) HeLa cells were cotransfected with an SiRNA directed against Luciferase (siLuc) or a mixture of siRNAs directed against ERM proteins (siERM), and empty vector or vector expressing Gem. The inset shows the level of ERM proteins in cells treated with SiRNAs. Cells were treated and observed as in C. The data represent the average of two separate experiments.
We next investigated whether the activation of endogenous Ezrin could substitute for its overexpression in cooperating with Gem to induce cell elongation. The stimulation of serum-starved HeLa cells with serum for 2 h led to a significant increase in the level of activated ERM proteins (not shown). This treatment also resulted in elongation of the cells ectopically expressing Gem (Figure 5C), though the effect was less pronounced than in serum-starved cells ectopically expressing both Gem and Ezrin; similarly, it required the expression of full-length and active Gem. Finally, we depleted HeLa cells from ERM proteins by treating them with a mixture of SiRNAs directed against each of these three proteins (Pust et al., 2005); as shown in the inset of Figure 5D, this resulted in a significant knockdown in the level of ERM proteins (70% reduction, relative to actin, not shown), as well as in a marked reduction of the elongation effect, thereby demonstrating that Ezrin is indeed required for the induction of cell elongation by Gem.
Gem Requires Ezrin To Affect the Actin Cytoskeleton and Cell Adhesion
HeLa cells stimulated with serum contain numerous actin stress fibers extending across the cell. In the elongated cells expressing Gem, stress fibers were no longer visible, yet the cortical actin network underlying the plasma membrane remained present and even reinforced in some cells (Figure 6A). Elongated serum-starved HeLa cells coexpressing Gem and Ezrin similarly displayed the absence of stress fibers and the concomitant appearance of a strong cortical actin network; it should be noted that despite this effect on the actin cytoskeleton, the network of microtubules appeared unaltered (not shown). Because the stimulation of cells with serum activates many signaling pathways, we investigated whether the elongation of cells and concomitant loss of actin stress fibers induced by Gem were dependent on the activity of Ezrin; to this end, we used the T567A mutant of Ezrin that was shown to act as a dominant-negative protein (Oshiro et al., 1998; Shaw et al., 1998; Gautreau et al., 2000; Tran Quang et al., 2000; Charras et al., 2006). Expression of this mutant alone did not affect actin structures nor focal adhesions (not shown). However, similarly to cells exhibiting SiRNA- mediated reduced expression of ERM proteins (Figure 5D), serum-stimulated HeLa cells ectopically expressing T657A Ezrin together with Gem retained their characteristic shape; as shown in Figure 6B, they also contained actin stress fibers similarly to control untransfected cells. Taken together these results show that Gem requires Ezrin to exert its morphological effects.
Figure 6.

Gem-induced cytoskeletal changes require active Ezrin. (A) HeLa cells were transfected with expression vectors for Gem or its S89A mutant. After a 16-h incubation in the absence of serum, cells were treated with 10% serum for 2 h, fixed, and stained with ALEXA-488-phalloidin to visualize F-actin (green) and anti-Gem antibodies (red). An overlay image is shown at the right. (B) HeLa cells were either transfected with Gem alone (top rows) or together with the dominant-negative Ezrin mutant T567A. After a 16-h incubation in the absence of serum, cells were treated with 10% serum for 2 h, fixed, and stained with ALEXA-488 phalloidin and antibodies to Gem (red) and VSVG (blue). Bar, 20 μm.
Elongated cells, whether expressing Gem alone and treated with serum (Figure 7A) or ectopically expressing both Gem and Ezrin in the absence of serum (not shown), only exhibited few focal adhesions, compared with their untransfected counterparts. The remaining adhesions, visualized by the presence of vinculin, were generally very large and were located at both extremities of the cells, as well as at few peripheral locations. Again, this phenotype was not observed when the inactive Gem S89A mutant was expressed, confirming that it is dependent on active Gem. Such a combination of the loss of stress fibers and disappearance of most focal adhesions explains the elongated morphology of the cells expressing Gem and Ezrin in the absence of serum, as well as those expressing Gem alone and treated with serum. At the biochemical level, this phenotype corresponded to a reduction in the level of active ERM proteins, as assessed by their C-terminal phosphorylation, as well as in the tyrosine phosphorylation of proteins involved in focal adhesions such as the focal adhesion kinase FAK and its close relative PYK2 (Figure 7B). Concomitantly, cells transfected with a Gem expression vector exhibited a reduction in their adhesion capacity to plastic or fibronectin (Figure 7C) compared with cells transfected with vector alone or the inactive Gem S89A mutant.
Figure 7.

Gem induces a reduction of cell adhesion. (A) HeLa cells were transfected with an expression vector for Gem (wt or S89A), starved for 16 h and stimulated with 10% serum for 2 h. They were stained for Vinculin (green) and Gem (red). Scale bar, 20 μm. (B) HeLa cells transfected with empty vector, or vector expressing Gem, were treated as above and lysates were analyzed by Western blotting for the presence of total and phosphorylated ERM, FAK, and PYK2 proteins. (C) HeLa cells transfected with vector alone, or vector expressing either wt or S89A Gem were assayed 24 h later for adhesion on plastic or fibronectin.
Gem Negatively Regulates Rho via the Rho-GAP Protein Gmip
Because Rho proteins play an important role in controlling the actin cytoskeleton, we investigated whether the physiological expression of Gem had any effect on the activity of these GTPases. To that end, we knocked down the expression of Gem in NIH 3T3 cells using RNA interference. The lack of antibodies able to detect the endogenous protein in NIH 3T3 cells led us to assess Gem expression at the level of its mRNA. As shown in Figure 8A, the treatment of cells with two different SiRNAs that efficiently reduced expression of the Gem mRNA resulted in a twofold increase in the level of active RhoA, however with no effect on the levels of active Cdc42 and Rac1. This is evidence that Gem specifically down-regulates the activity of RhoA, which is consistent with the disappearance of stress fibers from cells overexpressing Gem. Furthermore, we knocked down the expression of Gem in Hela cells using two different SiRNAs directed against human Gem; as shown in Figure 8B, this treatment was sufficient to induce the appearance of abundant stress fibers in serum-starved cells. Taken together, these results demonstrate that endogenous Gem down-regulates the activity of Rho and thereby controls its downstream effects on the actin cytoskeleton.
Figure 8.

Gem expression selectively down-regulates the level of active Rho. (A) NIH 3T3 cells were transfected with SiRNAs m1 or m2 directed against mouse Gem. Twenty-four hours later, (a) the level of remaining Gem mRNA was assessed by RT-PCR, and the levels of total and GTP-bound RhoA were measured by a pull-down assay. (b) Quantification of the relative levels of activated RhoA from A. (c and d) The levels of total and GTP-bound Rac and Cdc42 were quantified by pull-down assays in cells treated with anti-Gem SiRNAs. (B) HeLa cells were transfected with SiRNAs directed against human Gem or Luciferase (siLuc). After 24 h in the absence of serum, the level of remaining Gem protein was assessed by Western blotting, using actin as an internal standard. Cells seeded on coverslips were fixed and stained with ALEXA-488-phalloidin to visualize F-actin (green) and DAPI for nuclei. Scale bar, 20 μm, for all three micrographs.
We had previously shown that Gem interacts with Gmip, a protein carrying a Rho-GAP activity that acts specifically on RhoA, but neither on Rac nor on Cdc42 (Aresta et al., 2002); the selectivity of Gmip for RhoA suggested that it may constitute a good candidate to mediate the action of Gem on the Rho pathway and the actin cytoskeleton. To test this hypothesis, we knocked down Gmip in Hela cells using two different SiRNAs. As shown in Figure 9A, both SiRNAs efficiently reduced the expression of endogenous Gmip in the presence or absence of ectopically expressed Gem. Under these conditions, the overexpression of Gem in serum-stimulated HeLa cells was no longer able to induce cell elongation (Figure 9B), loss of stress fibers or the reduction of the number of focal adhesions (Figure 9D), nor the reduction in the level of phosphorylated ERMs characteristic of the down-regulation of the RhoA pathway by Gem (Figure 9C). Hence, we conclude that Gem requires Gmip to exert its biochemical and morphological effects. We then carried out fractionation experiments to assess whether there were any changes in the subcellular localization of Gmip consequential to the expression of Gem. As shown in Figure 10, endogenous Gmip is almost exclusively found in the cytosolic fraction of serum-starved as well as serum-stimulated cells. However, the overexpression of Gem and Ezrin in serum-starved cells enhanced the amount of endogenous Gmip present in the membrane fraction (Figure 10A); this effect was not seen when inactive mutants of either Gem or Ezrin were expressed (Ezrin T/A and Gem S89A, respectively). Similarly, a substantial enrichment of Gmip was found in the membranes of cells overexpressing Gem and stimulated with serum, an effect that was also dependent on the expression of active Gem (Figure 10B). Because the morphological effects of Gem require ERM proteins that are mainly active at the plasma membrane, our results suggest that thanks to its interaction with Ezrin, Gem may act to direct Gmip to the plasma membrane to down-regulate the activity of the Rho pathway.
Figure 9.

Gem requires Gmip to down-regulate stress fibers and focal adhesions. (A) Efficiency of Gmip SiRNAs. HeLa cells were first transfected with a control SiRNA directed against luciferase (Luc), or SiRNAs 1 or 2 directed against Gmip. Twenty-four hours later, they were transfected with empty vector or vector encoding Gem. The levels of Gmip, Gem, and α-adaptin (loading control) were assessed in lysates by Western blotting. (B) Gmip is required for cell elongation induced by Gem. HeLa cells were transfected as in A, and cell elongation was quantified as in Figure 5. (C) Cells transfected as above were starved for 16 h and stimulated with serum for 30 min. The levels of transfected Gem, endogenous Gmip, total Ezrin, and phosphorylated ERMs were assessed by Western blotting and quantified as indicated in Materials and Methods. (D) Cells were transfected as above with SiRNA directed against Gmip and Gem expression vector. Cells in the top panel were stained with antibodies against Vinculin and Gem. Cells in the bottom panel were stained with ALEXA-488 phalloidin and anti-Gem antibodies. Bar, 20 μm.
Figure 10.

Gem expression recruits Gmip to membranes. (A) HeLa cells were cotransfected with expression vectors encoding Gem (wild type [wt] or carrying the S89A mutation) and Ezrin (wt or the T567A mutation [T/A]) and maintained for 48 h in the absence of serum. (B) Cells were transfected as above with Gem only (wt or carrying the S89A mutation) and stimulated for 2 h with serum. In all cases, cells were lyzed and separated into cytosolic and membrane fractions as indicated in Materials and Methods. The contents of each fraction in Gmip, Gem, and actin was assessed by Western blotting.
DISCUSSION
There is converging evidence that the atypical Ras family protein Gem affects cell morphology and the actin cytoskeleton through pathways controlled by Rho GTPases. Indeed, Gem was described to interact with the Rho effector kinase ROCK I and to negatively regulate the Rho-ROCK pathway (Ward et al., 2002). In addition, we had shown that Gem interacts with Gmip, a Rho-specific GAP (Aresta et al., 2002), but the in vivo consequences of this interaction remained to be established. In this study, we show that Gem associates with the membrane–cytoskeleton linker protein Ezrin in vivo and that Gem requires Ezrin and Gmip to down-regulate the RhoA pathway, inhibit the formation of stress fibers, and reduce the level of focal adhesions. Although previous studies had documented that the ectopic expression of Gem severely affects the morphology of various cells growing in serum (Piddini et al., 2001; Ward et al., 2002), we show here that in the absence of serum, the expression of Gem has no morphological consequences. However, its coexpression with Ezrin results in the striking elongation of serum-starved cells, characterized by the disappearance of actin stress fibers, enhancement of the cortical actin network, and loss of most focal adhesions. A similar phenotype is obtained after the stimulation with serum of Gem-expressing cells, a treatment that, among many other consequences, enhances the level of activated endogenous ERM proteins as assessed by their phosphorylation at Thr 567 in Ezrin or the equivalent residue in Radixin or Moesin. The fact that cell elongation is markedly reduced in cells where the level of ERM proteins has been knocked down by treatment with SiRNAs demonstrates that Gem indeed requires ERM proteins to exert its cell elongation effect. However, the mere interaction of Gem with Ezrin is insufficient to trigger cell elongation, and this effect requires Ezrin activation. Indeed, two Ezrin mutants, incapable of undergoing activation in cells: i) an Ezrin mutant that is deficient in PIP2 binding and remains cytosolic (Barret et al., 2000) and ii) the T567A Ezrin mutant that is unable to undergo the C-terminal phosphorylation characteristic of its activated state (Gautreau et al., 2000), both fail to elicit cell elongation in cooperation with Gem (Figure 5B) though they are able to interact with Gem (not shown).
Conversely, the morphological effects require full-length and active Gem molecules: the S89N and S89A mutants of Gem, which by analogy to all other Ras-related proteins should be unable to switch to the active GTP-bound state (Opatowsky et al., 2006) and are unable to bind Ezrin and fail to cooperate with Ezrin for cell elongation. Furthermore, the C-terminal domain of Gem, which is necessary for its interaction with membranes (Maguire et al., 1994), but dispensable for its binding to Ezrin (Table 1 and Figure 1), is also required. These observations, taken together with our biochemical data showing that in the cellular fraction containing the bulk of active phosphorylated ERMs, presumably the plasma membrane, Gem is complexed with active ERMs, suggest that the Gem molecules that are biologically active in eliciting cell elongation might be those associated with Ezrin at the membrane. We have not elucidated how Gem switches to its GTP-bound form to be able to interact with Ezrin in cells, as suggested by the in vitro experiments shown in Figure 1; one hypothesis is that Ezrin might somehow elicit exchange of GDP for GTP on Gem or stabilize the GTP-bound form of the protein.
The elongation of cells induced by Gem and Ezrin does not involve rearrangements of the microtubule network, and contrarily to the induction by Gem of a dendritic-like phenotype described by Piddini et al. (2001), the effects that we observed are unaffected by the treatment of cells with the microtubule-targeted drugs nocodazole or cytochalasin D (Hatzoglou and de Gunzburg, unpublished observations). Elongation of the cells is likely to result from the observed rearrangements of the actin cytoskeleton, i.e., the disappearance of actin stress fibers concomitant with the enhancement of the cortical actin network and the loss of most focal adhesions except for those at the extremities of the cells and some at their periphery, processes that depend on the activity of the Rho pathway (Etienne-Manneville and Hall, 2002). Indeed, using RNA interference, we show that endogenous Gem down-regulates the level of active RhoA and of downstream actin stress fibers. Furthermore, we show that the Rho-GAP protein Gmip, that interacts with Gem, is responsible for the effects of Gem downstream from RhoA, i.e., reduction of ERM phosphorylation, loss of actin stress fibers, and drop in the number of focal adhesions. The fact that under the conditions that elicit cell elongation, i.e., coexpression of intact Gem and Ezrin, or expression of intact Gem in serum-stimulated cells, Gmip is enriched in membranes suggests that the effects of Gem on the actin cytoskeleton are mediated by those molecules recruited by active ERM proteins at the plasma membrane. We sought to test this hypothesis by directing Gem to the plasma membrane through the substitution of its seven C-terminal residues by the CAAX-containing C-terminal region of K-Ras; contrarily to the inactive control mutant where the Cys of the CAAX had been changed to a Ser, expression of Gem-CAAX led to the rapid rounding and detachment of nearly all transfected cells, indicating that targeting overexpressed Gem to the plasma membrane is highly toxic to cells (not shown).
In wake of the observations reported in this study, we propose as a working hypothesis that the interaction of Gem with active Ezrin at the plasma membrane leads, via the Rho-GAP protein Gmip, to a local inactivation of the Rho pathway resulting in the cytoskeletal effects described above. This mechanism could act synergistically with the previously reported ability of Gem to inhibit the formation of actin stress fibers induced by ROCK I (Ward et al., 2002). Hence Gem would exert a dual action on the Rho pathway, both down-regulating the activity of Rho and its downstream effectors via the Rho-GAP protein Gmip and inhibiting that of its effector ROCK I via direct interaction. This is reminiscent of the mechanism through which the Rnd3/RhoE protein acts as a Rho antagonist by inhibiting the activity of ROCK I (Riento et al., 2003), as well as by recruiting and activating p190Rho-GAP (Wennerberg et al., 2003), also leading to a loss of actin stress fibers and focal adhesions. Hence Gem and Rnd3/RhoE use similar biochemical mechanisms to control the Rho pathway, yet they may exert their actions in different cell types or at distinct subcellular locations. Indeed, Gem and Rnd3/RhoE are constitutively expressed at low levels in many cells (Chardin, 2006; de Gunzburg, unpublished observations) but exhibit regulated expression in response to distinct extracellular cues. The Rnd3/RhoE protein is induced in fibroblasts in response to PDGF (Riento et al., 2003) and DNA-damaging agents (Villalonga et al., 2004). Gem mRNA was reported to be induced in T lymphocytes stimulated via the TCR (Maguire et al., 1994), endothelial cells treated with inflammatory cytokines (Vanhove et al., 1997), and primary human fibroblasts in response to serum (Iyer et al., 1999). Little is known about the subcellular locations Rnd3/RhoE and Gem. However, the results presented here lead us to propose that a fraction of Gem molecules associate with active Ezrin at the plasma membrane–cytoskeleton interface where they would act via Gmip, to regulate the activity of the Rho pathway. The interplay between Ezrin, Gem, and Gmip may be highly dynamic because we were unable so far to isolate a complex simultaneously containing all three proteins. Further investigation will be required to understand how and in which circumstances two proteins of the Ras family, Gem and Rnd3/RhoE, exert their actions to regulate the activity of another Ras family protein and its downstream pathways, RhoA.
ACKNOWLEDGMENTS
We are indebted to Dr. Monique Arpin for Ezrin mutants, expression constructs, and antibodies; to Dr. Bruno Goud for P5D4 anti-VSVG antibodies; and to M. Martin, C. Roy, and A McClatchey for antibodies against Moesin, Radixin, and Merlin. We acknowledge the invaluable help of Dr. Jacques Camonis in construction of the placental cDNA library and its screening in the yeast two-hybrid system and are thankful to Dr. Corinne Leprince for help with confocal microscopy, as well as to Anne Ridley for her critical reading of the manuscript. A.H. and I.A. were supported by postdoctoral fellowships from Inserm and the Ligue Nationale contre le Cancer, respectively, and this work was funded in part by the Association de Recherche contre le Cancer.
Abbreviations used:
- ERM
Ezrin, Radixin, Moesin
- N-ERMAD
N-terminal ERM association domain
- C-ERMAD
C-terminal ERM association domain
- PIP2
phosphatidylinositol 4,5 bisphosphate
- GAP
GTPase-activating protein
- GST
glutathione S-transferase
- TEV
tobacco etch virus.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E06-06-0510) on January 31, 2007.
 The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
REFERENCES
- Algrain M., Turunen O., Vaheri A., Louvard D., Arpin M. Ezrin contains cytoskeleton and membrane binding domains accounting for its proposed role as a membrane-cytoskeletal linker. J. Cell Biol. 1993;120:129–139. doi: 10.1083/jcb.120.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreoli C., Martin M., Le Borgne R., Reggio H., Mangeat P. Ezrin has properties to self-associate at the plasma membrane. J. Cell Sci. 1994;107(Pt 9):2509–2521. doi: 10.1242/jcs.107.9.2509. [DOI] [PubMed] [Google Scholar]
- Aresta S., de Tand-Heim M. F., Béranger F., de Gunzburg J. A novel Rho GTPase-activating-protein interacts with Gem, a member of the Ras superfamily of GTPases. Biochem. J. 2002;367:57–65. doi: 10.1042/BJ20020829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barret C., Roy C., Montcourrier P., Mangeat P., Niggli V. Mutagenesis of the phosphatidylinositol 4,5-bisphosphate (PIP2) binding site in the NH2-terminal domain of Ezrin correlates with its altered cellular distribution. J. Cell Biol. 2000;151:1067–1080. doi: 10.1083/jcb.151.5.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner M., Sillman A. L., Blackwood E. M., Srivastava J., Madson N., Schilling J. W., Wright J. H., Barber D. L. The Nck-interacting kinase phosphorylates ERM proteins for formation of lamellipodium by growth factors. Proc. Natl. Acad. Sci. USA. 2006;103:13391–13396. doi: 10.1073/pnas.0605950103. Epub 2006 Aug 13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguin P., Mahalakshmi R. N., Nagashima K., Cher D.H.K., Takahashi A., Yamada Y., Seino Y., Hunziker W. 14-3-3 and calmodulin control subcellular distribution of Kir/Gem and its regulation of cell shape and calcium channel activity. J. Cell Sci. 2005;118:1923–1934. doi: 10.1242/jcs.02321. [DOI] [PubMed] [Google Scholar]
- Beguin P., Nagashima K., Gonoi T., Shibasaki T., Takahashi K., Kashima Y., Ozaki N., Geering K., Iwanaga T., Seino S. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature. 2001;411:701–706. doi: 10.1038/35079621. [DOI] [PubMed] [Google Scholar]
- Beranger F., Goud B., Tavitian A., de Gunzburg J. Association of the Ras-antagonistic Rap1/Krev-1 proteins with the Golgi complex. Proc. Natl. Acad. Sci. USA. 1991;88:1606–1610. doi: 10.1073/pnas.88.5.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A., Edwards K., Fehon R. G. ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 2002;3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- Chardin P. Function and regulation of Rnd proteins. Nat. Rev. Mol. Cell Biol. 2006;7:54–62. doi: 10.1038/nrm1788. [DOI] [PubMed] [Google Scholar]
- Charras G. T., Hu C. K., Coughlin M., Mitchison T. J. Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 2006;175:477–490. doi: 10.1083/jcb.200602085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S., Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Fievet B. T., Gautreau A., Roy C., Del Maestro L., Mangeat P., Louvard D., Arpin M. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J. Cell Biol. 2004;164:653–659. doi: 10.1083/jcb.200307032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlin B. S., Andres D. A. Rem is a new member of the Rad- and Gem/Kir Ras-related GTP-binding protein family repressed by lipopolysaccharide stimulation. J. Biol. Chem. 1997;272:21982–21988. doi: 10.1074/jbc.272.35.21982. [DOI] [PubMed] [Google Scholar]
- Finlin B. S., Crump S. M., Satin J., Andres D. A. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. Proc. Natl. Acad. Sci. USA. 2003;100:14469–14474. doi: 10.1073/pnas.2437756100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlin B. S., Mosley A. L., Crump S. M., Correll R. N., Ozcan S., Satin J., Andres D. A. Regulation of L-type Ca2+ channel activity and insulin secretion by the Rem2 GTPase. J. Biol. Chem. 2005;280:41864–41871. doi: 10.1074/jbc.M414261200. [DOI] [PubMed] [Google Scholar]
- Finlin B. S., Shao H., Kadono-Okuda K., Guo N., Andres D. A. Rem2, a new member of the Rem/Rad/Gem/Kir family of Ras-related GTPases. Biochem. J. 2000;347(Pt 1):223–231. [PMC free article] [PubMed] [Google Scholar]
- Fischer R., Wei Y., Anagli J., Berchtold M. W. Calmodulin binds to and inhibits GTP binding of the ras-like GTPase Kir/Gem. J. Biol. Chem. 1996;271:25067–25070. doi: 10.1074/jbc.271.41.25067. [DOI] [PubMed] [Google Scholar]
- Fromont-Racine M., Rain J. C., Legrain P. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat. Genet. 1997;16:277–282. doi: 10.1038/ng0797-277. [DOI] [PubMed] [Google Scholar]
- Gautreau A., Louvard D., Arpin M. Morphogenic effects of ezrin require a phosphorylation-induced transition from oligomers to monomers at the plasma membrane. J. Cell Biol. 2000;150:193–203. doi: 10.1083/jcb.150.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granes F., Urena J., Rocamora N., Vilaro S. Ezrin links syndecan-2 to the cytoskeleton. J. Cell Sci. 2000;113:1267–1276. doi: 10.1242/jcs.113.7.1267. [DOI] [PubMed] [Google Scholar]
- Hamada K., Shimizu T., Matsui T., Tsukita S., Tsukita S., Hakoshima T. Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 2000;19:4449–4462. doi: 10.1093/emboj/19.17.4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V. R., et al. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- Kaiser C., Michaelis S., Mitchell A. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Maguire J., Santoro T., Jensen P., Siebenlst U., Yewdell J., Kelly K. Gem: an induced, immediate early protein belonging to the Ras family. Science. 1994;265:241–244. doi: 10.1126/science.7912851. [DOI] [PubMed] [Google Scholar]
- Mangeat P., Roy C., Martin M. ERM proteins in cell adhesion and membrane dynamics. Trends Cell Biol. 1999;9:187–192. doi: 10.1016/s0962-8924(99)01544-5. [DOI] [PubMed] [Google Scholar]
- Matsui T., Maeda M., Doi Y., Yonemura S., Amano M., Kaibuchi K., Tsukita S., Tsukita S. Rho-Kinase phosphorylates COOH-terminal threonines of Ezrin/Radixin/Moesin (ERM) proteins and regulates their head-to-tail association. J. Cell Biol. 1998;140:647–657. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T., Yonemura S., Tsukita S. Activation of ERM proteins in vivo by Rho involves phosphatidyl-inositol 4-phosphate 5-kinase and not ROCK kinases. Curr. Biol. 1999;9:1259–1262. doi: 10.1016/s0960-9822(99)80508-9. [DOI] [PubMed] [Google Scholar]
- Moyers J. S., Bilan P. J., Zhu J., Kahn C. R. Rad and Rad-related GTPases interact with calmodulin and calmodulin- dependent protein kinase II. J. Biol. Chem. 1997;272:11832–11839. doi: 10.1074/jbc.272.18.11832. [DOI] [PubMed] [Google Scholar]
- Ng T., et al. Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility. EMBO J. 2001;20:2723–2741. doi: 10.1093/emboj/20.11.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opatowsky Y., Sasson Y., Shaked I., Ward Y., Chomsky-Hecht O., Litvak Y., Selinger Z., Kelly K., Hirsch J. A. Structure-function studies of the G-domain from human gem, a novel small G-protein. FEBS Lett. 2006;580:5959–5964. doi: 10.1016/j.febslet.2006.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro N., Fukata Y., Kaibuchi K. Phosphorylation of moesin by Rho-associated kinase (Rho-kinase) plays a crucial role in the formation of microvilli-like structures. J. Biol. Chem. 1998;273:34663–34666. doi: 10.1074/jbc.273.52.34663. [DOI] [PubMed] [Google Scholar]
- Pan J. Y., Fieles W. E., White A. M., Egerton M. M., Silberstein D. S. Ges, a human GTPase of the Rad/Gem/Kir family, promotes endothelial cell sprouting and cytoskeleton reorganization. J. Cell Biol. 2000;149:1107–1116. doi: 10.1083/jcb.149.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piddini E., Schmid J. A., de Martin R., Dotti C. G. The Ras-like GTPase Gem is involved in cell shape remodelling and interacts with the novel kinesin-like protein KIF9. EMBO J. 2001;20:4076–4087. doi: 10.1093/emboj/20.15.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietromonaco S. F., Simons P. C., Altman A., Elias L. Protein kinase C-theta phosphorylation of moesin in the actin-binding sequence. J. Biol. Chem. 1998;273:7594–7603. doi: 10.1074/jbc.273.13.7594. [DOI] [PubMed] [Google Scholar]
- Pust S., Morrison H., Wehland J., Sechi A. S., Herrlich P. Listeria monocytogenes exploits ERM protein functions to efficiently spread from cell to cell. EMBO J. 2005;24:1287–1300. doi: 10.1038/sj.emboj.7600595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X. D., Schwartz M. A. Determination of GTP loading on Rho. Methods Enzymol. 2000;325:264–272. doi: 10.1016/s0076-6879(00)25448-7. [DOI] [PubMed] [Google Scholar]
- Reynet C., Kahn C. R. Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science. 1993;262:1441–1444. doi: 10.1126/science.8248782. [DOI] [PubMed] [Google Scholar]
- Riento K., Guasch R. M., Garg R., Jin B., Ridley A. J. RhoE binds to ROCK I and inhibits downstream signaling. Mol. Cell. Biol. 2003;23:4219–4229. doi: 10.1128/MCB.23.12.4219-4229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander E. E., ten Klooster J. P., van Delft S., van der Kammen R. A., Collard J. G. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw R. J., Henry M., Solomon F., Jacks T. RhoA-dependent phosphorylation and relocalization of ERM proteins into apical membrane/actin protrusions in fibroblasts. Mol. Biol. Cell. 1998;9:403–419. doi: 10.1091/mbc.9.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield P., Garrard S., Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Express. Purif. 1999;15:34–39. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- Smith W. J., Nassar N., Bretscher A., Cerione R. A., Karplus P. A. Structure of the active N-terminal domain of Ezrin. Conformational and mobility changes identify keystone interactions. J. Biol. Chem. 2003;278:4949–4956. doi: 10.1074/jbc.M210601200. [DOI] [PubMed] [Google Scholar]
- Splingard A., et al. Biochemical and structural characterization of the Gem GTPase. J. Biol. Chem. 2007;282:1905–1915. doi: 10.1074/jbc.M604363200. [DOI] [PubMed] [Google Scholar]
- Tran Quang C., Gautreau A., Arpin M., Treisman R. Ezrin function is required for ROCK-mediated fibroblast transformation by the Net and Dbl oncogenes. EMBO J. 2000;19:4565–4576. doi: 10.1093/emboj/19.17.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S., Yonemura S. Cortical actin organization: lessons from ERM (ezrin/radixin/moesin) proteins. J. Biol. Chem. 1999;274:34507–34510. doi: 10.1074/jbc.274.49.34507. [DOI] [PubMed] [Google Scholar]
- Vanhove B., HoferWarbinek R., Kapetanopoulos A., Hofer E., Bach F. H., DeMartin R. Gem, a GTP-binding protein from mitogen-stimulated T cells, is induced in endothelial cells upon activation by inflammatory cytokines. Endothelium (New York) 1997;5:51–61. doi: 10.3109/10623329709044158. [DOI] [PubMed] [Google Scholar]
- Villalonga P., Guasch R. M., Riento K., Ridley A. J. RhoE inhibits cell cycle progression and Ras-induced transformation. Mol. Cell. Biol. 2004;24:7829–7840. doi: 10.1128/MCB.24.18.7829-7840.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward Y., Spinelli B., Quon M. J., Chen H., Ikeda S. R., Kelly K. Phosphorylation of critical serine residues in Gem separates cytoskeletal reorganization from down-regulation of calcium channel activity. Mol. Cell. Biol. 2004;24:651–661. doi: 10.1128/MCB.24.2.651-661.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward Y., Yap S.-F., Ravichandran V., Matsumura F., Ito M., Spinelli B., Kelly K. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J. Cell Biol. 2002;157:291–302. doi: 10.1083/jcb.200111026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennerberg K., Forget M. A., Ellerbroek S. M., Arthur W. T., Burridge K., Settleman J., Der C. J., Hansen S. H. Rnd proteins function as RhoA antagonists by activating p190 RhoGAP. Curr. Biol. 2003;13:1106–1115. doi: 10.1016/s0960-9822(03)00418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]