

Abstract
While several West Nile vaccines are being developed, none are yet available for humans. In this study aimed at developing a vaccine for humans, West Nile virus (WNV) envelope protein (E) and non-structural protein 1 (NS1) were produced in the Drosophila S2 cell expression system. The C-terminal 20% of the E protein, which contains the membrane anchor portion, was deleted, thus allowing for efficient secretion of the truncated protein (80E) into the cell culture medium. The proteins were purified by immunoaffinity chromatography (IAC) using monoclonal antibodies that were flavivirus envelope protein group specific (for the 80E) or flavivirus NS1 group specific (for NS1). The purified proteins were produced in high yield and used in conjunction with adjuvant formulations to vaccinate mice. The mice were tested for both humoral and cellular immune responses by a plaque reduction neutralization test and ELISA, and by lymphocyte proliferation and cytokine production assays, respectively. The results revealed that the 80E and the NS1 proteins induced both high-titered ELISA and neutralizing antibodies in mice. Splenocytes from immunized mice, cultured in vitro with the vaccine antigens as stimulants, showed excellent proliferation and production of cytokines (IFN-γ, IL-4, IL-5, and IL-10). The level of antigen-stimulated lymphocyte proliferation and cytokine production was comparable to the level obtained from mitogen (phytohemagglutinin or pokeweed) stimulation, indicating a robust cellular response as well. These findings are encouraging and warrant further in vivo studies to determine the protective efficacy of the WNV vaccine candidate.
Keywords: West Nile vaccine, Recombinant subunit vaccine
1. Introduction
Since the introduction of West Nile virus (WNV) into the United States in 1999, annual outbreaks have caused severe and fatal encephalitis in humans and equines and death in a variety of species of feral birds throughout the U.S. and parts of Canada [1,2]. In addition, more recent findings show evidence of WNV human and equine infection in several countries of tropical America [3]. While WNV disease generally results in moderate to severe flu-like symptoms in approximately 20% of infected individuals, about 1 out of 150 progress to severe neurological disease [4]. However, the proportion of reported cases that are neuroinvasive is much higher, about 40% in the past few years [2], because of biased reporting of neuroinvasive disease compared to uncomplicated WNV fever. About 5–14% of neuroinvasive cases are fatal. In a high percentage of the non-fatal cases permanent neurological disabilities have been reported. These clinical findings are significantly worse in elderly patients. In a recent study involving a group of 233 hospitalized patients during an outbreak of WNV infections in Israel, the overall case fatality rate was 14%. However, the case fatality rate was 29% among patients aged 70 or older [5]. Similar findings were also reported from recent epidemics in Romania [6] and Russia [7]. Thus, there is significant morbidity and mortality associated with the disease.
WNV is a member of the Flaviviridae family, genus Flavivirus. As an enveloped, positive strand RNA virus, the RNA genome comprises 10 genes, coding for three structural and 7 non-structural proteins [8]. The structural proteins include the core or capsid protein (C), and a pre-membrane protein (prM), which is cleaved to yield glycosylated membrane protein and envelope protein (E) in the mature virion. The envelope protein shares significant homology with the envelope proteins of other flaviviruses, particularly those of the other members of the Japanese encephalitis virus (JEV) serocomplex: JEV, St. Louis encephalitis virus (SLEV), and Murray Valley encephalitis virus (MVEV). Antibodies directed against particular epitopes of the envelope protein are capable of virus neutralization, i.e., the inhibition of virus infection of susceptible cells in vitro. The dominant neutralizing epitopes have been mapped to one of three domains of the envelope protein, domain III, using monoclonal antibodies for dengue virus [9], as well as JEV [10] and WNV [11]. Additional neutralizing epitopes have recently been identified on domains I and II of the envelope protein as well [12]. Neutralizing antibodies (recognizing domain III epitopes) are generally specific for each virus and do not cross-neutralize other viruses (or other serotypes of the same virus if multiple serotypes exist). Viral neutralizing antibodies are generally accepted as the best in vitro correlate of in vivo protection against viral infection or immunity to subsequent infection for flaviviruses [13,14]. Therefore, assessment of virus neutralizing responses induced by vaccine candidates represents a reasonable way to screen for potential vaccine efficacy.
Currently, there is no approved commercially available vaccine for prevention of WNV infection in humans, and therapy for disease is limited to only supportive and symptomatic treatment. Several candidate WNV vaccines are in various stages of research and development. These include: (i) a “naked” DNA vaccine encoding the prM and E genes [15]; (ii) a live, attenuated dengue serotype 4-WNV chimera [16]; (iii) a live attenuated Yellow Fever-WNV chimera [17]; (iv) a recombinant envelope protein vaccine expressed in E. coli [18] and Drosophila cells [19]; (v) a live, attenuated WNV (veterinary) vaccine [20]; (vi) a formalin-inactivated WNV (veterinary) vaccine [21]; and a canarypox virus vectored vaccine [22]. Scientists at Hawaii Biotech have successfully used a proprietary method of expression to produce recombinant envelope proteins from flaviviruses, such as dengue serotypes 1–4, JEV, hepatitis C, and WNV [23–26]. These proteins are truncated at the C-terminus, leaving 80% of the native envelope protein (80E). The truncation deletes the membrane anchor portion of the protein, thus allowing it to be secreted into the extracellular medium, facilitating recovery. Furthermore, the expressed proteins have been shown to be properly glycosylated and to maintain native conformation as determined by reactivity with conformationally sensitive monoclonal antibodies (Hawaii Biotech, unpublished data), and X-ray crystallography structure determination [24–26]. The expression system used to produce these recombinant proteins involves the construction of expression plasmids containing cDNAs which are then used to transform insect cells. The resulting transformant cell lines have been shown to be genetically stable by Southern blot analysis of restriction digests of DNA from serially passaged cell lines. This has been shown for at least 10 transformant cell lines (Clements, DE, et al., Hawaii Biotech, unpublished data).
Moreover, in addition to the envelope protein, we investigated inclusion of a non-structural protein (NS1) from WNV in the vaccine formulations. The purpose of including NS1 protein is the potential to enhance the ability of the vaccine to elicit a cell-mediated immune response, as well as an additional humoral component of immunity. Although non-structural proteins are not present in mature virions, they are produced as a necessary part of the enzymatic system for viral replication. Peptide epitopes processed from these proteins are displayed on the surface of infected antigen-presenting cells in association with MHC class I or class II molecules, and thus may be recognized by subsets of immune cell populations, i.e., CD8+ or CD4+ T lymphocytes. When activated, these cells can function as cytotoxic T cells, and thus are capable of eliminating cells infected with virus [27,28]. This cellular immune response may contribute significantly to the overall protective efficacy of a subunit vaccine. In addition, there is evidence that NS1 may elicit a humoral protective immune response involving the complement fixing activity of antibodies to this protein [29,30], through mechanisms, such as antibody-dependent, complement-mediated cytolysis, or Fc receptor mediated antibody-dependent cellular cytotoxicity [30]. Thus, the inclusion of NS1 in the vaccine formulation can be justified on the basis of a humoral as well as a cellular immune response. The same expression system used for production of recombinant envelope proteins has also been used successfully for the production of the NS1 protein from dengue virus, and is now being used successfully for the production of NS1 from WNV.
The purpose of this study is to describe the production, purification, and immunogenicity in mice of a WNV recombinant subunit vaccine. The protective efficacy of the vaccine in the golden hamster model of West Nile encephalitis is described in an accompanying paper [31].
2. Materials and methods
2.1. Animals
Balb/c or Swiss Webster mice, 6–8 week old females, were used in all experiments. Mice were obtained from Charles River Laboratories (Hollister, CA), and maintained in a specific pathogen-free (SPF) environment and given food and water ad libitum.
2.2. Vaccines
2.2.1. Expression of recombinant proteins
The West Nile envelope protein (E) and non-structural protein 1 (NS1) were produced in the Drosophila S2 expression system. The expression system consists of the Drosophila S2 cells and a series of broad host plasmid vectors that directed the expression of heterologous proteins [32–34]. The expression plasmid pMttbns (derived from pMttPA) contained the following elements: Drosophila melanogaster metallothionein promoter, the human tissue plasminogen activator secretion leader (tPAL) and the SV40 early polyadenylation signal. A 14 base pair BamHI fragment was excised from the pMttbns vector to yield pMttΔXho that contained a unique XhoI site in addition to an existing unique BglII site. This expression vector targets expressed proteins that were secreted into the culture medium. All WNV genome sequences were introduced into the pMttΔXho vector using these unique BglII and XhoI sites. For the expression of a carboxy-truncated WNV envelope protein, a synthetic gene encoding the prM protein and 80% of the E protein from WNV was synthesized (Midland Certified Reagent Co., Midland, TX). The nucleotide sequence of the synthetic gene followed the published sequences of WNV isolated in 1999 in New York City [35]. The C-terminal truncation of the E protein at amino acid 401 eliminated the transmembrane domain to allow the E protein to be secreted into the medium. For the expression of a full-length WNV NS1 protein, a gene fragment was generated by RT-PCR from viral RNA (gift from A. Barrett, Univ. of Texas Medical Branch, Galveston). The NS1 gene fragment represented nucleotides 2470–3525 on the genome [35] and coded for a product containing 352 amino acid residues. Both the synthetic prM80E and RT-PCR generated NS1 gene fragments were designed to include restriction endonuclease sites that were used for cloning and also included two stop codons immediately following the last WNV codon. The final prM80E plasmid construct was designated pMttWNprM80E and the NS1 plasmid construct was designated pMttWNNS1.
S2 cells were co-transformed with these pMttΔXho-based expression plasmids and the pCoHygro selection plasmid that encodes hygromycin resistance utilizing the calcium phosphate co-precipitation method or with Cellfectin (Invitrogen Kits, Carlsbad, CA) according to the manufacturer’s recommendations. Cells were co-transformed with 20 μg total DNA with a 20:1 ratio of expression plasmid to selection plasmid. Transformants were selected with hygromycin B (Roche Molecular Biochemicals, Indianapolis, IN) at 300 μg/ml. Following selection, cells were adapted to growth in the serum free medium Excel 420 (JRH, Lenexa, KS). For expression, cells were grown in Excel 420, 300 μg/ml hygromycin, and induced with 200 μM CuSO4. Cells were seeded at a density of 2 × 106 cells/ml and allowed to grow for 6–7 days, yielding cell densities of 1.5 to 2 × 107 cells/ml. The culture supernatant was examined for expressed protein by SDS-PAGE and Western blot.
For the detection of West Nile 80E on Western blots a rabbit polyclonal anti-WNV antibody (BioReliance Corp.) followed by an anti-rabbit IgG-alkaline phosphatase conjugated secondary antibody was used. For the detection of West Nile NS1 on Western blots the flavivirus group specific anti-NS1 monoclonal 7E11 followed by an anti-mouse IgG-alkaline phosphatase conjugated secondary antibody was used. The blots were developed with NBT/BCIP (Sigma Chem. Co.) solid phase alkaline phosphatase substrate.
2.2.2. Purification of recombinant proteins
The purification of both the 80E and the NS1 proteins was accomplished by immunoaffinity chromatography (IAC). For 80E, the monoclonal antibody (mab) 4G2 was utilized, while mab 7E11 was utilized for purification of NS1. Both mabs were purified from cell culture grown hybridomas using protein A chromatography. Antigen purification was performed by filtration of the insect cell culture medium using a What-man 1 filter. The crude material was then loaded onto the IAC column, which contains immobilized MAb that was covalently coupled via N-hydroxysuccinimide chemistry, at a linear flow rate of 2 cm/min. After the sample was loaded, the matrix was washed with 10 mM phosphate buffered saline (PBS, 140 mM NaCl), pH 7.2, containing 0.05% (v/v) tween-20 (PBST). Bound protein was eluted from the IAC column with 20 mM glycine buffer, pH 2.5. The eluent was neutralized and then the buffer was exchanged, using PBST for 80E, or PBS for NS1. The purified products were routinely analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie blue or silver staining, Western blot, UV absorption, and sandwich ELISA to determine purity, identity, quantity, and bioactivity. In addition, samples were analyzed by N-terminal amino acid sequencing and amino acid analysis. These analyses provided confirmation of identity and the quantity of the purified products (data not presented).
2.2.3. Adjuvant
ISCOMATRIX® adjuvant was obtained from CSL Ltd., Parkville, Melbourne, Victoria, Australia. This is a saponin-based adjuvant containing Quil A and cholesterol components [36].
2.3. Mouse immunogenicity analysis
2.3.1. Vaccinations and splenocyte preparations
Mice were vaccinated twice, subcutaneously, at a 4 week interval with the indicated amounts (see Tables 1 and 2) of viral antigens plus adjuvant. On days 4 and 7 post booster vaccination, splenectomies were performed on 3–5 mice from each group and splenocyte suspensions were prepared. Erythrocytes were lysed with an NH4Cl-based lysis solution, and the cell pellet was washed three times and resuspended in cell culture medium. Cell counts were performed on each suspension using a hemacytometer, and the suspensions were diluted to 4 × 106 cells/ml for lymphocyte proliferation and cytokine production assays.
Table 1.
WNV antibody titers produced in mice to 80E and to NS1 proteins
| Group # | 80E dose (μg) | NS1 dose (μg) | ELISA titer to 80Ea | ELISA titer to NS1a | PRNT90 titerb |
|---|---|---|---|---|---|
| 1 | 0.3 | 1 | ~500 | 6450 (4120–10,100) | 640 |
| 2 | 1 | 1 | 10,420 (2410–45,100) | 4730 (2060–10,800) | 1280 |
| 3 | 0 | 0 | <250 | <250 | <10 |
Dilution of serum yielding 50% maximal absorbance in the ELISA as determined by a sigmoidal dose response, variable slope, mathematical model (GraphPad Prizm). Titers given are the GMT of individual mice (n = 5). Values in parenthesis: lower limit of 95% confidence interval (CI) to upper limit of 95% CI of GMT.
Highest dilution of serum (using 2-fold serial dilutions) yielding ≧90% reduction in plaques. Assays performed on pooled serum aliquots from each mouse within a group.
Table 2.
WNV specific neutralizing antibody titers produced in Balb/c mice following vaccination with varying doses of the 80E ± NS1 proteins
| Group # | 80E dose (μg) | NS1 dose (μg) | Neutralizing antibody titera |
|---|---|---|---|
| 1 | 1 | 0 | 5120 |
| 2 | 1 | 0.3 | NTb |
| 3 | 1 | 1 | 5120 |
| 4 | 3 | 0 | 1280 |
| 5 | 3 | 0.3 | 2560 |
| 6 | 3 | 1 | 5120 |
| 7 | 10 | 0 | 5120 |
| 8 | 10 | 0.3 | NT |
| 9 | 10 | 1 | 5120 |
| 10 | 0 | 0 | <10 |
Highest dilution of serum (using 2-fold serial dilutions) yielding ≧90% reduction of the WNV plaque foming units. Assays performed on pooled serum aliquots from each mouse within a group.
Not tested.
2.3.2. Lymphocyte proliferation assays
Aliquots (100 μl) of each splenocyte suspension were dispensed into each well of a 96-well cell culture plate. Aliquots (100 μl) of the WNV antigens (80E and/or NS1) were then dispensed into the wells containing each of the cell suspensions (in quadruplicate), at a final concentration of 5 μg/ml of each antigen. Wells with unstimulated (antigen omitted) cell suspensions, as well as phytohemagglutinin (PHA) stimulated cell suspensions (as a positive control) were also included. Cultures were incubated at 37 °C/5% CO2/humidified for 7 days (3 days for PHA stimulated cultures), and then one microcurie of tritiated (methyl-3H) thymidine (60 Ci/mmol; ICN Biomedicals, Inc., Irvine, CA) was added to each well (in a volume of 10 μl), and incubation was continued for 18 h. After incubation, the cell cultures were harvested onto glass fiber filtration plates and washed extensively using a vacuum driven harvester system (Filtermate Plate Harvester, PerkinElmer Co.). The filtration plates were then analyzed for radioactivity using the Top-Count Microplate Scintillation and Luminescence Counter (PerkinElmer Co.).
2.3.3. Cytokine production assays
Aliquots (0.5 ml) of each splenocyte suspension were dispensed into wells of a 24-well cell culture plate. Aliquots (0.5 ml) of the same antigens used for lymphocyte proliferation were dispensed into the wells containing each of the cell suspensions. Unstimulated and pokeweed mitogen (PWM)-stimulated cell suspensions were also included. Cultures were incubated for 5 days at 37 °C/5% CO2/humidified. The culture supernatants were then harvested and frozen prior to analysis for specific cytokines. The cytokines interferon-gamma (IFN-γ), interleukin-4 (IL-4), IL-5, and IL-10 were assayed by commercially available enzyme-linked immunosorbent assay (ELISA) kits or reagents according to the directions of the vendor (BD Pharmingen, San Diego, CA, or Biosource International, Camarillo, CA), or by using a flow cytometric bead array assay (BD Biosciences).
2.3.4. Antibody titrations
The WNV antibody response was determined on serum samples collected from individual mice 14 days post booster vaccination, or on pooled aliquots from individual mice within a group. Sera were tested for antibodies to both the 80E and NS1 proteins by a standard ELISA technique using 96-well microplates coated with the individual WNV antigens. Briefly, plate wells were coated with antigen at 1 μg/well in PBS overnight at 4 °C, and blocked with PBS/1% bovine serum albumin (BSA) for 1 h at room temperature. The wells were then washed three times with PBST, incubated with serum dilutions prepared in PBST/0.1% BSA for 2 h at room temperature, washed three times, and incubated with goat anti-mouse IgG (H + L)-alkaline phosphatase conjugate (Southern Biotechnology Associates, Birmingham, AL) for 1 h at room temperature. In some experiments, IgG1 and IgG2a specific antibody titers were determined using mouse IgG subclass specific secondary antibody-alkaline phosphatase conjugates (Southern Biotechnology Associates, Birmingham, AL). After washing four times, 100 μl of the enzyme substrate p-nitrophenylphosphate (pNPP, Sigma Chemical Co., St. Louis, MO), dissolved in 0.1 M trishydroxymethylaminomethane (tris) buffer, 0.1 M NaCl, 5 mM MgCl2, pH 9.5, was added and the plates were incubated in the dark for 30 min at room temperature. Reactions were stopped by addition of 50 μl of 2.5N NaOH, and the absorbance at 405 nm (A405) was read in an automated microplate reader.
In addition, serum samples were also tested for WNV neutralizing antibody by a plaque reduction neutralization test (PRNT) according to a previously described technique [37,38]. The Egypt 101 strain of WNV was used and all tests were performed on VERO cell cultures propagated in 24-well microplates.
3. Results
3.1. Production and purification of vaccine antigens
3.1.1. Production of recombinant antigens
S2 cells were co-transformed, grown, and induced for protein expression as described above. SDS-PAGE and Western blots of crude 80E and NS1 are shown in Figs. 1 and 2. Comparison of Coomassie blue stained SDS-PAGE gels of known quantities of purified protein and crude material, revealed that the production of both proteins was ~10–25 mg/l of culture (data not shown).
Fig. 1.
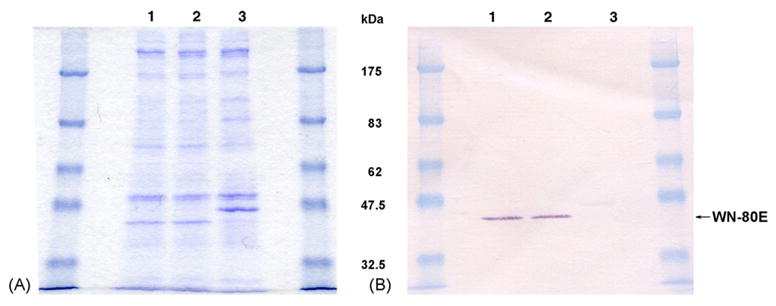
(A) Coomassie blue stained SDS-PAGE of WNV 80E protein expressed by Drosophila S2 cells under non-reducing conditions. Lane 1, Spinner Culture #1 of cell line WN-80E-1 harvested 2/19/03; Lane 2, Spinner Culture #2 of cell line WN-80E-1 harvested 2/10/03; Lane 3, culture of a dengue transformant cell line. The migration of the WNV 80E is faster than the dengue 80E due to differences in glycosylation and tertiary structure (samples are non-reduced). (B) Western blot of duplicate SDS-PAGE gel seen in A. The blot was probed with a commericially available WNV rabbit polyclonal from BioReliance. This antibody cross-reacts slightly with the Dengue 80E.
Fig. 2.
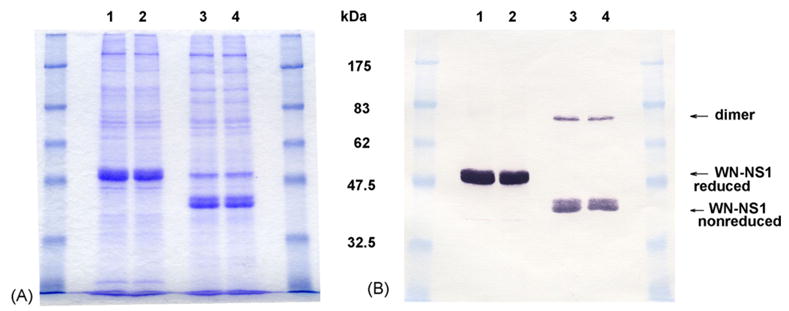
(A) Coomassie blue stained SDS-PAGE of WNV NS1 protein expressed by Drosophila S2 cells under reducing (Lanes 1 and 2) and non-reducing conditions (Lanes 3 and 4). Lanes 1 and 3, Spinner Culture #1 of cell line WN-NS1-5 harvested 7/6/03; Lanes 2 and 4, Spinner Culture #2 of cell line WN-NS1-5 harvested 7/6/03. (B) Western blot of duplicate SDS-PAGE gel seen in A. The blot was probed with the mouse monoclonal 7E11. The two approximately 40 kDa bands of WN-NS1 are two different glycoforms of the NS1 protein. The higher MW reactive band at about 80 kDa in Lanes 3 and 4 is a dimer. The 7E11 antibody reacts more strongly with reduced than non-reduced NS1.
3.1.2. Purification of expressed recombinant proteins
WNV 80E and NS1 proteins were purified by IAC. Representative SDS-PAGE and Western blot profiles of the two purified proteins are presented in Figs. 3 and 4. For the analysis, both samples were run under non-reducing conditions. The 80E molecule migrated as a single band with a relative molecular weight consistent with that determined from the amino acid composition (i.e., 43 kDa). This finding indicated that disulfide bonding did not occur between molecules, although multimers (e.g. dimers) stabilized by non-covalent interactions were still present in the native state. Trace contaminants (~5 bands) were visible in a 10 μg load on the Coomassie stained gel. Assuming a threshold of detection of 100 ng, the purity of the 80E was estimated to be >90%. In contrast, the NS1 migrates as two distinct forms: one with a relative molecular weight that was consistent with that expected for a monomeric form (40 kDa) and one with a relative molecular weight that was consistent with a dimeric form (80 kDa). Unlike the 80E preparation, a major contaminant was clearly visible in a 5 μg load with possibly 2–3 minor contaminants as well. Since the major contaminant was still visible in a 1 μg load but not in a 0.5 μg load, the purity of the NS1 preparations were estimated to be ~90%. The percent yield of the purified material recovered (based on amino acid analysis of purified protein) was about 75–80% for 80E and about 45–50% for NS1 (data not shown).
Fig. 3.
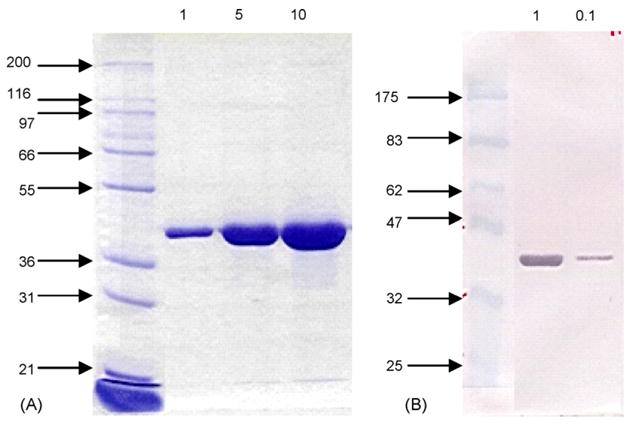
Coomassie stained SDS-PAGE gel (A) and Western blot (B) of purified WNV 80E. Both samples were run under non-reducing conditions on 10% gels. The Western blot was developed using a rabbit polyclonal antisera developed against formalin inactivated dengue virus. The sizes of the molecular weight markers (in kDa) are indicated to the left of the gel and blot. The sample loadings (in μg) are presented at the top of each.
Fig. 4.
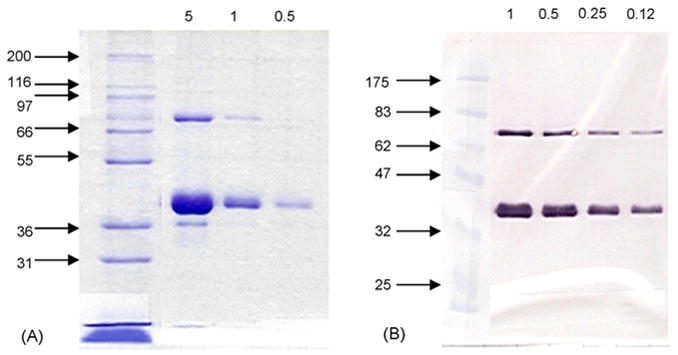
Coomassie stained SDS-PAGE gel (A) and Western blot (B) of purified WNV NS1. Both samples were run under non-reducing conditions on 10% gels. The Western blot was developed using a rabbit polyclonal antisera developed against purified dengue NS1. The sizes of the molecular weight markers (in kDa) are indicated to the left of the gel and blot. The sample loadings (in μg) are presented at the top of each.
3.2. Immunogenicity in mice
3.2.1. Humoral immunity
Swiss Webster mice were vaccinated with 0.3 or 1 μg of 80E, with 1 μg of NS1. ISCOMATRIX® adjuvant was used at 12 μg per mouse, including an adjuvant control group. The antibody responses to low immunizing doses of both 80E and NS1 proteins (1 μg of either antigen) as determined by ELISA and PRNT were characterized by high antibody titers (Table 1). The PRNT titers were high with an immunizing dose of 80E as low as 0.3 μg.
In a separate experiment, Balb/c mice were vaccinated with 1, 3, or 10 μg of 80E, in combination with 0, 0.3, or 1 μg of NS1. ISCOMATRIX® adjuvant was used at 5 μg per mouse, including an adjuvant control group. PRNT titers were also determined in this experiment and the results (Table 2) showed very high titers in all groups assayed, with no dose response evident.
In addition, an analysis of IgG subclass (IgG1 and IgG2a)-specific antibody titers to WNV 80E and NS1 was performed by ELISA. These results demonstrated that IgG1 titers to 80E were relatively constant within the range of immunizing doses from 1 to 10 μg (Table 3). Anti-80E IgG2a titers, however, appear to decrease within this range. This dose response result revealed a decreasing ratio of IgG2a/IgG1 titers with increasing doses of 80E (Fig. 5A). The addition of NS1 to the immunizing 80E vaccine did not affect the titers to 80E (Table 3).
Table 3.
WNV isotype specific antibody titersa produced in mice following vaccination with 80E and NS1 proteins
| Group # | 80E dose (μg) | NS1 dose (μg) | 80E IgG1 titer | 80E IgG2a titer | NS1 IgG1 titer | NS1 IgG2a titer |
|---|---|---|---|---|---|---|
| 1 | 1 | 0 | 61,500 | 38,800 | <250 | <250 |
| 2 | 1 | 0.3 | 92,600 | 36,900 | 27,100 | 2,700 |
| 3 | 1 | 1 | 47,600 | 32,400 | 35,900 | 10,500 |
| 4 | 3 | 0 | 43,000 | 12,300 | <250 | <250 |
| 5 | 3 | 0.3 | 25,100 | 12,300 | 2,300 | 800 |
| 6 | 3 | 1 | 66,400 | 13,600 | 25,800 | 18,500 |
| 7 | 10 | 0 | 61,500 | 10,000 | <250 | <250 |
| 8 | 10 | 0.3 | 37,800 | 7,900 | 500 | 600 |
| 9 | 10 | 1 | 28,500 | 10,000 | 3,400 | 3,100 |
| 10 | 0 | 0 | <250 | <250 | <250 | <250 |
Dilution of serum yielding 50% maximal absorbance in the ELISA as determined by a sigmoidal dose response, variable slope, mathematical model (GraphPad Prizm). Assays performed on pooled serum aliquots from each mouse within a group.
Fig. 5.
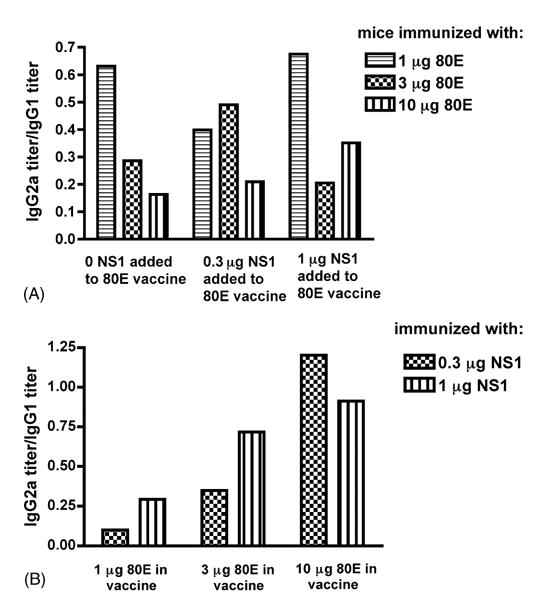
Ratio of IgG2a to IgG1 antibody titers. Panel A: antibody titers to WNV 80E. Panel B: antibody titers to WNV NS1.
Evaluation of antibody responses to NS1 revealed that both IgG1 and IgG2a antibody titers to NS1 increase with increasing immunizing doses of NS1 from 0.3 to 1 μg (Table 3). However, the increase in IgG2a was greater than the increase in IgG1, resulting in an increasing ratio of IgG2a/IgG1 titers to NS1 with increasing immunizing doses within this range (Fig. 5B). An increase in the amount of 80E in the vaccine, from 1 to 10 μg, also appears to decrease the antibody titers to NS1, but the decrease is relatively greater for IgG1 than for IgG2a (Table 3). This yields an increasing ratio of IgG2a/IgG1 antibody titers to NS1 when the amount of 80E added to the vaccine is increased (Fig. 5B).
3.2.2. Cellular immunity
Following immunization of Swiss Webster mice with 0.3 or 1 μg of 80E, in combination with 1 μg of NS1, antigen-stimulated lymphocyte proliferation and cytokine production from immune splenocytes in vitro were assayed as described above. The results of the lymphocyte proliferation assays (Fig. 6) demonstrate excellent proliferation in response to either 80E or NS1 stimulation. There was no difference between splenocytes from mice vaccinated with 0.3 or 1 μg of 80E when stimulated with 80E in vitro. A trend for greater proliferation was seen following stimulation with a cocktail containing both antigens compared to either antigen alone, but the trend was not statistically significant (t-test). Stimulation indices calculated from the data shown in Fig. 6 varied from 23 to 48 for the vaccine groups and were <2 for the adjuvant control group.
Fig. 6.
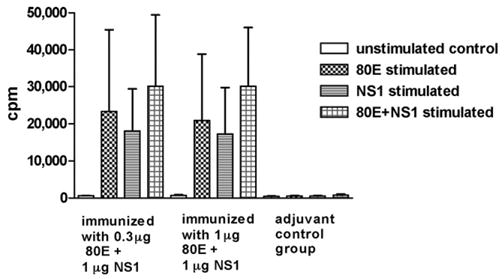
Antigen-stimulated lymphocyte proliferation. Splenectomies performed 4 days post booster vaccination. The total cpm recovered from tritiated thymidine incorporation per 4 × 105 cells (total number of cells cultured per well) is plotted. Values derived from quadruplicate wells per stimulant condition were averaged. Histograms represent the mean of individual mice (n = 3). Error bars represent the standard deviations. Tests of significance (p < 0.05) were performed between groups with the same stimulant and between stimulants within a group using an unpaired t-test with the aid of a commercially available statistical program (GraphPad Prizm).
The yields of the cytokines IFN-γ, IL-4, and IL-5 were also analyzed from cultures of antigen-stimulated immune splenocytes from these mice. The results demonstrated high levels of each of these cytokines from the stimulated immune cells (Fig. 7). Stimulation with either 80E, NS1, or a combination of both yielded about the same levels of IFN-γ. In contrast, production of IL-4 following stimulation by 80E or NS1 was additive in that stimulation with both antigens yielded much more cytokine than either antigen alone. Production of IL-5 also showed additive stimulation in some cases, but not as dramatic as IL-4. Production of IL-10 had a similar pattern to that of IL-5 (data not shown). There was no significant dose response to the immunizing dose of 80E.
Fig. 7.
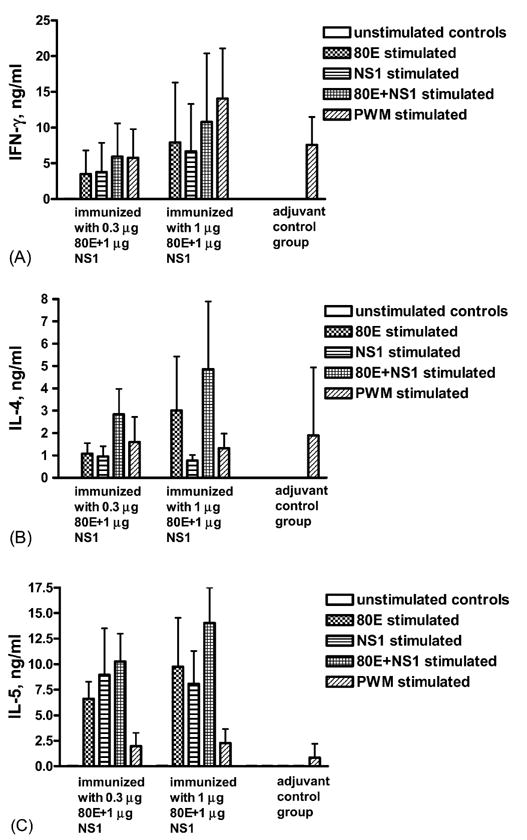
Antigen-stimulated cytokine production in vitro. Splenectomies performed 7 days post booster vaccination. Panels A, B, and C: IFN-γ, IL-4, and IL-5, respectively. Histograms represent the mean of individual mice (n = 5). Error bars represent the standard deviations. Tests of significance (p < 0.05) were performed between groups with the same stimulant and between stimulants within a group using an unpaired t-test with the aid of a commercially available statistical program (GraphPad Prizm).
4. Discussion
The results reported herein describe the preparation, properties, and immunogenicity in mice of a recombinant subunit candidate vaccine for the prevention of WNV encephalitis in humans. The vaccine is comprised of a truncated envelope protein (80E) and a non-structural protein (NS1) from the WNV (New York flamingo strain). The data presented document the production and purification of the specific antigens, and their use in vaccine formulations for the generation of both cellular and humoral immune responses in vaccinated animals. The results reported in the accompanying paper [31] document the ability of the vaccine to afford protection against lethal viral infection in a hamster model of WNV encephalitis highly relevant to human disease.
The Drosophila S2 expression system used to produce the WNV vaccine has proven to be capable of expressing and secreting native-like proteins with complex secondary and tertiary structure. This was shown in the crystal structure of a carboxy truncated dengue virus envelope protein which was determined using protein expressed in S2 cells [24–26]. Thus, the expression system is highly efficient both in terms of the quality of antigen produced as well as the quantity. In this study, the yields of purified WNV antigen, combined with the high expression levels resulted in an economical production system for the WNV vaccine.
Our initial evaluations indicated that the WNV vaccine in combination with adjuvants elicited both a humoral and cellular immune response in mice. The humoral response was characterized by high titers of antibodies to both 80E and NS1 antigens by ELISA as well as high neutralizing titers by the PRNT assay. There was a very steep dose response to 80E antigen when antibodies were determined by ELISA, but not by PRNT assays. The observation that the antigen-specific IgG2a/IgG1 ratio decreased with increasing dose of 80E suggests that the balance between a “Th1” or “Th2”-type immune response to a vaccine or immunization regimen based on the relative IgG2a to IgG1 isotype response is dependent on the dose of immunogen used. Thus, conclusions regarding this balance derived from data obtained in experiments using only a single dose level of immunogen may be misleading.
Cell-mediated immune responses were also assessed in mice vaccinated with 80E and NS1 by antigen-stimulated lymphocyte proliferation and cytokine production in vitro by splenocytes from immunized mice. The results of both assays demonstrated robust responses to both antigens. The production of IFN-γ in particular is considered to be a hallmark of the “Th1” cell-mediated immune response [39,40]. Thus, the vaccine formulations described produce a “balanced” Th1/Th2 immune response, with high levels of specific, functional (viral neutralizing) antibodies, as well as a robust cellular response. The inclusion of NS1 in the vaccine may provide further benefit by generating additional antibodies and/or T cells capable of protective activities. A recent report has documented both complement and Fcγ receptor-dependent and -independent protective activities of antibodies to WNV NS1 [41]. In our ongoing studies on a recombinant subunit vaccine for dengue virus, the addition of NS1 to envelope protein in the vaccine increased the production of IFN-γ from antigen-stimulated immune mouse splenocytes in vitro (Lieberman, MM et al., unpublished data). Also, partial protection of hamsters against WNV encephalitis was obtained by vaccination with a formulation containing NS1 alone as the only immunogen [31].
Other investigators have developed WNV candidate vaccines based on live viruses [16,17,20,22], inactivated virus [21], DNA molecules [15], or envelope proteins produced in bacterial [18] and insect (Drosophila) cell expression systems [19]. Except for the latter system, there are intrinsic difficulties associated with each of these candidate vaccines. Safety concerns, of course, are paramount with all live viral vaccines, particularly in the case of a virus disease with relatively low prevalence, in which the vaccine is given to healthy subjects. Under-attenuation of the virus may result in disease manifestation, whereas over-attenuation may abrogate vaccine efficacy. Also, reversion to wild type or mutation to increased virulence (or decreased efficacy) may occur. Moreover, even if properly attenuated, live viral vaccines are contra-indicated for specific patient populations, such as immune deficient or immune suppressed patients, as well as particular segments of the normal population, such as pregnant women or elderly individuals. Inactivated whole virus vaccines may present production problems at commercial scale, in terms of growth of the virus to sufficiently high titers for economical yield, as well as hazardous containment issues for large scale growth of non-attenuated live virus. Naked DNA vaccines are unproven for any infectious disease at this time, and the issue of potential immunopathology due to the induction of an autoimmune reaction to the DNA over the long term is unresolved. Finally, the expression of recombinant proteins in bacterial systems has often resulted in aberrant tertiary structure of the expressed protein due to inadequate disulfide bond formation and/or improper glycosylation.
Production of a recombinant subunit-based vaccine as reported herein offers an important alternative to these approaches. High level expression of WNV envelope and non-structural proteins with native-like conformation has produced a vaccine candidate which is a potent immunogen capable of inducing high titer virus neutralizing antibodies and good cellular immune responses.
Acknowledgments
This work was supported in part by contracts NO1-AI 25489 and NO1-AI30027, and grants 1 R43 AI052600-01A1 and 9 R44 NS52139-02A1, from the National Institutes of Health.
References
- 1.Petersen LR, Roehrig JT, Hughes JM. West Nile virus encephalitis. N Engl J Med. 2002;347:1225–6. doi: 10.1056/NEJMo020128. [DOI] [PubMed] [Google Scholar]
- 2.CDC West Nile Virus website: Statistics, Surveillance, and Control. http://www.cdc.gov/ncidod/dvbid/westnile/surv&controlCaseCount06detailed.htm.
- 3.Komar N, Clark GG. West Nile virus activity in Latin America and the Caribbean. Rev Panam Salud Publica. 2006;19:112–7. doi: 10.1590/s1020-49892006000200006. [DOI] [PubMed] [Google Scholar]
- 4.Mostashari F, Bunning ML, Kitsutani PT, Singer DA, Nash D, Cooper MJ, et al. Epidemic West Nile encephalitis, New York, 1999, results of a household-based seroepidemiological survey. Lancet. 2001;358:261–4. doi: 10.1016/S0140-6736(01)05480-0. [DOI] [PubMed] [Google Scholar]
- 5.Chowers MY, Lang R, Nassar F, Ben-David D, Giladi M, Rubinshtein E, et al. Clinical characteristics of the West Nile fever outbreak in Israel. Emerg Infect Dis. 2001;7:675–8. doi: 10.3201/eid0704.010414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai TF, Popovici F, Cernescu C, Campbell GL, Nedelcu NI. West Nile encephalitis epidemic in southeastern Romania. Lancet. 1998;352(9130):767–71. doi: 10.1016/s0140-6736(98)03538-7. [DOI] [PubMed] [Google Scholar]
- 7.Platonov AE, Shipulin GA, Shipulina OY, Tyutyunnik EN, Frolochkina TI, Lanciotti RS, et al. Outbreak of West Nile virus infection, Volgograd region, Russia, 1999. Emerg Infect Dis. 2001;7:128–32. doi: 10.3201/eid0701.010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersen LR, Roehrig JT. West Nile virus: a reemerging global pathogen. Emerg Infect Dis. 2001;7:611–4. doi: 10.3201/eid0704.010401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crill WD, Roehrig JT. Monoclonal antibodies that bind to domain III of Dengue Virus E glycoprotein are the most efficient blockers of virus adsorption to Vero cells. J Virol. 2001;75:7769–73. doi: 10.1128/JVI.75.16.7769-7773.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin C-W, Wu S-C. A functional epitope determinant on domain III of the Japanese encephalitis virus envelope protein interacted with neutralizing-antibody combining sites. J Virol. 2003;77:2600–6. doi: 10.1128/JVI.77.4.2600-2606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beasley DWC, Barrett ADT. Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein. J Virol. 2002;76:13097–100. doi: 10.1128/JVI.76.24.13097-13100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gould LH, Sui J, Foellmer H, Oliphant T, Wang T, Ledizet M, et al. Protective and therapeutic capacity of human single-chain Fv-Fc fusion proteins against West Nile virus. J Virol. 2005;79:14606–13. doi: 10.1128/JVI.79.23.14606-14613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markoff L. Points to consider in the development of a surrogate for efficacy of novel Japanese encephalitis virus vaccines. Vaccine. 2000;18:26–32. doi: 10.1016/s0264-410x(00)00038-4. [DOI] [PubMed] [Google Scholar]
- 14.Zent O, Beran J, Jilg W, Mach T, Banzhoff A. Clinical evaluation of a polygeline-free tick-borne encephalitis vaccine for adolescents and adults. Vaccine. 2003;21:738–41. doi: 10.1016/s0264-410x(02)00592-3. [DOI] [PubMed] [Google Scholar]
- 15.Chang G-JJ, Davis BS, Hunt AR, Holmes DA, Kuno G. Flavivirus DNA vaccines. Ann New York Acad Sci. 2001;951:272–85. [PubMed] [Google Scholar]
- 16.Pletnev AG, Putnak R, Speicher J, Wagar EJ, Vaughn DW. West Nile virus/dengue type 4 virus chimeras that are reduced in neurovirulence and peripheral virulence without loss of immunogenicity or protective efficacy. Proc Natl Acad Sci. 2002;99:3036–41. doi: 10.1073/pnas.022652799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monath TP, Arroyo J, Miller C, Guirakhoo F. West Nile virus vaccine. Curr Drug Targets Infect Disord. 2001;1:37–50. doi: 10.2174/1568005013343254. [DOI] [PubMed] [Google Scholar]
- 18.Wang T, Anderson JF, Magnarelli LA, Wong SJ, Koski RA, Fikrig E. Immunization of mice against West Nile virus with recombinant envelope protein. J Immunol. 2001;167:5273–7. doi: 10.4049/jimmunol.167.9.5273. [DOI] [PubMed] [Google Scholar]
- 19.Ledizet M, Kar K, Foellmer HG, Wang T, Bushmich SL, Anterson JF, et al. A recombinant envelope protein vaccine against West Nile virus. Vaccine. 2005;23:3915–24. doi: 10.1016/j.vaccine.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Lustig S, Olshevsky U, Ben-Nathan D, Lachmi BE, Malkinson M, Kobiler D, et al. A live attenuated WNV strain as a potential veterinary vaccine. Viral Immunol. 2000;13:401–10. doi: 10.1089/vim.2000.13.401. [DOI] [PubMed] [Google Scholar]
- 21.Ng T, Hathaway D, Jennings N, Champ D, Chiang YW, Chu HJ. Equine vaccine for West Nile virus. Dev Biol (Basel) 2003;114:221–7. [PubMed] [Google Scholar]
- 22.Minke JM, Siger L, Karaca K, Austgen L, Gordy P, Bowen R, et al. Recombinant canarypoxvirus vaccine carrying the prM/E genes of West Nile virus protects horses against a West Nile virus-mosquito challenge. Arch Virol Suppl. 2004;(18):221–30. doi: 10.1007/978-3-7091-0572-6_20. [DOI] [PubMed] [Google Scholar]
- 23.Cuzzubbo AJ, Endy TP, Nisalak A, Kalayanorooj S, Vaughn DW, Ogata SA, et al. Use of recombinant envelope proteins for serological diagnosis of dengue virus infection in an immunochromatographic assay. Clin Diagn Lab Immunol. 2001;8:1150–5. doi: 10.1128/CDLI.8.6.1150-1155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Modis Y, Ogata S, Clements D, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci USA. 2003;100(12):6986–91. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427(6872):313–9. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- 26.Modis Y, Ogata S, Clements D, Harrison SC. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J Virol. 2005;79:1223–31. doi: 10.1128/JVI.79.2.1223-1231.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livingston PG, Kurane I, Dai L-C, Okamoto Y, Lai CJ, Men R, et al. Dengue virus-specific, HLA-B35-restricted, human CD8+ cytotoxic T lymphocyte (CTL) clones. J Immunol. 1995;154:1287–95. [PubMed] [Google Scholar]
- 28.Mathew A, Kurane I, Rothman AL, Zeng LL, Brinton MA, Ennis FA. Dominant recognition by human CD8+ cytotoxic T lymphocytes of Dengue virus nonstructural proteins NS3 and NS1. 2a J Clin Invest. 1996;98:1684–92. doi: 10.1172/JCI118964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlesinger JJ, Foltzer M, Chapman S. The Fc portion of antibody to yellow fever virus NS1 is a determinant of protection against YF encephalitis in mice. Virology. 1993;192:132–41. doi: 10.1006/viro.1993.1015. [DOI] [PubMed] [Google Scholar]
- 30.Chung KM, Nybakken GE, Thompson BS, Engle MJ, Marri A, Fremont DH, et al. Antibodies against West Nile virus nonstructural protein NS1 prevent lethal infection through Fcγ receptor-dependent and -independent mechanisms. J Virol. 2006;80:1340–51. doi: 10.1128/JVI.80.3.1340-1351.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watts DM, Tesh RB, Siirin M, da Rosa AT, Newman PC, Clements DE, et al. Efficacy and durability of a recombinant subunit West Nile vaccine candidate in protecting hamsters from West Nile encephalitis. Vaccine. doi: 10.1016/j.vaccine.2006.08.008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider J. Cell lines derived from late embryonic stages of Drosophila melanogaster. J Embryol Exper Morphol. 1972;27:353–65. [PubMed] [Google Scholar]
- 33.Johansen H, van der Straten A, Sweet R, Otto E, Maroni G, Rosenberg M. Regulated expression at high copy number allows production of a growth-inhibitory oncogene product in Drosophila Schneider cells. Genes Dev. 1989;3:882–9. doi: 10.1101/gad.3.6.882. [DOI] [PubMed] [Google Scholar]
- 34.Culp JS, Johansen H, Hellmig B, Beck J, Matthews TJ, Delers A, et al. Regulated expression allows high level production and secretion of HIV gp120 envelope glycoprotein in Drosophila Schneider cells. Bio/technology. 1991;9:173–7. doi: 10.1038/nbt0291-173. [DOI] [PubMed] [Google Scholar]
- 35.Lanciotti RS, Roehrig JT, Deubel V, Smith J, Parker M, Steele K, et al. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science. 1999;286:2333–7. doi: 10.1126/science.286.5448.2333. [DOI] [PubMed] [Google Scholar]
- 36.Ronnberg B, Fekadu M, Morein B. Adjuvant activity of non-toxic Quillaja saponaria Molina components for use in ISCOM matrix. Vaccine. 1995;13:1375–82. doi: 10.1016/0264-410x(95)00105-a. [DOI] [PubMed] [Google Scholar]
- 37.Xiao SY, Guzman H, Zhang H, Travassos da Rosa APA, Tesh RB. West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg Infect Dis. 2001;7:714–21. doi: 10.3201/eid0704.010420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tesh RB, Arroyo J, Travassos da Rosa APA, Guzman H, Xiao SY, Monath TP. Efficacy of killed virus vaccine, live attenuated chimeric virus vaccine, and passive immunization for prevention of West Nile virus encephalitis in hamster model. Emerg Infect Dis. 2002;8:1392–7. doi: 10.3201/eid0812.020229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–73. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 40.Borish L, Rosenwasser L. Update on cytokines. J Allergy Clin Immunol. 1996;97:719–30. doi: 10.1016/s0091-6749(96)80146-1. [DOI] [PubMed] [Google Scholar]
- 41.Chung KM, Nybakken GE, Thompson BS, Engle MJ, Marri A, Fremont DH, et al. Antibodies against West Nile virus nonstructural protein NS1 prevent lethal infection through Fcγ receptor-dependent and -independent mechanisms. J Virol. 2006;80:1340–51. doi: 10.1128/JVI.80.3.1340-1351.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]