

Abstract
An overview is presented on the application of surface-enhanced infrared absorption (SEIRA) spectroscopy to biochemical problems. Use of SEIRA results in high surface sensitivity by enhancing the signal of the adsorbed molecule by approximately two orders of magnitude and has the potential to enable new studies, from fundamental aspects to applied sciences. This report surveys studies of DNA and nucleic acid adsorption to gold surfaces, development of immunoassays, electron transfer between metal electrodes and proteins, and protein–protein interactions. Because signal enhancement in SEIRA uses surface properties of the nano-structured metal, the biomaterial must be tethered to the metal without hampering its functionality. Because many biochemical reactions proceed vectorially, their functionality depends on proper orientation of the biomaterial. Thus, surface-modification techniques are addressed that enable control of the proper orientation of proteins on the metal surface.
Figure.
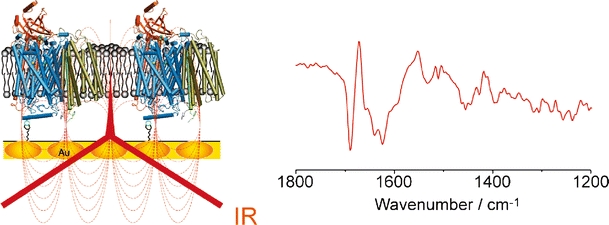
Surface enhanced infrared absorption spectroscopy (SEIRAS) on the studies of tethered protein monolayer (cytochrome c oxidase and cytochrome c) on gold substrate (left), and its potential induced surface enhanced infrared difference absorption (SEIDA) spectrum
Keywords: FTIR, Protein, Membrane, Electron transfer, Self-assembled monolayer
Introduction
The seminal discovery of surface-enhanced Raman scattering (SERS) in the early 70s opened the field of surface-enhanced spectroscopy [1, 2]. The phenomenon has subsequently also been observed at longer wavelengths and, ultimately, led to the realization of surface-enhanced infrared absorption spectroscopy (SEIRAS) [3]. Several reports appeared in the 90s on both practical and theoretical aspects of the phenomenon [4–6]. SERS and SEIRAS have lately received attention in the field of biochemistry and biophysics, because of growing interest in bio-nanotechnology [7]. Typical approaches of bio-nanotechnology are constructions of hybrid devices in which bio-molecules, e.g. DNA or proteins, are combined with a solid sensing and/or actuating substrate, for example as an electrode. With this architecture the whole bandwidth of biological functions can be addressed by exchange of signals with the sensor/actuator. The concept of the hybrid bio-device is key to the development of biosensors for DNA or proteins, or for immunoassays on a chip, etc. This concept is, moreover, valuable not only for technological progress but also for fundamental studies on proteins and other biologically active materials. Triggering the properties of the adsorbate on the substrate enables functional studies of biomolecules. A critical issue in the design of such a device is assessment of the interface between biomaterial and substrate in which the essential signal relay between the two different materials occurs. This signal relay comprises only small amounts of monolayer molecules at the interfaces which are difficult to detect by conventional Raman and IR techniques. Spectroscopic distinction from the strong background of the bulk is also difficult. This obstacle is overcome by exploiting the “optical near-field effect” of surface-enhanced spectroscopy in which the signal enhancement is restricted to the interface. Characteristic of vibrational techniques, SERS and SEIRA provide a wealth of molecular information on the level of a single chemical bond.
SERS and SEIRA are complementary techniques, and each has its own advantages and disadvantages. SERS takes advantage of its enormous enhancement factor (of the order of 106–1012). The strongest enhancement occurs as a result of the resonance condition if the biomolecule carries a chromophoric co-factor. The fluorescence which often accompanies this may render detection of the Raman spectrum difficult, however. Although the latter is not a problem with SEIRA, the surface-enhancement is only modest (∼101–103). SEIRAS probes almost all bands of the adsorbed species as long as the vibrational mode includes a dipole component perpendicular to the surface (surface-selection rule) [5]. Although the enhancement factor of SEIRAS is smaller than that of SERS, the cross-section for IR absorption is several orders of magnitude higher than the corresponding Raman cross-section. Thus, the modest enhancement of SEIRAS may be sufficient for many applications.
To study the functionality of proteins by IR spectroscopy, the difference technique has provided an unprecedented amount of molecular information [8–10]. The IR spectrum of a protein is recorded in one state—often the resting state—and subtracted from the IR spectrum of another state—an active state or reaction intermediate. The difference spectrum then contains only the vibrational bands associated with the transition from one state to the other. All of the other vibrational bands are cancelled which drastically simplifies interpretation of the vibrational changes. As a consequence, the amplitudes of the difference bands are much smaller than the absorption bands of the entire protein (difference bands may be smaller by a factor of 10−4, depending on the size of the protein). Resolving the small difference bands requires acute spectroscopic sensitivity, in particular when surface-enhanced infrared difference spectroscopy (SEIDAS) is performed on a protein monolayer.
In this report, we review applications of SEIRA in which biochemical processes were studied. Because SEIRAS was introduced to the field of biomolecules only recently, we consider it worthwhile to start with practical aspects of the method.
Experimental considerations
Preparation of thin metal-film substrates
Preparation of the thin metal film is the critical part of a successful SEIRA experiment. Enhancement by SEIRA is very dependent on the size, shape, and particle density of the selected metal-island film. These properties are easily affected by the experimental conditions during film fabrication, e.g. rate of film deposition, type of the substrate, the substrate temperature, etc. [11].
SEIRA-active metal islands are usually prepared by high-vacuum evaporation of the metal on to a supporting substrate. Metals such as Au, Ag, Cu, and Pt are vapor-deposited by Ar sputtering, electron-beam heating, or resistive thermal heating of a tungsten basket. The thickness and the rate of deposition are monitored by use of a quartz crystal microbalance (QCM). Controlling of the deposition rate is essential for optimum enhancement. Slow deposition (0.1 nm s−1 or less for deposition of Au or Ag on Si or CaF2) generally results in greater enhancement [11]. This condition also depends on the type of metal, the type of substrate, and the metal film thickness, however, and must therefore be optimized for the system being used. As the morphology of the metal film affects the extent of surface enhancement, templates such as a periodic particle-array film prepared by nanosphere lithography have been used [12, 13]. This approach not only increases the enhancement factor but the reproducibility of signal enhancement will enable quantitative SEIRAS.
Although vacuum evaporation is routinely used, the equipment is costly and not readily available. An alternative means of forming a metal thin film is by chemical (electroless) deposition. Stable SEIRA-active thin films of Au, Pt, Cu, and Ag have been reported on Si or Ge substrates [14–17]. The procedure for preparing a thin Au film on a silicon surface is described below:
The surface of the Si is covered with 40% w/v NH4F for a few minutes (typically 1–3 min) to remove the oxide layer and to terminate the surface with hydrogen.
-
After rinsing with water, a freshly prepared 1:1:1 mixture of:
- 0.03 mol L−1 NaAuCl4
- 0.3 mol L−1 Na2SO3+0.1 mol L−1 Na2S2O3+0.1 mol L−1 NH4Cl, and
- 2% w/v HF
is put on the Si surface for 60–90 s.
Although a shiny Au film is formed, the Au surface may still be contaminated with thio compounds from the plating chemicals. These are removed by electrochemical cycling of the potential between 0.1 and 1.4 V in 0.1 mol L−1 H2SO4 until the cyclic voltammogram of polycrystalline gold appears (broad oxidation peak above 1.1 V and a sharp reduction peak at 0.9 V relative to the SCE). One can, instead, apply a dc voltage of +1.5 V between the Au film and the counter electrode (e.g. a Pt wire) for ca. 1 min.
Atomic force microscope (AFM) images of the chemically deposited Au film reveals an island structure similar to that of the vacuum-evaporated Au films, albeit with somewhat larger average diameter of the metal islands [16, 17]. Another advantage of the chemical method is the stronger adhesion of the deposited metal layer to the substrate. This property helps significantly when long-term stability of the metal film is required, as is typical for preparation of biomimetic devices (vide infra).
An interesting option is the use of colloidal gold nano particles [18, 19]. Colloidal gold is prepared by reducing tetrachloroauric(III) acid with sodium citrate. It is also commercially available in different particle sizes. Typically, 10–50 nm colloidal gold is chosen. A major difference from the metal thin-film method is that the sample is attached to the colloidal gold suspension before measurement. The colloidal gold is then collected on an optical substrate by filtration or by centrifugation. The sample/colloid gold is measured either in the transmission configuration (with an IR card) or dried on an ATR prism.
Removal of the metal film from the supporting substrate
The metal film prepared by vacuum-evaporation is usually poorly adhesive. It is easily wiped off with ethanol or acetone. A metal film prepared by the chemical deposition method adheres much more strongly, and hence may not be removed by wiping or even by polishing with aluminium powder. Such metal films can be dissolved by immersion in a boiling solution of a 1:1:1 mixture of HCl (32%), H2O2 (30%), and H2O.
Geometry and optical configuration
The most widespread optical arrangement employs the so-called metal underlayer configuration (sample/metal film/supporting substrate; Fig. 1a). IR-transparent materials are usually used as supporting substrates. Highly refractive materials, for example Si, Ge, and ZnSe, are suitable for internal reflection optical geometry (Fig. 1a, (ii)) and CaF2 and BaF2 are more suitable for transmission geometry (Fig. 1a (i)). For the former, relatively thick metal films (in the range of ten to several hundred nanometers) can be used whereas for the latter geometry the thickness of metal film should be kept to less than 10 nm, because the island structure starts to merge at higher thickness. As a result, the metal film is no longer transmissive [11, 20].
Fig. 1.
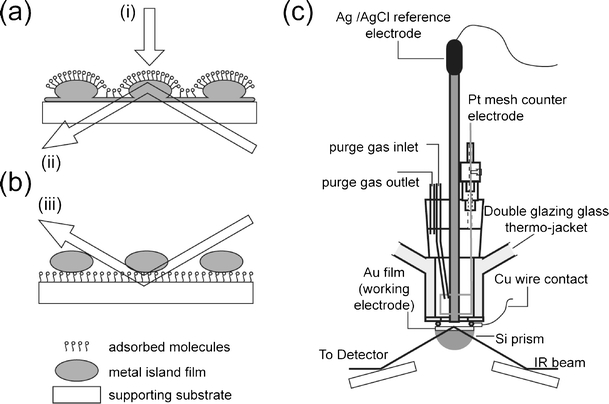
Schematic diagram of the optical configuration of SEIRAS. (a) Metal underlayer configuration. (b) Metal overlayer configuration. The arrows denote the optical pathway of the IR beam in (i) transmission, (ii) attenuated total reflection, and (iii) external reflection geometry. (c) Spectro-electrochemical cell for SEIRA spectroscopy
For internal reflection geometry (Kretschmann attenuated total-reflection geometry), half-cylindrical, half-spherical, or trapezoidal prisms are commonly used. The former two types are advantageous for optical reasons, because the position of the focus on the metal film is not affected by changes in the angle of incidence of the IR beam. Si is commonly used as reflection element, because of its high chemical stability. One disadvantage is that the available spectral range is then limited to >1000 cm−1. Although use of Ge enables use of a wider spectral window (>700 cm−1), it is not suitable for electrochemical experiments at potentials above 0.0 V (relative to the SCE) in acidic solution (pH<4.0), in which it starts to dissolve. Although Ge matches well with Ag or Cu, combination with Au may cause a decrease in the enhancement factor, because of the formation of a Ge–Au alloy at the boundary which strongly affects the morphology of the island structure of the film.
Non-IR-transparent material, for example glass, polymers, carbon, and metal, can be used as a substrate only with external reflection which uses the metal overlayer configuration (metal particle/sample/supporting substrate, Fig. 1b). In this configuration the thin metal layer (ca. 4–7 nm) is directly evaporated on the sample/support surface. It should be noted that poorly reflective material is optically favorable, because of reduced distortion of band shape in the corresponding SEIRA spectrum [21, 22]. This configuration is advantageous for detection in trace analysis of a sample adsorbed on glass or a polymer [22]. It is pointed out, however, that external reflection IR spectroscopy is not very useful for functional studies of biomaterial, because of the required presence of bulk water that strongly absorbs the IR probe beam.
Chemical modification of the metal film surface
It is crucial to chemically modify the bare metal if direct contact with the metal surface hampers the structure and function of the biomaterial. Strong interaction forces may even induce denaturation of proteins, because their secondary structure is very dependent on weak hydrogen-bonds between amino acids. The use of a chemically modified electrode (CME) is an approach used to tackle these obstacles [23]. The surface of the metal electrode is modified by heterobifunctional crosslinkers that comprise:
a thiol group that is spontaneously adsorbed by the metal electrode;
a spacer group with different lengths of alkyl chain; and
a functional headgroup pointing toward the bulk solution.
The CME surface can be conveniently prepared by the so-called “SAM (self-assembled monolayer) method” in which the SEIRA-active metal substrate is immersed in a solution containing the crosslinker molecule (normally for between 10 min and 2 h, depending on size of the molecule). The SAM of the crosslinker is formed spontaneously by quasi-covalent bonding between the sulfur and the metal surface. The substrate is then thoroughly rinsed with the solvent so that a SAM of the crosslinker remains on the metal substrate.
Adsorption of the protein by the chemically modified electrode (CME) is a suitable model for protein–protein interaction. By varying the properties of the functional headgroup it is possible to mimic the specific conditions of the substrate during the protein–protein or protein–ligand interaction process. For example, use of a carboxyl headgroup mimics the side-chain terminus of Asp or Glu. The negatively charged carboxyl headgroup can specifically interact with positively charged amino acids, for example Lys, His, and Arg residues, on the protein surface. Another approach is to exploit the specific interaction of Ni2+-chelated nitrilotriacetic acid (NTA) with a sequence of histidine residues genetically introduced into the target protein (His-tag) [24, 25]. Details of this approach are given below.
Applications
Studies of nucleic acids and DNA
SEIRAS studies on nucleic acid bases have provided information about the adsorption and orientation of thymine on silver island film [26], and of cytosine [27] and uracil [28] on gold electrodes. These experiments have been expanded to studies of the hydrogen-bonding interaction between complementary bases (adenine (A)–thiamine (T) and guanine (G)–cytosine (C)) as model systems of DNA and RNA. Sato et al. used SEIRAS to study base-pairing between thymidine and 6-amino-8-purinethiol, a thio-derivatized adenine, immobilized on a gold electrode surface [29]. They observed a characteristic thymidine band at 1571 cm−1 adsorbed on the adenine-modified surface by hydrogen-bonding. This base-pairing occurs at potentials higher than 0.1 V (relative to the SCE), at which the N1 of adenine is deprotonated and orientated toward the solution. This result implies that hydrogen-bonding between the base pairs can be controlled by the applied electrode potential.
As an interesting extension of the method, SEIRAS has been used as a diagnostic criterion in cancer research [30, 31]. Although the reported enhancement factor of 3–5 only modest [31], the enhanced bands are impossible to observe with conventional IR spectroscopy but are crucial for determining the properties of nucleic acids of tumor tissues and their interaction with anticancer drugs.
Immunoassays based on SEIRAS
There is substantial interest in the development of biosensors in which the biological component is associated with a transducer to provide a means of detecting changes in the biological component. Most current biosensors consist of an enzyme or an antibody which interacts with the substrate or the respective antigen. The enzymes or antibodies are usually immobilized on a platform and the interaction between the enzyme and the substrate (or the antibody and the antigen) is monitored. Biosensors based on surface plasmon resonance (SPR) have received considerable attention in recent years. In this method, the biomaterial is immobilized on the surface of a metal-film-covered optical prism (ATR–Kretschmann configuration), i.e. the same optical principle is applied as with SEIRAS. The advantage of SEIRAS compared with SPR is that the chemical information derived from the vibrational spectrum identifies the nature of the adsorbed species yet the sensitivity of SEIRAS is comparable with that of SPR. The sensitivity of SPR is poor when the refractive index of the adsorbed species is close to that of the solvent, as it is for small organic molecules dissolved in an organic solvent. SEIRAS detects the individual vibrational modes of functional groups of the adsorbate, and the S/N ratio does not depend on the type of solvent if the vibrational bands of the solvent do not overlap with those of the adsorbate. SEIRAS can, moreover, be used to detect even minute structural changes that occur in the adsorbed layer during the adsorption process.
Brown et al. have applied SEIRAS to biosensor analysis for determination of antibody–antigen interactions [32]. In this experiment, antibodies for Salmonella (anti-SAL) were immobilized on a gold surface deposited on a silicon wafer. After taking the spectrum of anti-SAL, this “dipstick” sensor was immersed in a solution containing the antigen (SAL). The SEIRA spectra of anti-SAL without SAL has characteristic antibody peaks at 1085 and 990 cm−1. Addition of SAL results in an additional band at 1045 cm−1 which was assigned to the P–O stretching vibration of the phospholipid in the cell wall, which indicates the presence of SAL in the solution.
Initially, the analyte adsorbed by the “dipstick” sensor was probed by external reflection SEIRA spectroscopy. Later, the authors developed a new method for SEIRAS in which they used a colloidal gold surface as adsorption platform [19]. The colloidal gold–antibody–antigen complexes are left to assemble in solution, taking advantage of the tight binding of the antibodies to colloidal gold which stabilizes the colloid solution. When the sample has been assembled the complexes are collected, either by filtration on a porous polyethylene membrane [19] or by centrifugation on an ATR substrate [18], and are then readily mounted in the FTIR spectrometer. Both “dipstick” and “colloidal gold” furnish similar spectral quality, although the enhancement of the latter approach is not mentioned. The colloidal gold system is more convenient and less expensive means of preparation than coating by sputtering. Filtration of the colloidal particles on disposable IR cards is also a rapid, easy, and cost-effective means of producing a SEIRA substrate which operates in transmission mode and does not require complicated optical arrangements.
SEIRAS to probe protein functionality
The metal used for surface enhancement, can also be used as an electrode. The enzymatic reactions of many biological systems, especially those of membrane proteins, are driven by an electrochemical gradient across the cell membrane. Such a system can be artificially reproduced on an electrode surface to mimic the physiological properties of a biological membrane.
Electrochemically induced oxidation and reduction of a monolayer of cytochrome c (Cc), a protein that mediates single-electron transfer between the integral membrane protein complexes of the respiratory chain, is the model system for electron transfer within and between proteins [33]. Electrons are directly injected to and/or withdrawn from Cc after proper contact has been established with the electrode by means of a suitable surface modifier. Proteins can be attached to such a chemically modified electrode (CME) by electrostatic attraction or covalent interaction. Figure 2 depicts potential-induced difference spectra of Cc that has been electrostatically adsorbed by different types of CME. The positive and negative peaks are indicative of conformational changes associated with the transition from the fully oxidized to the fully reduced state. Details of band assignment have been published [14]. The difference spectra reveal subtle changes of the secondary structure induced by rearrangement of the hydrogen-bonded network among the internal amino acid side-chains surrounding the heme chromophore. Note that the peak positions of the observed Cc bands do not depend on the type of CME layer whereas the relative intensities are strongly dependent on the CME. The former suggests that the internal conformational changes of Cc are not affected by interaction with the modifiers. The latter observation is attributed to the different surface structure, i.e. the orientation or position of the Cc relative to the CME underlayer [14].
Fig. 2.

Potential-induced redox difference spectra of a cytochrome c monolayer adsorbed on a variety of chemically modified electrodes: (a) mercaptopropionic acid, (b) mercaptoethanol, (c) cysteine, (d) dithiodipyridine. Minor spectral contributions from the surface modifier have been subtracted to selectively reveal the vibrational spectra of the adsorbed cytochrome c [14]
Studies of molecular and protein recognition
The acute sensitivity of SEIRAS is particularly useful when the technique is applied to studies of membrane proteins. Handling of the membrane proteins usually requires great care, because of their sensitivity to degeneration as soon as they are separated from the native lipid bilayer. The large size of the proteins also makes it difficult to control their orientation.
A successful strategy for immobilization of membrane proteins is to attach the purified protein, by using the selective affinity of a genetically introduced histidine tag (His-tag), to a nickel-chelating nitrilotriacetic acid (Ni-NTA) monolayer (SAM) self-assembled on a chemically modified gold surface. The lipid environment of the surface-anchored membrane protein is subsequently restored by in-situ dialysis of the detergent around the amphiphilic membrane protein. This approach results in orientated immobilization and reconstitution of the native matrix, which enhances the stability of the membrane protein and restores full functionality.
The success of the method has been demonstrated by use of cytochrome c oxidase (CcO) [24, 25]. Recombinant CcO (solublized by detergent) from Rhodobacter sphaeroides was immobilized on the stepwise-formed Ni-NTA SAM bound to the gold surface. It is advantageous to follow each surface-modification step by in-situ SEIRAS, which ensures qualitative and quantitative control of each reaction step. The protein is subsequently embedded in the lipid layer by removing the detergent by addition of Bio-Beads. SEIRA spectra reveal the increase of several lipid bands during the reconstitution without any dissociation of protein from the surface. Atomic-force microscopy (AFM) provides direct evidence of the formation of the lipid bilayer. Spots of size ∼7 nm appear in the AFM image; this is in accordance with the diameter of a single CcO molecule [34]. It is remarked that the same approach can be used to bind CcO to a rough Ag surface. Integrity of the native structures of the heme cofactors (heme a and heme a3) was shown by surface-enhanced resonance Raman spectroscopy (SERRS) [35, 36].
Full functionality of the lipid-reconstituted CcO electrode is demonstrated by the fact that a catalytic current resulting from reduction of oxygen, the native property of CcO, is observed when an electron is donated by the adsorbed Cc. In such an experiment orientation of the CcO is controlled by the position of the His-tag on either side of the membrane surface CcO. With molecular genetic techniques, the His-tag is introduced either in the C-terminal tail of subunit I or in the C-terminus of subunit II. By binding CcO through the respective His-tag, the former orients CcO such that the binding site of Cc is exposed to the bulk solution. The latter orientation obstructs the binding site for Cc, because of the barrier imposed by the lipid bilayer. Indeed, SEIRAS, in combination with electrochemistry, reveals that Cc binds and initiates the catalytic reaction of CcO only when the Cc binding site faces the bulk aqueous phase and that Cc does not interact with the oppositely oriented CcO [24, 25].
Interestingly, the electrochemically induced redox difference surface-enhanced infrared difference absorption (SEIDA) spectrum of the Cc–CcO complex is remarkably similar to that of Cc adsorbed on a carboxy-terminated SAM (mercaptoundecanoic acid, MUA) (Fig. 3c and d). It is noted that SEIDA spectra of Cc are sensitive to the properties of the terminal headgroup of the SAM that it interacts with [14]. The similarity of the spectra of Cc–CcO and Cc–MUA suggests a preponderance of carboxylate residues at the physiological docking site of Cc, i.e., those of the side-chains of aspartate or glutamate units. Ferguson-Miller et al. have, indeed, identified residues Glu148, Glu157, Asp195, and Asp214 (all in subunit II of CcO from R. sphaeroides) as the major interaction partners of Cc [37].
Fig. 3.

Potential-induced IR difference spectra of: (a) cytochrome c bound to a monolayer of cytochrome c oxidase tethered to a gold surface via His-tag/Ni-NTA interaction; (b) tethered monolayer of cytochrome c oxidase alone; (c) difference between (a) and (b) which recovers the vibrational contribution from cytochrome c only; (d) cytochrome c adsorbed to a monolayer of mercaptoundecanoic acid
The same strategy has recently been applied to immobilization of photosystem 2 (PS2) on an Au surface with the purpose of developing a semi-artificial device for production of hydrogen by photosynthetic oxidation of water [38]. In this report, SEIRA was used to determine the adsorption kinetics of PS2 on the Ni-NTA-modified Au surface. On illumination of the surface-bound PS2 a photocurrent was generated. The action spectrum corresponds to the absorption spectrum of PS2, indicating that the observed photocurrent is caused by photoreaction of PS2.
This methodology is a general approach for immobilization of proteins, because the introduction of affinity tags is routine with modern genetic techniques. Other tags beyond the His-tag may also be an option. Thus, orientational control of protein adsorption on a solid surface can be conveniently achieved. An oriented sample is mandatory when the vectorial function of membrane proteins is addressed. Many membrane proteins are asymmetric in their functionality, because they translocate ions or solutes preferentially in one direction or because their stimulant, e.g. ligand, binding partner protein, membrane potential, etc., affects the protein from one side only.
Summary and outlook
Despite the accomplishments reported in this article, studies of biomaterials by SEIRA is still in its infancy. Potential-induced difference spectroscopy using SEIRA is a promising possibility for study of the functionality of proteins. The stimulus in such studies is not limited to the electric trigger, as reviewed here, but can also be light illumination, temperature jump, or chemical induction. Many protein functions can be addressed by these means and the mechanism of action of these molecular machines may be resolved down to the level of a single bond.
The sensitivity may not be sufficient for functional difference spectroscopy on any protein with SEIDAS, however. As the enhancement factor for SEIRA is modest compared with that for SERS, optimization of the enhancement by proper design of the metal surface is crucial. Although enhancement factors are usually in the range 10–100, factors as high as 1000 have sometimes been reported [16]. To obtain the latter enhancement reproducibly, however, development of homogeneous island metal film preparation under strict topological control will be mandatory.
References
- 1.Fleischmann M, Hendra PJ, McQuilla AJ (1974) Chem Phys Lett 26:163–166 [DOI]
- 2.Jeanmaire DL, Van Duyne RP (1977) J Electroanal Chem 84:1–20 [DOI]
- 3.Hartstein A, Kirtley JR, Tsang JC (1980) Phys Rev Lett 45:201–204 [DOI]
- 4.Aroca RF, Ross DJ, Domingo C (2004) Appl Spectrosc 58:324A–338A [DOI] [PubMed]
- 5.Osawa M (1997) Bull Chem Soc Jpn 70:2861–2880 [DOI]
- 6.Osawa M (2001) Near-Field Optics and Surface Plasmon Polaritons 81:163–187
- 7.Willner I, Katz E (2000) Angew Chem Int Ed 39:1180–1218 [DOI] [PubMed]
- 8.Garczarek F, Gerwert K (2006) Nature 439:109–112 [DOI] [PubMed]
- 9.Hellwig P, Mogi T, Tomson FL, Gennis RB, Iwata J, Miyoshi H, Mäntele W (1999) Biochemistry 38:14683–14689 [DOI] [PubMed]
- 10.Rödig C, Chizhov I, Weidlich O, Siebert F (1999) Biophys J 76:2687–2701 [DOI] [PMC free article] [PubMed]
- 11.Nishikawa Y, Nagasawa T, Fujiwara K, Osawa M (1993) Vib Spectrosc 6:43–53 [DOI]
- 12.Haynes CL, McFarland AD, Smith MT, Hulteen JC, Van Duyne RP (2002) J Phys Chem B 106:1898–1902 [DOI]
- 13.Haynes CL, McFarland AD, Van Duyne RP, Godwin HA (2005) J Chem Educ 82:768A
- 14.Ataka K, Heberle J (2004) J Am Chem Soc 126:9445–9457 [DOI] [PubMed]
- 15.Berna A, Delgado JM, Orts JM, Rodes A, Feliu JM (2006) Langmuir 22:7192–7202 [DOI] [PubMed]
- 16.Miyake H, Ye S, Osawa M (2002) Electrochem Commun 4:973–977 [DOI]
- 17.Rodes A, Orts JM, Perez JM, Feliu JM, Aldaz A (2003) Electrochem Commun 5:56–60 [DOI]
- 18.Kamnev AA, Dykman LA, Tarantilis PA, Polissiou MG (2002) Biosci Rep 22:541–547 [DOI] [PubMed]
- 19.Seelenbinder JA, Brown CW, Pivarnik P, Rand AG (1999) Anal Chem 71:1963–1966 [DOI] [PubMed]
- 20.Osawa M, Ataka K, Yoshii K, Nishikawa Y (1993) Appl Spectrosc 47:1497–1502 [DOI]
- 21.Nishikawa Y, Fujiwara K, Ataka K, Osawa M (1993) Anal Chem 65:556–562 [DOI]
- 22.Nishikawa Y, Fujiwara K, Osawa M, Takamura K (1993) Anal Sci 9:811–815
- 23.Moss D, Nabedryk E, Breton J, Mäntele W (1990) Eur J Biochem 187:565–572 [DOI] [PubMed]
- 24.Ataka K, Giess F, Knoll W, Naumann R, Haber-Pohlmeier S, Richter B, Heberle J (2004) J Am Chem Soc 126:16199–16206 [DOI] [PubMed]
- 25.Ataka K, Richter B, Heberle J (2006) J Phys Chem B 110:9339–9347 [DOI] [PubMed]
- 26.Aroca R, Bujalski R (1999) Vib Spectrosc 19:11–21 [DOI]
- 27.Ataka K, Osawa M (1998) Langmuir 14:951–959 [DOI]
- 28.Pronkin S, Wandlowski T (2003) J Electroanal Chem 550:131–147 [DOI]
- 29.Sato Y, Noda H, Mizutani F, Yamakata A, Osawa M (2004) Anal Chem 76:5564–5569 [DOI] [PubMed]
- 30.Chekhun VF, Solyanik GI, Kulik GI, Tryndiak VP, Todor IN, Dovbeshko GI, Repnytska OP (2002) J Exp Clin Cancer Res 21:599–607 [PubMed]
- 31.Dovbeshko GI, Chegel VI, Gridina NY, Repnytska OP, Shirshov YM, Tryndiak VP, Todor IM, Solyanik GI (2002) Biopolymers 67:470–486 [DOI] [PubMed]
- 32.Brown CW, Li Y, Seelenbinder JA, Pivarnik P, Rand AG, Letcher SV, Gregory OJ, Platek MJ (1998) Anal Chem 70:2991–2996 [DOI] [PubMed]
- 33.Fedurco M (2000) Coord Chem Rev 209:263–331 [DOI]
- 34.Mayer D, Ataka K, Heberle J, Offenhäusser A (2005) Langmuir 21:8580–8583 [DOI] [PubMed]
- 35.Friedrich MG, Giess F, Naumann R, Knoll W, Ataka K, Heberle J, Hrabakova J, Murgida DH, Hildebrandt P (2004) Chem Commun 2376–2377 [DOI] [PubMed]
- 36.Hrabakova J, Ataka K, Heberle J, Hildebrandt P, Murgida DH (2006) Phys Chem Chem Phys 8:759–766 [DOI] [PubMed]
- 37.Zhen YJ, Hoganson CW, Babcock GT, Ferguson-Miller S (1999) J Biol Chem 274:38032–38041 [DOI] [PubMed]
- 38.Badura A, Esper B, Ataka K, Grunwald C, Wöll C, Kuhlmann J, Heberle J, Rögner M (2006) Photochem Photobiol [DOI] [PubMed]