

Abstract
The human pregnane X receptor (PXR) recognizes a range of structurally- and chemically-distinct ligands and plays a key role in regulating the expression of protective gene products involved in the metabolism and excretion of potentially harmful compounds. The identification and development of PXR antagonists is desirable as a potential way to control the up-regulation of drug metabolism pathways during the therapeutic treatment of disease. We present the 2.8 Å resolution crystal structure of the PXR ligand binding domain (LBD) in complex with T0901317 (T1317), which is also an agonist of another member of the orphan class of the nuclear receptor superfamily, the liver X receptor (LXR). In spite of differences in the size and shape of the receptors' ligand binding pockets, key interactions with this ligand are conserved between human PXR and human LXR. Based on the PXR-T1317 structure, analogues of T1317 were generated with the goal of designing an PXR antagonist effective via the receptor's ligand binding pocket. We find that selectivity in activating PXR vs. LXR was achieved; such compounds may be useful in addressing neurodegenerative diseases like Niemann-Pick C. We were not successful, however, in producing a PXR antagonist. Based on these observations, we conclude that the generation of PXR antagonists targeted to the ligand binding pocket may be difficult due to the promiscuity and structural conformability of this xenobiotic sensor.
Keywords: nuclear receptor, xenobiotics, LXR, antagonism, structure-activity relationships, structure-based design
Introduction
The human pregnane X receptor (PXR; alternatively SXR, PAR) responds to a variety of endogenous and exogenous compounds in liver, intestine and other tissues, and is a key regulator of the expression of genes central to xenobiotic metabolism and excretion 1-3. PXR is also responsible for an important class of drug interactions caused by the efficient up-regulation of chemoprotective pathways that lead to the elimination of a wide range of therapeutics 4-7. It has also recently been shown that T0901317 (T1317), the ligand described here in complex with PXR, protects against the development of the neurodegenerative disease Niemann-Pick C in a PXR-dependent fashion within a mouse model of this condition 8.
Like other members of the orphan class of the nuclear receptor (NR) superfamily, PXR contains DNA-binding and ligand-binding domains (DBD, LBD, respectively), acts as a heterodimer with the retinoid X receptor-α (RXRα), and binds to a range of direct- and everted-repeat elements in the regulatory region of target genes 9. Upon association with an activating agonist, a transcriptional coactivator protein like the steroid receptor coactivator-1 (SRC-1) is recruited to the activation function-2 (AF-2) region of the PXR LBD, which facilitates changes in chromatin structure and activation of the basal transcriptional machinery. A leucine-rich LxxLL motif in transcriptional coactivators (where x is any amino acid) has been shown to interact with a groove present in the active orientation of NR LBDs 10, 11. Crystal structures of the human PXR LBD have been determined in complexes with a variety of small (e.g., SR12813, hyperforin) and large (e.g., rifampicin) ligands, and with fragments of the human transcriptional coactivator SRC-1 12-15. These structures have revealed that PXR's ligand binding promiscuity is a function of its large and conformable ligand binding pocket, which is framed in part by sequence elements novel to PXR relative to other NR LBDs. In addition, the PXR LBD forms a unique homodimer mediated by a tryptophan zipper-like motif, and it has been shown that this interface plays a role in receptor function and association with coactivators 16.
The liver X receptor (LXR), another member of the orphan class of NRs that functions as a heterodimer with RXRα, plays an important role in monitoring the levels of oxysterols in hepatocytes and regulates the expression of genes essential for cholesterol homeostasis 17-21. The LBDs of the two LXR isoforms, α and β, both share 31% sequence identity with the human PXR LBD (and 77% with one another), as well as the conserved overall structural fold common to NR ligand binding domains 22-26. The PXR LBD deviates from that of LXR in its ∼60-residue α1- α3 insert that adds one helix (α2) and two strands (β1, β1') and frame a significantly larger ligand binding pocket relative to LXR 10. The synthetic NR ligand T0901317 (T1317) is an established agonist for LXR, exhibiting robust upregulation of target gene expression 19. The structural basis of T1317 binding to LXR isoform β has been elucidated previously 22, 24, 26.
In addition to their association with transcriptional coactivators, NR LBDs bind to transcriptional corepressors (e.g., NCoR, SMRT) that exert opposite effects on gene transcription by mechanisms that include enhancing the condensed structure of chromatin 4. Interactions between NRs and corepressors can occur in the absence of ligand, but are enhanced in the presence of antagonizing ligands. A paradigm for this effect is provided by efficient down-regulation of transcription caused by the association of the estrogen receptor (ER) with the established antagonist tamoxifen. It has been shown structurally that corepressors contain an extended leucine-rich region that binds to an inactive LBD conformation distinct in structure from that of an active LBD, particularly in the position of the terminal α-helix in the LBD fold (αAF in PXR) 27. In ER, tamoxifen sterically blocks the active orientation of the terminal helix in that fold, producing a state that preferentially binds to transcriptional corepressors and down-regulates gene expression 28. In the case of PXR, most ligands are found to act as agonists of this receptor, in line with its established role in protecting tissues from potentially harmful chemicals. A small number of antagonists have been described, however, including the ET-743 29 and the antifungal ketoconazole 30, 31. Indeed, ketoconazole has been shown recently to repress the binding of both transcriptional coactivators and corepressors to PXR 30. The structural basis of the antagonism of PXR by these compounds has not been established, however.
The identification or design of new antagonists of human PXR are desirable because of the important role this receptor plays in drug metabolism, and because the current antagonists exhibit weak EC50 values relative to established agonists 29-31. Repressing the ability of PXR to recognize the presence of therapeutic compounds may allow lower doses to be administered with higher efficacy and fewer side effects. Here we present the 2.8 Å resolution crystal structure of the PXR LBD in complex with T1317. Using this structure as a guide, and inspired by selective ER modulators produced from ER agonists, we generate T1317 analogues designed to act as PXR antagonists by disrupting the active conformation of the receptor's αAF. We find, however, that compounds based on this scaffold either retain their ability to bind to PXR, and thus serve as agonists, or simply do not bind to the receptor. We conclude that the design of effective pocket-targeted PXR antagonists may be difficult due to the promiscuous and conformable nature of the receptor's ligand binding pocket.
Results
PXR-T1317 Structure
Crystals of the human PXR LBD were grown in the presence of 10-fold molar excess T0901317 (T1317), x-ray diffraction data to 2.8 Å resolution were collected, and the structure was determined and refined to R and Rfree values of 0.216 and 0.279, respectively (Table I). Two ternary complexes were observed in the asymmetric unit, each containing one human PXR LBD, one orientation of bound T1317, and one fifteen amino acid stretch of the human transcriptional coactivator steroid receptor coactivator 1 (SRC-1, residues 682-696) (Figure 1). The PXR LBD is expressed with an 88-amino acid fragment of SRC-1 (residue 623-710) to improve protein stability. This is the first structure of PXR in which the coactivator fragment remains attached during crystallization. The retention of this fragment is likely due to the relatively high affinity and potent agonist activity of T1317. The 88-amino acid region of human SRC-1 employed contains two leucine-rich NR box motifs (at 633-637, LVQLL, and 690-694. LHRLL). Only the second NR box was observed associated with the AF-2 region of the PXR LBD in both molecules in the crystallographic asymmetric unit. This implies that the sequence around or including LHRLL may have higher affinity for PXR relative to the region around the LVQLL motif.
Table I.
Crystallographic Statistics for the PXR-T0901317 Complex
| Resolution (Å; highest shell) | 50-2.8 Å (2.9-2.8) |
| Space Group | P212121 |
| Asymmetric Unit | two molecules |
| Cell Constants (Å, °) | a = 83.9 b = 90.6 |
| c = 105.7 | |
| α = β = γ = 90 | |
| Data Collection Facility | SER-CAT (APS) |
| Total Reflections | 119,110 |
| Unique Reflections | 20,332 |
| Mean Redundancy (highest shell) | 5.8 (5.4) |
| Rsym* (%; highest shell) | 13.5 (46.7) |
| Completeness (%; highest shell) | 97.6 (91.1) |
| Mean I/σ (highest shell) | 25.9 (5.1) |
| Rcryst† | 21.6 |
| Rfree‡ | 27.9 |
Rsym = Σ|I-<I>| / ΣI, where I is the observed intensity and <I> is the average intensity of multiple symmetry-related observations of that reflection.
Rcryst = Σ∥Fobs|-|Fcalc∥ / Σ|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively.
Rfree = Σ∥Fobs|-|Fcalc∥ / Σ|Fobs| for 10% of the data not used at any stage of structural refinement.
Figure 1.
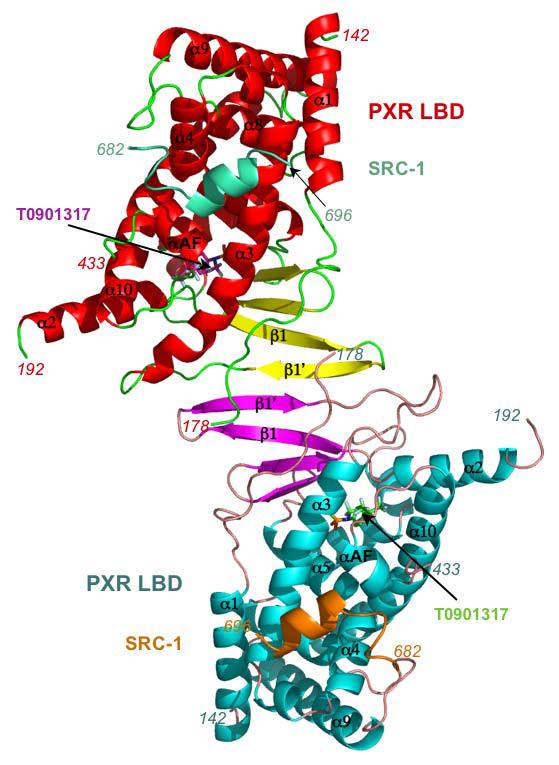
Crystal structure of the homodimer in the asymmetric unit of the ligand binding domain of human PXR (PXR LBD) in complex with T0901317 (T1317). The PXR LBD in one monomer is rendered in red, yellow and green, and in the other monomer in cyan, magenta and pink. The fragments of the steroid receptor coactivator 1 (SRC-1) are shown in aqua and orange, and the T1317 ligands in magenta and green.
The PXR LBD in this T1317 complex retains the same overall structure observed in previous PXR structures, sharing, for example, 1 Å root-mean-square deviation (rmsd) over Cα positions with the structure of the apo (unliganded) PXR LBD 15. The LBDs also form a homodimer consistently observed either via crystallographic or, as in this case, non-crystallographic symmetry. This homodimer interaction is mediated largely by interdigitating aromatic residues from β1' in each monomer, and its formation has been shown to be central to transcriptional activity and coactivator recruitment by PXR 16. Only a small number of shifts in the positions of amino acids that line the ligand binding pocket of PXR were observed between the T1317 and apo structures. For example, Leu-209 and Met-323 undergo a rotamer changes and shifts in position of 6.0 Å. It was also noted that the side chains of both His-407 and Phe-429 shift 1.0 Å toward the bound ligand relative to the apo structure, and in doing so form an 3.8 Å aromatic edge-to-edge van der Waals contact (compared to 5.3 Å for the same atoms in the apo structure). Because Phe-429 is located on the αAF of the PXR's AF-2 region, this interaction likely stabilizes the active form of the receptor during the upregulation of gene transcription.
PXR-T1317 Interactions
T1317 forms three polar and twelve van der Waals contacts with amino acid side chains that line the PXR ligand binding pocket (Figure 2; Table II). His-407 is positioned 2.4 Å from the ligand's hydroxyl group, while the polar groups of the Gln-285 side chain are 2.9 and 3.2 Å from one sulfoxy oxygen and His-327 is 3.3 Å from the other. This is the first time His-327 has been observed within hydrogen bonding distance (generously defined) of a bound ligand in any of the PXR complexes determined to date. Similarly, Tyr-306, which forms an edge-to-face with the free benzyl ring of T1317, has also not been observed to contact ligand in previous structures. The same T1317 benzyl ring forms parallel and edge-to-face aromatic stacking interactions with Phe-288 and Trp-299, respectively. The two CF3 groups of the ligand form van der Waals contacts with five residues, including a 3.5 Å interaction with Met-425 located on the receptor's αAF that likely helps to stabilize the active conformation of the AF-2 surface. The volume of the PXR ligand binding pocket in this structure was measured to be 1,334 Å3 and it was further found that the T1317 ligand occupied all but 442 Å3 of that space. In total, the fifteen residues contacted by T1317 is the largest observed for a small ligand in PXR's pocket, but does not exceed the eighteen residues contacted by the large macrolide antibiotic rifampicin 12.
Figure 2.
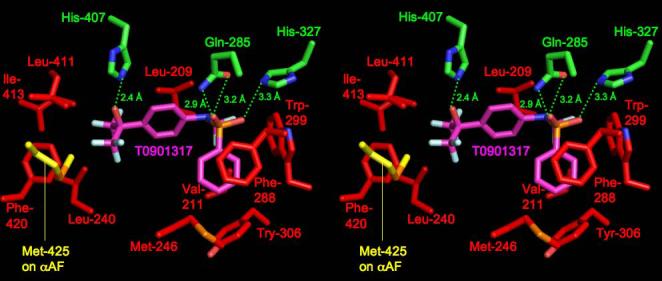
Stereoview of the binding of T0901317 within the ligand binding pocket of the human PXR LBD. Residues making polar contacts are rendered in green, those making hydrophobic interactions are in red, with Met-425 from αAF highlighted in yellow.
Table II.
Comparison of Residues Contacting T0901317 in PXR and LXR. Amino acids forming hydrogen bonding interactions with ligand are listed in bold, those on the terminal αAF helices of the receptors are underlined, and those not contacting ligand are in italics and parentheses.
| PXR | LXR |
|---|---|
| Gln-285 | Leu-313 |
| His-327 | (Ser-357) |
| His-407 | His-435 |
| Leu-209 | --* |
| Val-211 | --* |
| Leu-240 | Phe-268 |
| Met-243 | Phe-271 |
| Met-246 | Leu-274 |
| Phe-288 | Thr-316 |
| Trp-299 | Ile-327 |
| Tyr-306 | (Tyr-335) |
| Leu-411 | Val-439 |
| Ile-413 | Leu-442 |
| Phe-420 | Leu-449 |
| Met-425 | Leu-453 |
| (Cys-284-Ser)† | Met-312 |
| (Cys-301)‡ | Phe-329 |
| (Leu-206) | Leu-345 |
| (Met-323) | Ile-353 |
| (Phe-429) | Trp-457 |
Residues 209 and 211 are on a sequence insert novel to PXR; thus, no equivalent side chains exist in LXR.
Cys-284 was mutated to serine to improve the crystallization behavior of PXR; see Materials and Methods.
While Cys-301 corresponds in sequence to Phe-329, the PXR side chain Phe-288 overlaps structurally with this LXR phenylalanine; see Figure 4.
T1317 Binding by LXR vs. PXR
We next compared the structure of the human PXR LBD-T1317 complex to that of the human LXRβ LBD complexed to the same ligand 22, 24-26. The LBDs exhibited the same overall fold, sharing 2.2 Å rmsd over Cα positions (and 27% sequence identity), although the secondary structural elements present on the α1- α3 insert novel to PXR, including β1, β1', and α2, are not present in the LXR LBD (Figure 3). LXR's ligand binding pocket is roughly half the size of PXR's (650 Å3) and the observed binding of T1317 within the pocket uses essentially all the available space. The distinct and limited shape of the LXR pocket causes the T1317 ligand to bind in a position rotated by ∼30° and shifted by up to 5.3 Å relative to the position observed in the PXR complex. Sixteen LXR side chains contact T1317, two of which form hydrogen bonds with the ligand (His-435 and Thr-316). His-435 in LXR corresponds in both sequence and structure to His-407 in PXR; however, Gln-285, which forms two polar contacts with T1317 in the PXR complex, is replaced by a leucine in LXR (Table II, Figure 4). Thr-316, which forms the second polar interaction in LXR, is related in sequence to Phe-288 in PXR, but corresponds in structure to His-327, albeit shifted in position by ∼5.5 Å. The benzyl ring of T1317 forms aromatic contacts with phenylalanines 271 and 329 in LXR, which helps to position the ligand distinctly within the pocket when compared to PXR. It is also noted that distinct histidine-aromatic interactions are observed in PXR relative to LXR. An edge-to-face aromatic interaction between His-435 and Trp-457 had been noted previously for LXR; this contact is replaced in PXR by a 3.8 Å edge-to-edge interaction between His-407 and Phe-429. A second histidine aromatic contact is observed in PXR: a 3.3 Å face-to-edge interaction between Trp-299 and His-327 (Figure 4). In total, PXR and LXR share one conserved polar contact and eight conserved hydrophobic interactions in their respective complexes with T1317 (Table II). In addition, both receptors directly contact corresponding residues located on the αAF helices of their AF-2 surfaces (Met-425 in PXR, Leu-453 in LXR). LXR apparently further stabilizes its AF-2 region by the formation of the aromatic contact between His-435 and Trp-457, which is located on αAF. Taken together, these observations show that, in spite of differences in pocket shape and ligand orientation, numerous key contacts are conserved between these related receptors, leading to the efficient upregulation of targets genes by T1317.
Figure 3.
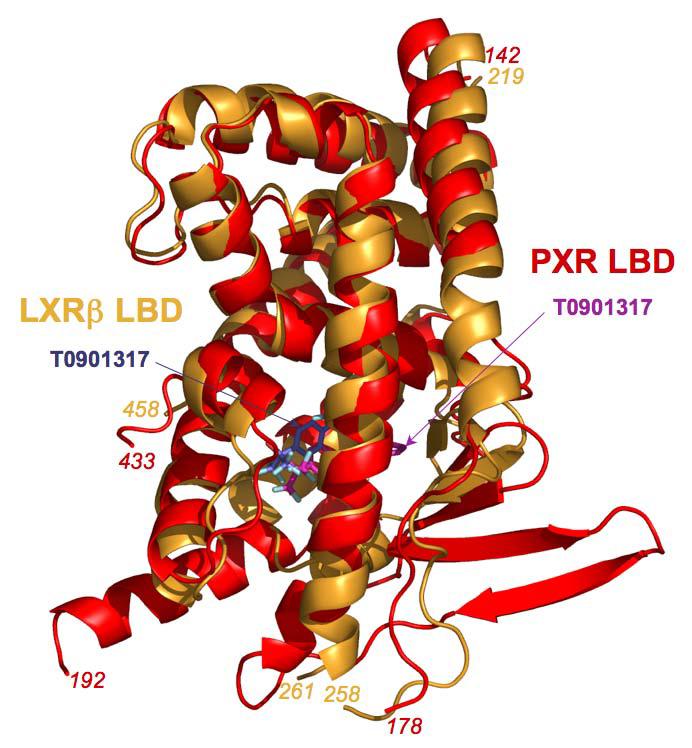
Superposition of the human PXR LBD monomer (red) on that of the human LXRβ LBD (gold), with the T0901317 ligands present in both structures shown in magenta for PXR and blue for LXR.
Figure 4.
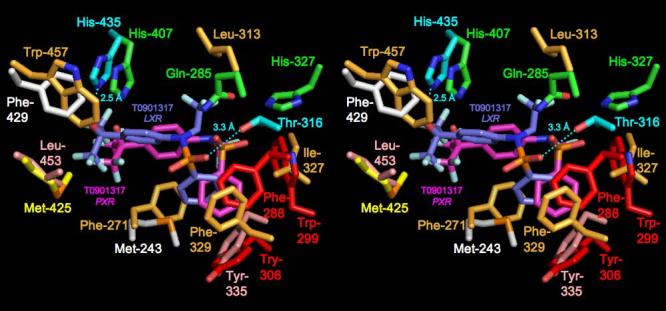
Stereoview comparing the binding of T0901317 to the LBDs of human PXR and LXRβ. Human PXR residues and T1317 ligand are colored as in Figure 1B, except Phe-429 and Met-243, which are shown in white. In LXR, residues forming polar ligand contacts are shown in cyan, while those forming hydrophobic interactions are in gold or, for Leu-452 and Tyr-335, in pink.
Design and Examination of Putative Antagonists
Because T1317 appears to bind particularly strongly to PXR, we next sought to design a PXR antagonist using the T1317 structure as a scaffold. It is known that antagonists of other nuclear receptors (e.g., tamoxifen for the estrogen receptor) appear to function by sterically blocking the active position of αAF 28. Thus, we chose to change the structure of the CF3-containing moiety in T1317, which binds adjacent to αAF in PXR (Figure 2). Eleven analogues were synthesized that included both smaller and larger groups at this position (Figure 5; Scheme 1). These compounds, along with T1317, were tested for their ability to bind to PXR in an in vitro ligand-competition assay, and to activate PXR-mediated transcription in transient transfection assays in cultured cells (Table III). Compounds 2-5, which retained the hydroxyl group and one CF3 group, all exhibited good binding to and activation of PXR. In contrast, compounds 6 and 7, which retain the hydroxyl group but not a CF3, are poor binders and activators of PXR. Similarly, compound 8, which contains a acetyl group, was the least efficacious compound examined. These data support the importance of the hydrogen bond donated by the T1317 hydroxyl group to the His-407 side chain in PXR, in particular the electron withdrawing character of the CF3 groups that polarize the hydroxyl group and improve its ability to share its hydrogen atom 25.
Figure 5.
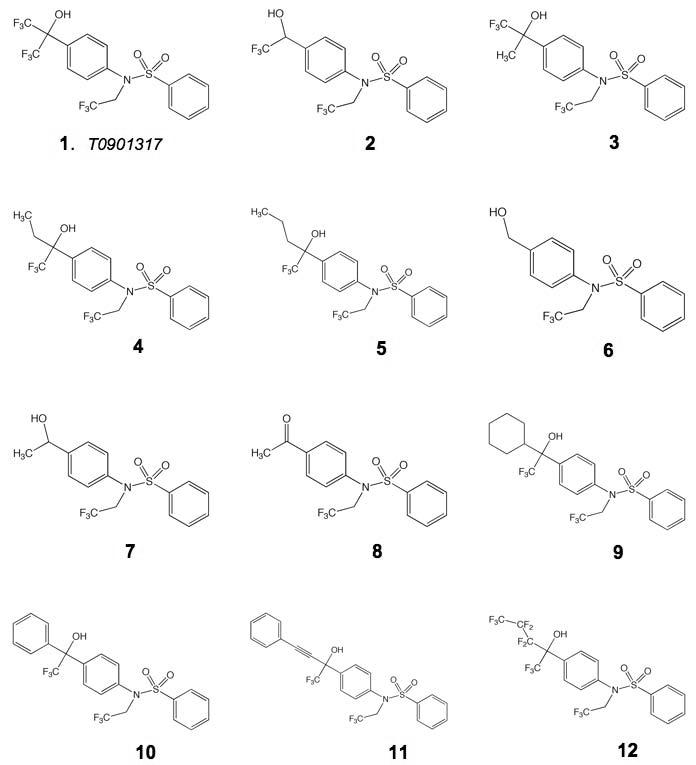
Structures of analogues of T0901317 (compound 1) examined for their impact on human PXR and human LXRβ.
Scheme 1.

Reaction conditions: (a) benzenesulfonyl chloride, pyridine, 60°C; (b) 2,2,2-trifluoroethyl trifluoromethylsulfonate, K2CO3, MeCN, 80°C; (c) oxalyl chloride, DMSO, Et3N, rt; (d) trifluoromethyltrimethylsilane, TBAF, THF, 50°C; (e) Dess-Martin periodinane, pyridine, DCM, rt; (f) RMgBr, THF, −78°C to rt; (g) phenylacetylene, n-BuLi, THF, −78°C to rt; (h) vinylmagnesiun bromide, THF, −78°C to rt.
Table III.
T0901317 Analogues and Their Impact on Human PXR and LXRβ. See Figure 5 for analogue structures. Compound 1 is T0901317 present in the PXR complex structure presented here.
| Compound | PXR pIC50 | PXR pEC50 | PXR %max | LXR pEC50 | LXR %max |
|---|---|---|---|---|---|
| 1 | 7.4 | 7.9 | 93 | 7.0 | 100 |
| 2 | 5.0 | 6.0 | 90 | <4.5 | 19 |
| 3 | 5.4 | 6.3 | 104 | 5.8 | 50 |
| 4 | 6.6 | 6.9 | 101 | 5.4 | 73 |
| 5 | 6.5 | 6.8 | 96 | 5.6 | 27 |
| 6 | 5.1 | 5.0 | 23 | <4.5 | 3 |
| 7 | 5.6 | 5.0 | 45 | <4.5 | 3 |
| 8 | 5.8 | 5.0 | 11 | <4.5 | 3 |
| 9 | 7.6 | 8.5 | 103 | <4.5 | 9 |
| 10 | 7.2 | 7.8 | 99 | 5.5 | 30 |
| 11 | 6.2 | 6.0 | 139 | <4.5 | 5 |
| 12 | 7.7 | 8.0 | 98 | 6.2 | 58 |
Compounds 9-12 were designed to include large adducts adjacent to the hydroxyl and CF3 groups, with the goal of sterically disrupting αAF position. We were surprised to find, however, that each compound bound well to PXR and served as an effective agonist rather than an antagonist (Table III). For example, the presence of cyclohexyl or benzyl rings (compounds 9, 10) were apparently accommodated by the receptor's ligand binding pocket. Indeed, even a benzyl group contained on an extended and rigid two-methylene linker (compound 11) did not disrupt either ligand binding or receptor activation. These observations suggest that the αAF and AF-2 region of PXR is reasonably conformable and capable of accommodating larger adducts than was previously appreciated. The structural basis of these effects may be based on the mobile α2 region of PXR, which is present on a sequence insert unique to this receptor relative to other members of the nuclear receptor superfamily. This stretch of the PXR LBD structure has been observed to shift in position by several Å between different ligand-bound complexes, and, in the case of the PXR structure bound to the large macrolide antibiotic rifampicin, to become completely disordered 12-15. The large adducts placed in the T1317 scaffold in compounds 9-12 may be directed toward this region of PXR and may create space for themselves by disrupting the position of α2 rather than αAF. In addition, they could extend from the receptor's pocket into solvent by creating a pore adjacent to Leu-240 (Figure 2). In either case, these observed effects underscore the fundamentally promiscuous nature of PXR in terms of ligand binding – the receptor contains a polypeptide insert that allows its binding pocket to expand and contract, facilitating the productive binding of a wide range of chemical structures.
The activation of gene expression by T1317 analogues was also examined via LXRβ by transient transfection assays in cultured cells (Table III). Similar to PXR, compounds 6-8, which do not retain the hydroxyl-group proximal to a CF3 moiety, were poor ligands for LXR, highlighting the importance of the His-435 hydrogen received from the polarized ligand hydroxyl group. In contrast to PXR, however, compounds 9 and 11 were poor agonists of LXR, while compounds 10 and 12 were relatively effective. Considering the structure of the LXRβ-T1317 complex, the benzyl group in 10 may stack upon the aromatic Phe-268 side chain, an interaction not possible with compounds 9 or 11. For 12, the flexible nature of its CF3-rich extension may facilitate productive binding by protruding past Phe-268 into solvent. Taken together, these observations regarding the impact of T1317 analogues on LXR-mediated gene expression reveal that this receptor, with its smaller ligand binding pocket and more narrow agonist profile, is more accommodating to changes in chemical structure than was expected. This highlights the plasticity present in the conserved nuclear receptor LBD fold, particularly for members of the former orphan class.
Discussion
T1317 is an efficacious activator of gene transcription mediated by both the nuclear receptors PXR and LXRβ 19. We show, by comparing the 2.8 Å resolution crystal structure of the PXR-T1317 complex to the LXRβ-T1317 complexes reported previously 22, 24-26, that these two LBDs share some analogous contacts to the ligand, but exhibit key differences as well. The packing of aromatic side chains against the benzyl group of T1317, as well as the distinct placement of the proximal hydrogen-bonding residue Thr-316, cause the ligand to adopt a position in LXR rotated ∼30° and shifted by ∼5 Å relative to that observed in PXR. Still, nearly half the contacts between protein and ligand are conserved in both complexes, including the close hydrogen bond between a histidine side chain and a polarized hydroxyl group of the ligand. Both LBDs also utilize an interaction between the same histidine side chain and an aromatic residue located on AF-helices of the receptors' AF-2 regions: Trp-457 in LXR and Phe-429 in PXR. Indeed, it was noted in this PXR complex that the presence of the ligand appears to mediate the formation of a direct 3.8 Å aromatic contact between these side chains caused by their shift by 1 Å in position toward the bound T1317. It is likely that the additional stabilization of the active conformation of αAF by this interaction plays in important role in the control of gene expression by both nuclear receptors.
We were surprised both by the difficulty we experienced in designing an antagonist to either receptor, and by the plasticity the LXR receptor exhibits upon derivatization of the efficacious ligand T1317. Given the numerous interactions observed between the T1317 and the ligand binding pockets of both receptors, it was expected that adding bulky groups to the region adjacent to the hydroxyl and dual CF3 moieties on the ligand would produce compounds that sterically block the active orientation of the AF-helix (as seen in the estrogen receptor with tamoxifen, for example) 28. Instead, we found for PXR that compounds either bound and were agonists, or appeared incapable of binding to the receptor. In addition, in the cases where reasonably large groups were added to the T1317 scaffold, the region of PXR's ligand binding pocket adjacent to αAF was remarkably amenable to accommodating bulky additional atoms. This is perhaps not surprising for PXR, given its well established promiscuous ligand binding character and the structural flexibility of unique regions (such as α2) in its ligand binding pocket. It would appear that for PXR, in contrast to more rigid and specific steroid receptors like the estrogen receptor, ligands can bridge between rigid portions of the pocket, while more flexible regions of the protein can shift to accommodate a variety of distinct chemical structures.
While the promiscuity of PXR is well known, we were not expecting to find that LXRβ would also show evidence for conformability in accommodating larger T1317 analogues. For example, compounds 10 and 12 were relatively effective agonists for LXR, in spite of the presence of large groups attached adjacent to T1317's hydroxyl moiety (Figure 5). The structural basis of this is likely centered on the region around Phe-268 in the LXR pocket, which may stack with the aromatic group in 10 and provide a pore for the conformable group in 12. LXR did exhibit more specificity for the types of groups it can accommodate in this position relative to PXR, however, as non-aromatic or longer rigid adducts in 9 and 11, respectively, that still activated PXR were clearly not agonists for LXRβ. Thus, the ligand binding domains of numerous members of the nuclear receptor superfamily, particularly the former orphan receptors, are likely to posses some degree of structural flexibility to assist in the accommodation of chemically-distinct ligands. It would appears that PXR, though, with its large and mobile α1-α3 insert, is at the extreme of this flexibility continuum with these unique features that are central to its significant promiscuity.
We conclude that the unique aspects of the PXR ligand binding pocket may make antagonist design particularly difficult for this member of the nuclear receptor superfamily. It is noted that compounds 9 and 11 appear to be selective for PXR relative to LXRβ; as such, they may be useful as leads to address neurodegenerative diseases like Niemann-Pick C 8. However, it may be necessary to look outside the ligand binding pocket of PXR to find sites effective at antagonizing the receptor using small molecules. Candidate sites on the LBD include the AF-2 surface 30 and the PXR homodimer interface, the disruption of which has been shown to impact productive coactivator binding by the receptor 16. An effective PXR antagonist, if identified, may be of significant clinical use to reduce the activation of xenobiotic metabolism pathways during the therapeutic treatment of disease.
Materials and Methods
Protein Expression and Purification
Generation of human PXR LBD in complex with the 88-amino acid fragment of human SRC-1 was accomplished as described previously 12-16. To prevent the formation of covalent complexes with reducing agent during crystallization, as has been seen with previous structures (data not shown), Cys-284 within the ligand binding pocket of the human PXR LBD was replaced with serine.
Crystallization
The human PXR ligand-binding domain/SRC-1 complex (hPXRLBD/SRC-1) was concentrated in the presence of 10-fold molar excesses T0901317 (T1317; synthesized in-house) to a final concentration of 5 mg/mL. Crystallization was achieved by hanging-drop vapor diffusion against the following conditions at 22 °C: 50 mM imidazole at pH 7.1, 10% 2-propanol, v/v.
Data Collection and Structure Determination
The structure of the T1317-bound form of the ligand binding domain of human PXR was determined by molecular replacement using the crystal structure of the apo (unliganded) PXR as a search model 15. Rotation and translation function searches were performed using AMoRe 32; clear solutions for each monomer of the dimer in the asymmetric unit were obtained in the proper space group, P212121. The structure was refined using CNS with the maximum likelihood function as a target, and included an overall anisotropic B-factor and a bulk solvent correction 33. Ten percent of the observed data were set aside for cross-validation using the free-R statistic prior to any structural refinement 34. Manual adjustments and building of the model (including the placement of the T0901317 ligands and SRC-1 coactivator fragments) were performed using O 35 and σA-weighted electron density maps 36. The structure exhibits good geometry (Table I) with no Ramachandran outliers.
Compound Syntheses
See Scheme 1 for steps involved in compound generation. 1H-NMR spectra were recorded on a Varian Gemini 400 MHz NMR spectrometer. 1H-NMR spectra are reported as chemical shift δ, number of protons, multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet; br s, broad singlet) and coupling constant (J) in Hertz. Electron Spray (ES) or Chemical Ionization (CI) was recorded on a Hewlett Packard 5989A mass spectrometer. Mass spectrometry results are reported as the mass over charge. Purity by HPLC [Luna 20 × 4 mm 3.0 micron C18(2) column, water (+0.1% v/v formic acid)/MeOH (+0.075% v/v formic acid) gradient: 50% MeOH to 100 % MeOH for 5 minutes, holding at 100% MeOH for final 1 minute, flow rate = 2.0 mL/min]. Starting material are either available from commercial sources or via literature procedures. Abbreviations used in the examples below have their accepted meanings in the chemical literature. For example, DCM (dichloromethane), THF (tetrahydrofuran), MeCN (acetonitrile), DMSO (dimethylsulfoxide) and TBAF (tetrabutylammonium fluoride).
Compound 6: 4-Aminobenzyl alcohol (10 g, 80 mmol) and benzenesulfonyl chloride (11.5 mL, 89 mmol) in pyridine were heated at 60°C overnight. Water was added and extracted with DCM. The organic layer was separated and evaporated to dryness. The residue was purified by silica gel chromatography using ethyl acetate to give 17 g (80%) of intermediate sulfonamide: 1H-NMR (DMSO-d6) δ: 7.8 (s, 1H), 7.65 (m, 3H). 7.10 (d, J=8.8Hz, 2H), 7.01 (d, J=8.8Hz, 2H), 5.04 (br s, 1H), 4.45 (s, 2H). The intermediate sulfonamide (1.2 g, 4.6 mmol), 2,2,2-trifluoroethyltrifluoro-methane sulfonate (1.97 g, 9.1 mmol) and K2CO3 (1.25 g, 9.1 mmol) in MeCN (20 mL) was heated at 80°C overnight. The solvents were removed under reduced pressure. The residue was purified by silica gel chromatography using 50% ethyl acetate in hexanes to give 6 (1.0 g, 80%): 1H-NMR (DMSO-d6) δ: 7.5-7.8 (m, 5H), 7.26 (d, J=8.5 Hz, 2H), 6.99 (d, J=8.5 Hz, 2H), 5.30 (br s, 1H), 4.62 (m, 2H), 4.43 (s, 2H); LC/MS (ES+): m/e 346 (M+H), 100% purity.
Compound 8: Prepared in a similar fashion from 4-aminoacetophenone (5.0 g, 37.0 mmol): 1H-NMR (DMSO-d6) δ: 7.90 (d, J=8.4 Hz, 2H), 7.56-7.71 (m, 5H), 7.27(d, J=8.4 Hz, 2H), 4.64 (m, 2H), 2.48 (s, 3H), LC/MS (ES+) m/e 358 (M + H), 95% purity)
Compound 2: DMSO (5.5 mL, 28 mmol) was added dropwise to 2M oxalyl chloride in DCM (19 mL)at −78°C under nitrogen followed by sequential addition of compound 6 (10 g, 30 mmol) in DMSO:DCM (1:1) (20 mL) and neat triethylamine (75 mL). The reaction mixture was gradually warmed to room temperature overnight whereupon the solvents were removed under reduced pressure. The residue was purified by silica gel chromatography with DCM to give an intermediate aldehyde (8.5 g, 85%): 1H-NMR (DMSO-d6) δ: 9.8 (s, 1H), 7.8 (m, 2 H), 7.52-7.81 (m, 5H), 7.38 (m, 2H).
The intermediate aldehyde (5.0 g, 14.6 mmol) and trifluoromethyltrimethylsilane (2.5 g, 17.5 mmol) in anhydrous THF (15 mL) was treated with a catalytic amount of TBAF at 0°C. The reaction mixture was gradually warmed to room temperature overnight. The reaction mixture was treated with 1N hydrochloric acid at room temperature for 1h. Water was added and extracted with DCM. The organic layer was separated and evaporated to dryness. The residue was purified by silica gel chromatography using 20% ethyl acetate in hexanes to give 2 (5.0 g, 85%): 1H-NMR (MeOH-d4) δ: 7.65 (m, 1H), 7.58 (d, J=8.6 Hz, 2 H), 7.52 (m, 2H), 7.45 (d, J=8.2 Hz, 2H), 7.11 (d, J=8.6 Hz, 2H), 5.03 (m, 1H), 4.43 (m, 2H); LC/MS (ES+) m/e 414 (M+H), 96% purity.
The following compounds were prepared in an analogous fashion:
Compound 12: Treatment of intermediate aldehyde with heptafluoropropyl-trimethylsilane (0.06 mL, 0.29 mmol) and purification by reverse phase chromatography on RP-C18 with 50-100% MeCN in water provided compound 12 (20 mg, 15%) as a racemic mixture: 1H-NMR (MeOH-d4) δ: 7.72 (m, 3H), 7.52 (m, 4H) 7.17 (d, J=8.6 Hz, 2H), 4.41 (m, 2H), 2.22 (m, 1H), 1.99 (m, 1H), 1.38 (m, 2H), 0.91 (m, 3H); LC/MS (ES+) m/e 456 (M+H), 98% purity.
Compound 3: Compound 3 was prepared in an analogous fashion from compound 8 to given 30 mg (25%) as a racemic mixture: 1H-NMR (MeOH-d4) δ: 7.49-7.65 (m, 7H), 7.10 (d, J=8.6 Hz, 2H), 4.40 (m, 2H), 1.70(s, 3H), LC/MS (ES+) m/e 428 (M + H), 98% purity.
Compound 4: Compound 2 (2.5 g, 6.0 mmol), Dess-Martin periodinane (5.2 g, 12.0 mmol) and pyridine (4mL, 48 mmol) in DCM (60 mL) was stirred at room temperature overnight. The reaction was treated with 60 mL of an aqueous solution of NaHCO3:Na2S2O3 (5:1) and stirred at room temperature for 2 h. The organic layer was separated and evaporated to dryness. The residue was purified by silica gel chromatography using 50% ethyl acetate in hexanes to give an intermediate trifluoromethyl ketone (2.2 g, 90%): 1H-NMR (MeOH-d4) δ: 7.46-7.62 (m, 7H), 7.21 (d, J=8.6 Hz, 2H), 4.45 (m, 2H). 1M ethyl magnesium bromide in THF (0.5 mL) was slowly added to a solution of the above ketone (100 mg, 0.24 mmol) in THF (3mL) at −78°C. The reaction mixture was gradually warmed to room temperature overnight. Saturated aqueous NH4Cl was added, and the aqueous layer was extracted with ethyl acetate. The organic layer was separated and evaporated to dryness. The residue was purified by reverse phase chromatography on RP-C18 using 50-100% MeCN in water to give 4 (5 mg, 10%) as a racemic mixture: 1H-NMR (MeOH-d4) δ: 7.54-7.71 (m, 7H), 7.11 (d, J=8.6Hz, 2H), 4.45 (m, 2H), 2.21 (m, 1H), 1.98 (m, 1H), 0.87 (m, 3H); LC/MS (ES+) m/e 445 (M+H), 98% purity.
The following compounds were prepared in an analogous fashion from the above intermediate trifluoromethyl ketone:
Compound 5: Using 2M n-propyl magnesium bromide in diethyl ether followed by purification by reverse phase chromatography on RP-C18 with 50-100% MeCN in water gave rise to compound 5 (7 mg, 10%) as a racemic mixture: 1H-NMR (MeOH-d4) δ: 7.47-7.65 (m, 7H), 7.11 (d, J=8.6 Hz, 2H), 4.41 (m, 2H), 2.22 (m, 1H), 1.99 (m, 1H), 1.38 (m, 2H), 0.91 (m, 3H); LC/MS (ES+) m/e 456 (M+H), 95% purity.
Compound 9: Using 2M cyclohexyl magnesium bromide in diethyl ether (0.13 mL) followed by purification by reverse phase chromatography on RP-C18 with 50-100% MeCN in water provided compound 9 (20 mg, 20%) as a racemic mixture: 1H-NMR (MeOH-d4) δ: 7.43-7.89 (m, 7H), 7.11 (d, J=8.6 Hz, 2H), 4.41 (m, 2H), 2.61-2.05 (4m, 5H), 1.03 (m, 3H), 0.93 (m, 3H); LC/MS (ES+) m/e 496 (M+H), 94% purity.
Compound 10: Using 3M phenyl magnesium bromide in diethyl ether (0.09 mL) followed by purification via reverse phase chromatography on RP-C18 with 50-100% MeCN in water gave rise to compound 10 (70 mg, 70%) as a racemic mixture: 1H-NMR (MeOH-d4) δ: 7.33-7.69 (m, 12H), 7.11 (d, J=8.5 Hz, 2H), 4.42 (m, 2H), LC/MS (ES+) m/e 490 (M+H), 100% purity.
Compound 11: To a solution of phenylacetylene (83 mg, 0.75 mmol) in THF (2 mL) was added 1.6M n-BuLi in hexanes (0.315 mL, 0.5 mmol) at −78°C over 30 minutes. After this time, a solution of the above intermediate trifluoromethylketone (100 mg, 0.25 mmol) in THF (2 mL) was added. The reaction mixture was gradually warmed to room temperature overnight. Saturated aqueous NH4Cl was added, and the aqueous layer was extracted with DCM. The organic layer was separated and evaporated to dryness. The residue was purified by reverse phase chromatography on RP-C18 using 50-100% MeCN in water to give compound 11 (77 mg, 75%) as a racemic mixture: 1H-NMR (MeOH-d4) δ: 7.43-7.80 (m, 12H), 7.17 (d, J=8.6 Hz, 2H), 4.42 (m, 2H); LC/MS (ES+) m/e 514 (M + H), 98% purity.
Compound 7: Compound 7 was prepared from 8 (100 mg, 0.28 mmol) and 1M vinyl magnesium bromide (0.84mL, 0.84 mmol) using the procedure described for the preparation of 4. Purification by reverse phase chromatography on RP-C18 using 50-100% MeCN in water provided 7 (30 mg, 30%) as a racemic mixture: 1H-NMR (MeOH-d4) δ: 7.49-7.67 (m, 5H), 7.32 (d, J=8.4 Hz, 2H), 7.03 (d, J=8.4 Hz, 2H), 4.79 (m, 1H), 4.39 (m, 2H), 1.40(s,2H); LC/MS (ES+) m/e 358 (M +H), 98% purity.
Transient Transfections
Transient transfection and reporter gene assays using full-length human PXR and LXRβ were performed as described previously 12.
Competition Ligand Binding Assay
Competitive ligand binding assays using [N-methyl-3H]-GW0438X were preformed as described elsewhere 16.
Footnotes
Supported by: National Institutes of Health grant DK62229 (M.R.R).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Backman M, Ohlsson R, Postlind H, Blomquist P, Berkenstam A. Proc Natl Acad Sci U S A. 1998;95(21):12208–13. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blumberg B, Sabbagh W, Jr., Juguilon H, Bolado J, Jr., van Meter CM, Ong ES, Evans RM. Genes & Develop. 1998;12(20):3195–205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM. Cell. 1998;92(1):73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 4.Carnahan VE, Redinbo MR. Curr Drug Metab. 2005;6(4):357–67. doi: 10.2174/1389200054633844. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. J Clin Invest. 1998;102(5):1016–23. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore JT, Kliewer SA. Toxicology. 2000;153(13):1–10. doi: 10.1016/s0300-483x(00)00300-0. [DOI] [PubMed] [Google Scholar]
- 7.Moore LB, Goodwin B, Jones SA, Wisely GB, Serabjit-Singh CJ, Willson TM, Collins JL, Kliewer SA. Proc Natl Acad Sci U S A. 2000;97(13):7500–2. doi: 10.1073/pnas.130155097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langmade SJ, Gale SE, Frolov A, Mohri I, Suzuki K, Mellon SH, Walkley SU, Covey DF, Schaffer JE, Ory DS. Proc Natl Acad Sci U S A. 2006;103(37):13807–12. doi: 10.1073/pnas.0606218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orans J, Teotico DG, Redinbo MR. Mol Endocrinol. 2005;19(12):2891–900. doi: 10.1210/me.2005-0156. [DOI] [PubMed] [Google Scholar]
- 10.Ingraham HA, Redinbo MR. Curr Opin Struct Biol. 2005;15(6):708–15. doi: 10.1016/j.sbi.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Nature. 1998;395(6698):137–43. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 12.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. Mol Endocrinol. 2005;19(5):1125–34. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 13.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. J Mol Biol. 2003;331(4):815–28. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 14.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. Biochemistry. 2003;42(6):1430–8. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 15.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. Science. 2001;292(5525):2329–33. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 16.Noble SM, Carnahan VE, Moore LB, Luntz T, Wang H, Ittoop OR, Stimmel JB, Davis-Searles PR, Watkins RE, Wisely GB, Lecluyse E, Tripathy A, McDonnell DP, Redinbo MR. Biochemistry. 2006;45(28):8579–8589. doi: 10.1021/bi0602821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. Nature. 1996;383(6602):728–31. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 18.Kalaany NY, Gauthier KC, Zavacki AM, Mammen PP, Kitazume T, Peterson JA, Horton JD, Garry DJ, Bianco AC, Mangelsdorf DJ. Cell Metab. 2005;1(4):231–44. doi: 10.1016/j.cmet.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Laffitte BA, Repa JJ, Joseph SB, Wilpitz DC, Kast HR, Mangelsdorf DJ, Tontonoz P. Proc Natl Acad Sci U S A. 2001;98(2):507–12. doi: 10.1073/pnas.021488798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cell. 1998;93(5):693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Repa JJ, Gauthier K, Mangelsdorf DJ. J Biol Chem. 2001;276(46):43018–24. doi: 10.1074/jbc.M107823200. [DOI] [PubMed] [Google Scholar]
- 22.Hoerer S, Schmid A, Heckel A, Budzinski RM, Nar H. J Mol Biol. 2003;334(5):853–61. doi: 10.1016/j.jmb.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 23.Jaye MC, Krawiec JA, Campobasso N, Smallwood A, Qiu C, Lu Q, Kerrigan JJ, De Los Frailes Alvaro M, Laffitte B, Liu WS, Marino JP, Jr., Meyer CR, Nichols JA, Parks DJ, Perez P, Sarov-Blat L, Seepersaud SD, Steplewski KM, Thompson SK, Wang P, Watson MA, Webb CL, Haigh D, Caravella JA, Macphee CH, Willson TM, Collins JL. J Med Chem. 2005;48(17):5419–22. doi: 10.1021/jm050532w. [DOI] [PubMed] [Google Scholar]
- 24.Svensson S, Ostberg T, Jacobsson M, Norstrom C, Stefansson K, Hallen D, Johansson IC, Zachrisson K, Ogg D, Jendeberg L. EMBO J. 2003;22(18):4625–33. doi: 10.1093/emboj/cdg456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams S, Bledsoe RK, Collins JL, Boggs S, Lambert MH, Miller AB, Moore J, McKee DD, Moore L, Nichols J, Parks D, Watson M, Wisely B, Willson TM. J Biol Chem. 2003;278(29):27138–43. doi: 10.1074/jbc.M302260200. [DOI] [PubMed] [Google Scholar]
- 26.Farnegardh M, Bonn T, Sun S, Ljunggren J, Ahola H, Wilhelmsson A, Gustafsson JA, Carlquist M. J Biol Chem. 2003;278(40):38821–8. doi: 10.1074/jbc.M304842200. [DOI] [PubMed] [Google Scholar]
- 27.Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, McKee DD, Galardi CM, Plunket KD, Nolte RT, Parks DJ, Moore JT, Kliewer SA, Willson TM, Stimmel JB. Nature. 2002;415(6873):813–7. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- 28.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. Cell. 1998;95(7):927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 29.Synold TW, Dussault I, Forman BM. Nat Med. 2001;7(5):584–90. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 30.Huang H, Wang H, Sinz M, Zoeckler M, Staudinger J, Redinbo MR, Teotico DG, Locker J, Kalpana GV, Mani S. Oncogene. 2006 doi: 10.1038/sj.onc.1209788. in press. [DOI] [PubMed] [Google Scholar]
- 31.Takeshita A, Taguchi M, Koibuchi N, Ozawa Y. J Biol Chem. 2002;277(36):32453–8. doi: 10.1074/jbc.M111245200. [DOI] [PubMed] [Google Scholar]
- 32.Navaza J, Saludjian P. Methods Enz. 1997;276A:581–594. doi: 10.1016/S0076-6879(97)76079-8. [DOI] [PubMed] [Google Scholar]
- 33.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr. D. 1998;54(Pt 5):905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 34.Brünger AT. Acta Crystallogr. D Biol. Crystallogr. 1993;49:24–36. doi: 10.1107/S0907444992007352. [DOI] [PubMed] [Google Scholar]
- 35.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 36.Read RJ. Acta Crystallogr. A. 1986;42:140–149. [Google Scholar]