

Abstract
Parkinson’s disease (PD) is associated with loss of total glutathione (GSH) which may contribute to progressive cell death. Peripheral GSH administration has been used clinically with reported benefits. Despite this, there is little specific information to characterize its cellular uptake or clearance, brain elevation with peripheral delivery or neuroprotective efficacy in PD models. The current study was carried out to provide this information using in vitro and in vivo approaches. In rat mesencephalic culture, the monoethyl-ester of GSH (GEE), but not GSH (1–10mM, 24hr) produced a dose-dependent elevation in GSH. The half-life for clearance was 10.14hr and was not different in cells depleted of GSH prior to loading. Elevation of GSH with GEE protected neurons from oxidative stress with H2O2 or metabolic stress with the complex I and II inhibitors, MPP+ and malonate, respectively. To determine if peripheral administration of GEE could elevate brain GSH levels, rats were administered 0.1–50mg/kg/d GEE via osmotic minipump either subcutaneously (sc) or via a cannula placed into the left cerebral ventricle (icv) for 28d. Only central delivery of GEE resulted in significant elevations of brain GSH. Elevation of brain GSH by icv infusion of GEE was examined for its neuroprotective effects against chronic central delivery of MPP+. Infusion of 0.142mg/kg/d MPP+ for 28d caused a selective ipsilateral loss of striatal dopamine. Co-infusion of MPP+ with 10mg/kg/d GEE significantly protected against striatal dopamine loss. These findings show that the ethyl ester of GSH but not GSH per se can elevate intracellular GSH, that brain elevation of GSH requires central delivery of the ethyl-ester and that this elevation provides neuroprotection against oxidative stress or chronic mitochondrial impairment.
Keywords: oxidative stress, glutathione ethyl ester, neurodegeneration, neuroprotection
The underlying etiology of sporadic Parkinson’s disease (PD) remains elusive, although much evidence implicates oxidative stress in the process. The substantia nigra (SN) from autopsied brain material has been shown to contain increased levels of protein carbonyls (Alam et al. 1997), malondialdehyde or 4-hydroxynonenol (Dexter et al. 1994a; Yoritaka et al. 1996) and 8-hydroxy-2-deoxyguanosine (Sanchez-Ramos et al. 1994), indicative of oxidative damage to proteins, lipids and DNA, respectively. One of the earliest biochemical derangements observed in the SN of patients with PD is loss of total glutathione levels (Perry & Yong 1986; Jenner et al. 1992; Sian et al. 1994), without a corresponding increase in oxidized glutathione levels (Sian et al. 1994). This decrease is also observed in incidental Lewy Body disease a condition thought to presage PD (Jenner et al. 1992; Dexter et al. 1994b). Glutathione is a major antioxidant in the cell and logically it would follow that the loss of intracellular GSH would contribute to the increase in free radical damage to cellular constituents. Thus, supplementation with antioxidants should be neuroprotective. Clinical studies, with vitamins E or C, however, have not supported this proposal (Fahn 1992; Shoulson 1998). More recent studies with co-enzyme Q10, have shown more promise (Shults et al. 2002), but it is unclear if protective benefits derive from its antioxidant properties or from other factors (Echtay et al. 2000; Echtay et al. 2002; Papucci et al. 2003). In addition to direct removal of reactive oxygen species by GSH, a number of associated enzymes confer varied roles for GSH in the cell. Glutathione peroxidase, reductase, transferase(s) and glutaredoxin all utilize GSH in reactions that remove peroxide, as well as potential endogenous and exogenous toxins, control the redox state of the cell and regulate protein function through thiolation and dethiolation. This network of enzymes using GSH as substrate have been implicated in DNA synthesis and repair, protein synthesis, amino acid transport, and enzyme activation or inactivation (Lomaestro & Malone 1995). GSH may also act as a redox modifier of some ionotropic currents (Ogita et al. 1995). The multifaceted functions served by GSH raise the possibility that replacement therapy with an antioxidant other than GSH may not carry out all of the important functions of GSH per se. Thus, elevation of brain levels of GSH may be a more efficacious approach to neuroprotection in diseases such as PD.
One trial using GSH in patients with PD reported clinical improvement (Sechi et al. 1996), but the small number of patients and lack of controls limited the usefulness of the investigation. Anecdotal reports of improvement of rigidity, coordination, speech and depression with intravenous administration of GSH can also be found from non-journal reports (www.1whey2health.com/parkinsons_glutathione.htm). A short term, double blinded clinical trial of intravenous (i.v.) glutathione in PD patients is currently being conducted at the University of South Florida and results are expected in 2006 (see Official Journal of the National Parkinson Foundation, vol. XVI, Fall 2005). Despite the use of peripheral administration of GSH clinically for PD, nothing is known regarding the ability of peripheral administration of GSH to elevate brain levels of GSH or the time for clearance of loaded GSH from cells. In addition, while a number of studies in vitro have indicated that elevation of intracellular GSH can protect against oxidative stress, little is known regarding its efficacy in vivo in chronic models of PD.
The present work was, therefore, carried out to determine if GSH or its ethyl ester crosses the plasma membrane of cells or the blood brain barrier to elevate intracellular or brain levels of GSH, the half life for clearance of loaded GSH from cells and the ability of elevated intracellular GSH to protect against oxidative stress or mitochondrial dysfunction in vitro or in vivo.
Material and Methods
Mesencephalic Culture and Treatment Conditions
Embryonic day 15 Sprague Dawley rats (Charles River and Co.) were used for dissection and culturing of ventral mesencephalon as routinely done in the laboratory (Zeevalk & Bernard 2005). Briefly, dissociated cells were plated at 2 x 105 cells/cm2 onto polyornithine coated wells in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% heat inactivated horse and fetal bovine serums (DMEM/serum) and incubated at 37C with 5% CO2. GEE (1–10 mM) or reduced glutathione (GSH) were added to cultures on d8 in vitro. To inhibit glutathione synthesis, buthionine sulfoximine (BSO, 1μM), was added to some cultures on d7 in vitro. Determinations for intracellular GSH content were carried out immediately following loading with GEE or GSH or in some studies 1–48hr after loading. For toxicity studies, cultures were exposed to either 75 μM H2O2 for 6 hr, 0.5μM MPP+ for 24 hr or 50 mM malonate for 24 hr in the presence or absence of GEE. GEE, when present, was given 24 hr before toxin treatment. At the end of exposure, the cultures were returned to conditioned medium. and reincubated for 72 hr prior to assessment of toxicity by either high affinity uptake or counts of TH immunoreactive cells as described below. Conditioned medium was obtained from untreated cultures grown in DMEM/serum for 7–9d.
In Vivo Studies: Pump Implantation and Cannula Placement
Sprague Dawley rats (300gm, Harlan; Indianapolis, IN) were housed in pairs and maintained on a 12 hr light-dark cycle with food and water available ad libitum. Experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were anesthetized (ketamine 50 mg/kg; xylazine 2.5 mg/kg; acetyl promazine 0.625 mg/kg; i.m.) prior to pump and cannula implantation. An Alzet osmotic minipump (Cupertino, CA; model 2ML4) was implanted subcutaneously on the back. For animals receiving infusions into the left lateral ventricle, cannula placement was made stereotaxically following the coordinates of Paxinos and Watson (1986): anterior +8.7, lateral left +1.4, depth −3.5. GEE (0.1–50 mg/kg/d) plus or minus MPP+ (0.142 mg/kg/d) was delivered at a rate of 2.5 μl/hr for 28d. At the end of the exposure, animals were sacrificed by decapitation and 2 min prior to euthanasia, the animals received a tail vein injection of 3-mercaptopropionate (160mg/kg) to inhibit glutamic acid decarboxylase activity (Korf & Venema 1983). The brains were rapidly removed and dissected. Wet weights were determined and the tissues maintained at −80C until analyzed for neurochemistry.
Toxicity: dopamine and GABA high affinity transport
The simultaneous measurement of transport of 3H-dopamine and 14C-GABA is as previously described (Zeevalk et al. 2000) with minor modifications. Cultures were incubated with 20 nM 3H-dopamine plus 5 μM 14C-GABA in the presence of 1 mM ascorbate, 100 μM pargyline (to prevent dopamine oxidation and metabolism), 10 μM aminoxyacetic acid ( to preserve GABA) and 1 mM ß-alanine (to block glial uptake of GABA) for 15 min at 37C. Intracellular radioactivity was extracted with 95% ethanol and quantitated by scintillation counting. Uptake at 4C was subtracted from other determinations to obtain energy dependent uptake.
Tyrosine Hydroxylase Immunocytochemistry and Cell Counts
Immunocytochemistry for TH was carried out as previously described (Zeevalk & Bernard 2005). Cultures were immunostained with mouse monoclonal anti-TH (1:6000, Diasorin, Inc.). Primary antibody was detected with the Avidin Biotin Peroxidase system (Vector Labs). Cells were viewed and counted at 100x magnification. For counting, the bottom of each well was marked at the center and in each of 4 quadrants. Marks were located and immunopositive cells to the right and left of the mark were counted in a 1 mm2 reticle by an observer blind to the treatment condition. Each condition was run in duplicate. Counts from 40, 1 mm2 fields per condition (20 fields/well x 2 wells x 2 replicate experiments) were averaged and reported as mean counts per square mm ± S.D.
HPLC Determination of Glutathione, Glutamate and GABA
Amino acids and total GSH were measured by HPLC as previously published (Zeevalk et al. 2000). Cells were extracted with 0.2N perchloric acid (PCA). For tissue samples, tissue was homogenized in 0.2N PCA at a wet weight of 10mg/ml. Samples were frozen at −80C until used for measurement. The extract was neutralized to pH 5.8 with 2M potassium carbonate, reacted with σ-phthalaldehyde and separated by reverse phase HPLC with fluorescent detection. Quantitation was obtained by comparison to known standards.
HPLC Determination of Dopamine and 5-HT
Tissue samples were homogenized in 0.2N PCA on the day of measurement. Samples were centrifuged and the supernatant used for detection of monoamines with HPLC using electrochemical detection as described elsewhere (Yazdani et al. 2006).
Statistics
Most data were analyzed using a one-way analysis of variance (ANOVA, Prism 3.0) with the different treatments as the independent factor. Comparisons between means were tested by the Tukey-Kramer Multiple Comparisons post-hoc test. In the case of left versus right differences from the same animal, data were analyzed using a two-tailed paired Students t test. In all cases, significance was indicated by p< 0.05.
Results
Elevation of glutathione with glutathione ethyl ester in mesencephalic culture
Preloading mesencephalic cultures for 24 hr with glutathione ethyl ester produced a dose-dependent increase in intracellular levels of GSH (Fig. 1A). Basal levels of GSH in the cultures were 1.75 ± 0.11 μmol/100mg protein ± sem. GSH levels were elevated 66, 144 and 191% of basal with 2.5, 5 and 10 mM GEE, respectively. In contrast, preloading for 24 hr with 1–10 mM reduced-GSH did not elevate intracellular glutathione levels (Fig. 1B). To better replicate conditions in Parkinson’s disease brain with regards to lowered GSH levels which could alter loading characteristics, cultures were partially depleted of GSH by a 24hr pretreatment with buthionine sulfoximine (BSO, 1μM) prior to GEE loading for 24 hr in the continued presence of BSO. Treatment of cultures with 1 μM BSO in the absence of GEE reduced intracellular GSH levels from a basal level of 1.62 ± 0.11 to 0.72 ± 0.10 μmol/100mg protein ± sem, n= 7–8 determinations per condition. This is a decrease of approximately 55% and similar to the 40–50% loss observed in PD (Jenner et al. 1992). When compared with GSH depleted cells, GEE loading significantly elevated GSH levels with ≥ 1 mM GEE although absolute intracellular levels of GSH were always less than those observed in cultures not treated with BSO at comparable GEE concentrations (Fig. 1C and compare with Fig. 1A).
Figure 1. Glutathione monoethyl ester but not glutathione elevates intracellular glutathione levels.
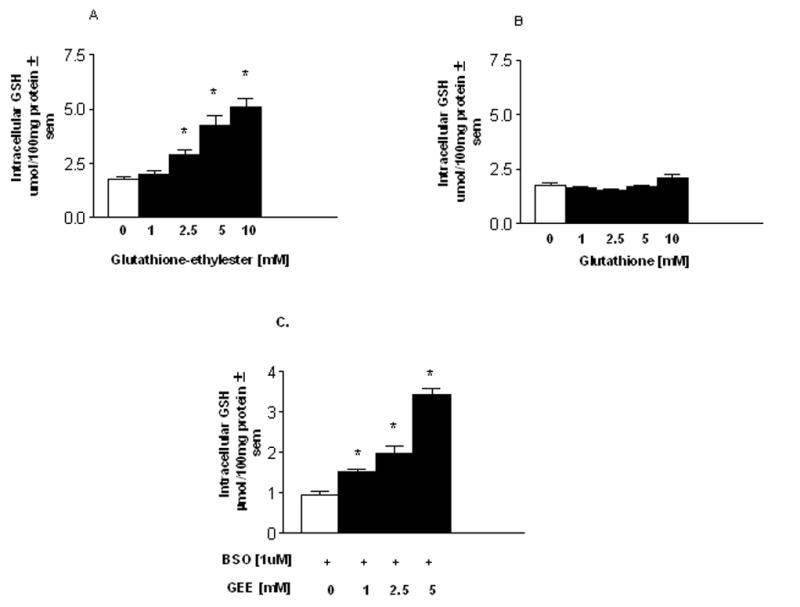
Mesencephalic cultures were preloaded for 24hr with either the monoethyl ester of glutathione (A) or with reduced glutathione (B).(C) Some cultures had glutathione partially depleted prior to loading by treatment for 24hr with buthionine sulfoximine (BSO, 1μM). GEE loading was carried out in the presence or absence of BSO. Following loading, cultures were washed, intracellular content extracted and glutathione quantified by HPLC. The n is from 4–5 separate experiments run in duplicate. *Different from zero GEE addition.
To determine whether there was any toxicity associated with GEE loading, cultures were loaded with 1–10 mM GEE for 24hr, switched to GEE free medium and allowed to recover for 72hr. Toxicity was determined by a functional assay for the high affinity transport of dopamine and GABA which allows assessment of toxicity in both these mesencephalic populations. Since toxicity is determined 3d after removal of GEE, the results reflect irreversible damage and are not a pharmacological consequence of treatment. GEE at 10 mM significantly decreased dopamine uptake by 38% indicating toxicity to this population (Fig. 2A). There was a trend towards a decrease in GABA uptake, but this was not statistically significant (Fig. 2B). Loading with 10 mM reduced-GSH did not produce toxicity to dopamine or GABA mesencephalic populations suggesting that the ethyl ester moiety was responsible for toxicity (data not shown).
Figure 2. Toxicity associated with glutathione monoethyl ester loading.
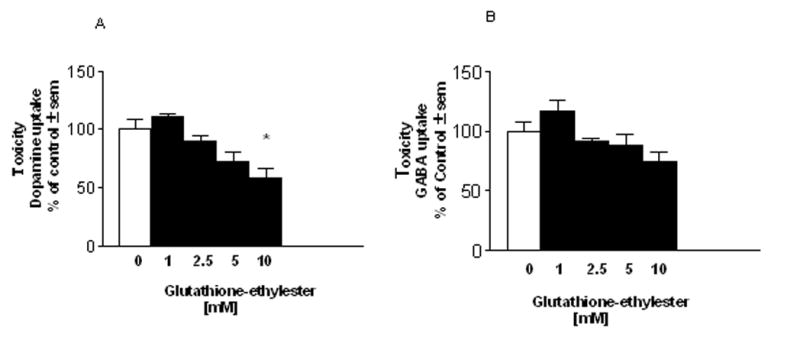
Cultures were exposed to glutathione monoethyl ester (GEE) for 24hr then allowed to recover for 3d prior to measuring toxicity by a functional assay of high affinity uptake for (A) 3H-dopamine or (B) 14C-GABA. Exposure to similar concentrations of GSH was not toxic. The n is from 3 experiments run in duplicate. *Different from zero GEE.
Half-life For Clearance Of Glutathione From Preloaded Glutathione Ethyl Ester
Cells closely regulate intracellular glutathione levels. We, therefore, wanted to determine how rapidly cells cleared elevated intracellular GSH when derived from exogenous sources. This would be important to know if trying to augment GSH levels in cells as a neuroprotective strategy. Studies were carried out to determine the half-life for clearance of GSH from preloaded GEE under normal and GSH-depleted conditions. Cultures were treated with 5 mM GEE or 5 mM GEE plus 1 μM BSO for 24hr, switched to fresh medium and intracellular levels of GSH were monitored immediately following loading and at 8, 16, 24 and 48hr post loading. The half-life was determined from a one-phase exponential decay curve where t1/2 = ln2/decay rate. The half-life for clearance of loaded GEE was 10.14 and 13.86 in normal and GSH-depleted cultures, respectively and was not statistically different from one another (Fig. 3).
Figure 3. Half-life for clearance of loaded glutathione.
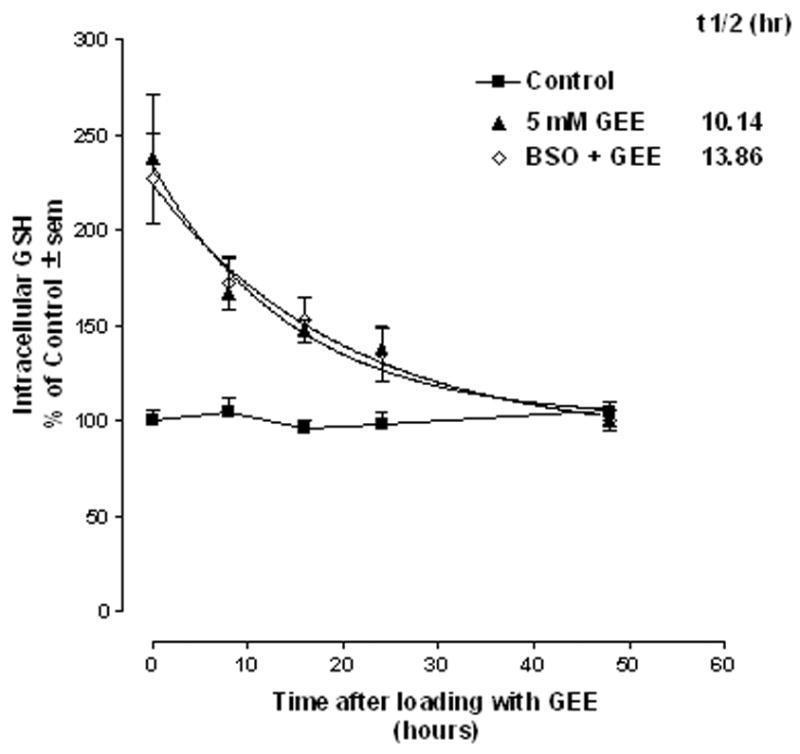
Mesencephalic cultures were treated with 5 mM glutathione monoethyl ester (GEE) plus or minus 1μM buthionine sulfoximine (BSO) for 24hr, switched to fresh medium plus or minus BSO and intracellular levels of GSH were monitored immediately following loading (0 time) and at 8,16,24, and 48hr post loading. The half-life was determined from a one phase exponential decay curve. The n is from 4 experiments run in duplicate.
Glutathione Ethyl Ester Preloading Protects Against Mitochondrial Inhibition And Oxidative Stress In Vitro
In order to determine if preloading cells with glutathione prior to exposure to an oxidative challenge with H2O2 (75 μM, 6 hr) or mitochondrial impairment with the complex II inhibitor malonate (50 mM, 24 hr) or complex I inhibitor MPP+ (0.5μM, 24 hr) protected mesencephalic neurons, cultures were exposed to GEE for 24 hr prior to the toxin challenge. After 3d recovery, toxicity was assessed by measurement of high affinity uptake for dopamine and GABA. GEE provided significant protection against H2O2 for both the dopamine and GABA populations with 2.5 and 5 mM GEE preloading (Fig. 4A). To provide further evidence that oxidative insult resulted in cell loss that was protected by GEE preloading, some cultures were examined for tyrosine hydroxylase (TH)-positive cell counts 3d after exposure to H2O2. As shown in Fig. 4B, 75 μM H2O2 produced a significant loss of TH-positive cells and this was fully protected by GEE preloading. Protection against malonate (Fig. 5A) and MPP+ (Fig. 5B) in the dopamine population was also observed with 1 mM GEE. Significant toxicity in the GABA population was not observed with either mitochondrial inhibitor.
Figure 4. Neuroprotection with glutathione loading against oxidative stress in vitro.
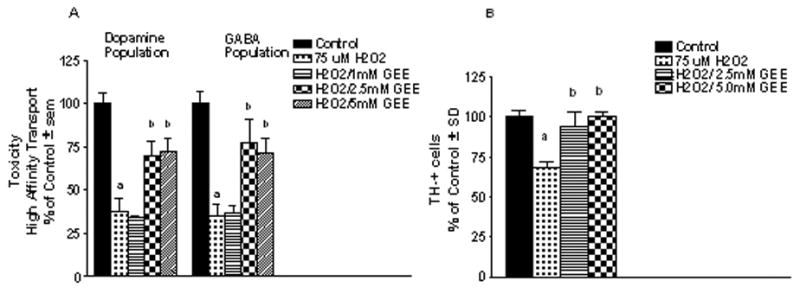
Cultures were exposed to 1–5 mM GEE for 24hr prior to exposure to 75μM H2O2 for 6hr. Cells were allowed to recover for 3d and toxicity assessed by (A) a functional assay for high affinity uptake for radiolabeled dopamine and GABA and (B) counts of tyrosine hydroxylase (TH) positive cells. The n is from 3–4 experiments per condition. aDifferent from control; bdifferent from H2O2 alone.
Figure 5. Neuroprotection with glutathione loading against mitochondrial inhibiton in vitro.
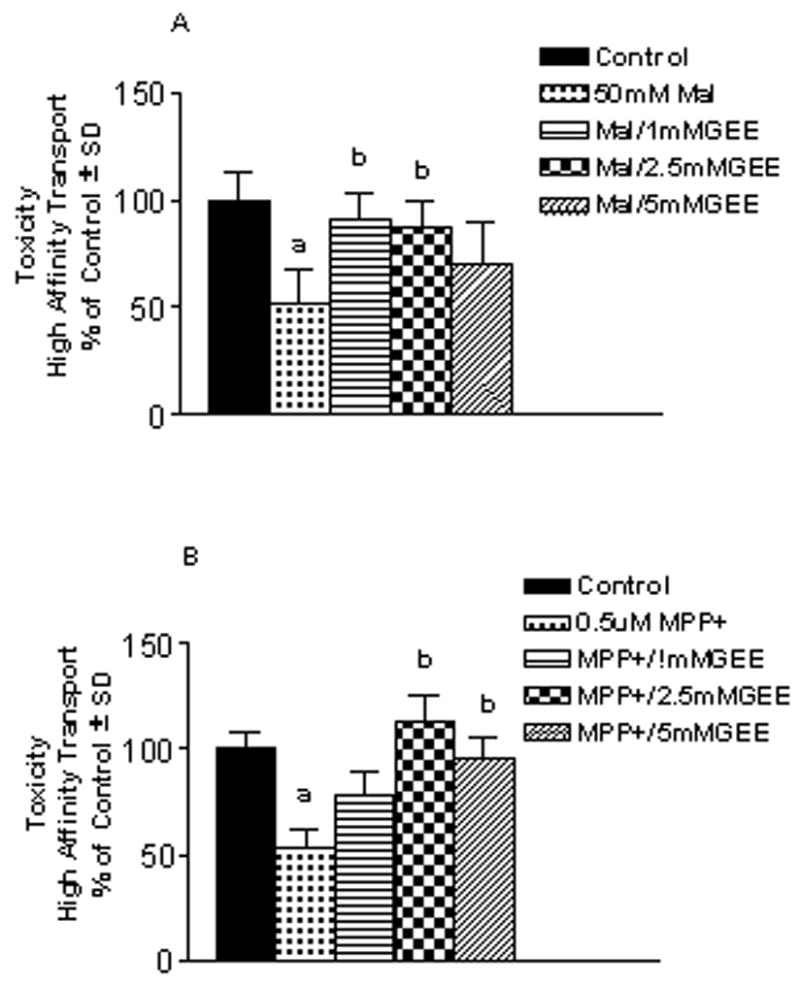
Mesencephalic cultures were preloaded for 24hr with 1–5 mM glutathione monoethyl ester (GEE) prior to an additional 24 hr incubation with (A) the complex II inhibitor malonate (50 mM) or (B) the complex I inhibitor MPP+ (0.5μM) . Cells recovered for 3d before assessing toxicity by measurement of the high affinity transport of dopamine. Toxicity was only observed in the mesencephalic dopamine population. The n is from 3–4 experiments for (A) and 2 experiments for (B) run in duplicate per condition. aDifferent from control; bdifferent from mitochondrial inhibitor alone.
Elevation Of Brain Glutathione In Vivo
GSH is only modestly taken up across the blood brain barrier (BBB) (Cornford et al. 1978; Kannan et al. 1990). Little is known regarding the transport of GEE and if peripherally administered GEE can elevate brain tissue levels of GSH. To study this, GEE was delivered either centrally into the left cerebral ventricle or peripherially via subcutaneous administration for 28d in Sprague Dawley rats using an Alzet osmotic minipump implanted under the skin at the back. Prior to initiation of these studies, the stability of GEE was tested by incubating a 50mg/ml solution of GEE in the dark at 37C for 0–28d and monitoring for the appearance of breakdown products by HPLC. No peaks for GSH, cysteine, glutamate, glycine or any dipeptides were observed at any time points indicating that GEE remains stable over the course of the experiment. MPP+ stability was demonstrated previously (Yazdani et al. 2006). Intraventricular delivery of GEE significantly elevated brain levels of GSH on the side ipsilateral to infusion (Figs. 6A–C). GEE at ≥ 5 mg/kg/d significantly increased striatal levels of GEE, whereas cortical levels were elevated with 10 mg/kg/d. There was a trend towards increased GEE in hippocampus, but results were not statistically significant. The elevation of GSH was restricted to the side of infusion. In contrast, peripheral administration of 0.2 – 50 mg/kg/d GEE subcutaneously delivered by osmotic pump for 28d did not result in elevation of GSH levels in any brain region examined (Figs. 7A–C).
Figure 6. Glutathione levels following icv infusion of GEE.
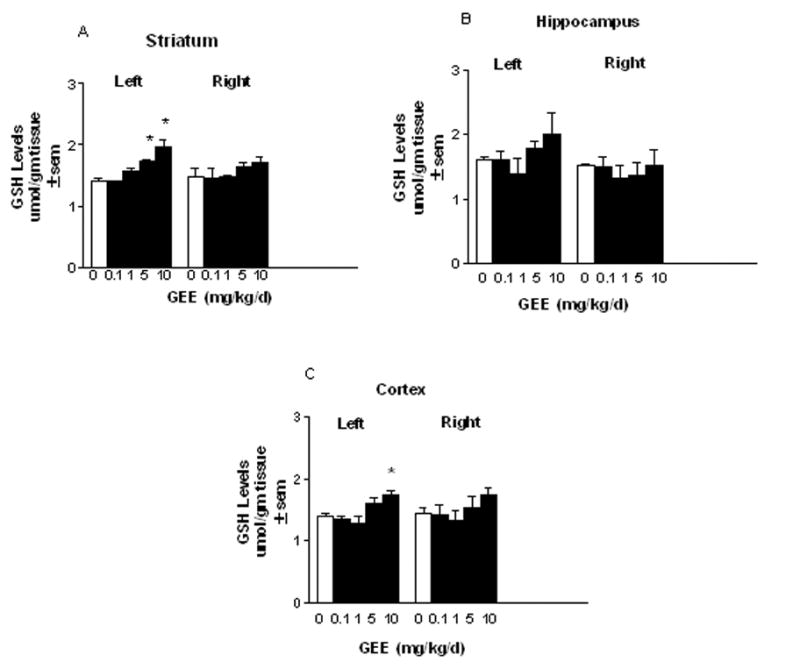
Sprague Dawley rats were implanted with an Alzet osmotic minipump under the skin at the back. A cannula was stereotaxically placed into the left cerebral ventricle (icv). Glutathione monoethyl ester (GEE) was dissolved in saline and delivered by the pump at 0.1–10 mg/kg/d. Animals were examined at the end of 28d and the indicated brain areas removed for HPLC determination of GSH. The n is from 4–6 animals per group. *Different from saline treated controls.
Figure 7. Glutathione levels following subcutaneous infusion of GEE.
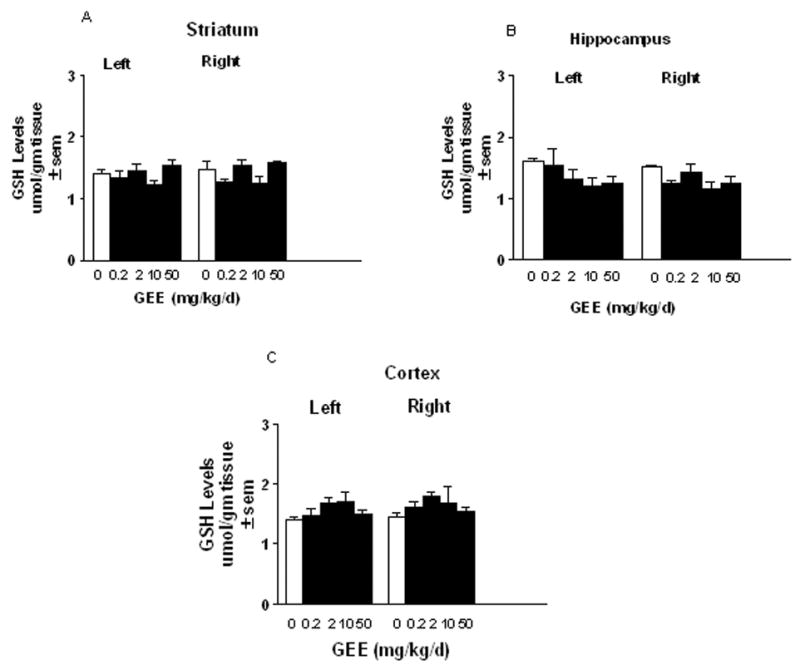
Rats were implanted with osmotic minipumps under the skin at the back. Pumps were loaded with 0.2–50 mg/kg/d glutathione monoethyl ester (GEE) for delivery for 28d. At the end of pump delivery, the brains were removed and left and right brain regions were analyzed for total GSH by HPLC. The n is from 4–6 animals per group.
Glutathione Ethyl Ester Provides Protection Against Dopamine Loss Due To Chronic Mitochondrial Impairment In Vivo
Mitochondrial deficits are found in the sporadic PD population and are thought to be a contributing factor to dopamine cell degeneration. While acute models of mitochondrial impairment have been used in many past studies to model PD, chronic mitochondrial perturbation is thought to better replicate the ongoing pathophysiology in the disease. To this end, our laboratory has developed a chronic model of mitochondrial impairment in rat using central delivery of MPP+ (Yazdani et al. 2006). Chronic delivery of 0.142 mg/kg/d MPP+ for 28d into the left lateral ventricle caused a selective loss of striatal dopamine with no significant loss of 5-HT, GABA or glutamate (Fig. 8A). As shown previously (Yazdani et al. 2006), this loss is only observed ipsilateral to infusion and results in the progressive degeneration of substantia nigra neurons. Striatal dopamine levels were 10.45 ± 0.31 and 10.77 ± 0.46 μg/gm tissue ± sem in left and right sides, respectively in saline-I treated controls and 3.82 ± 0.49 and 11.05 ± 0.32 μg/gm tissue ± sem in left and right striata, respectively from 0.142 mg/kg/d MPP+ treated animals.
Figure 8. Chronic MPP+ produces selective loss of striatal dopamine that is partially protected with co-infusion of GEE Sprague.
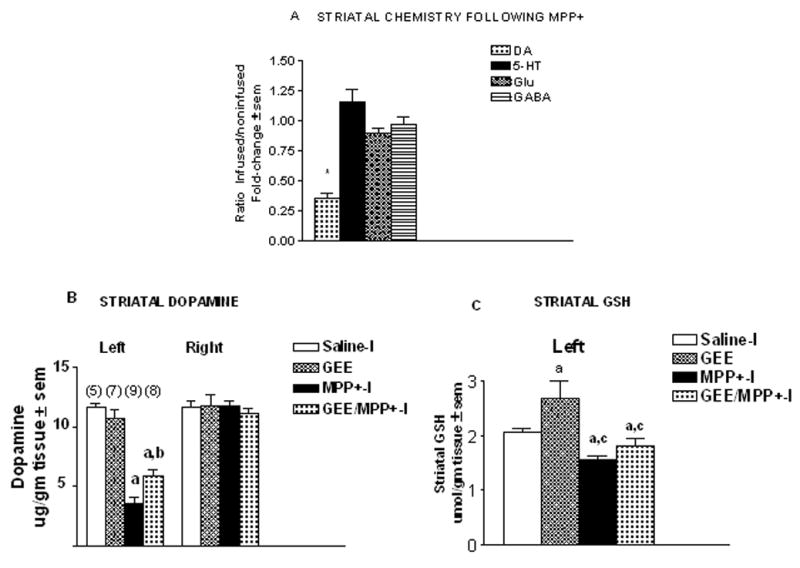
Dawley rats were administered either saline-I (0.108 mg/kg/d), glutathione monoethyl ester (GEE, 10 mg/kg/d), MPP+-I (0.142 mg/kg/d, free base), or GEE plus MPP+-I for 28d into the left cerebral ventricle via an osmotic minipump. (A) Selectivity of damage was assessed by HPLC measurement of left and right striatal levels of dopamine (DA), 5-HT, glutamate (Glu) and GABA 28d following MPP+-I infusion. Only DA levels were found to be reduced. The n is from 9 animals. *Different from non-infused side and saline treated control. (B) Striatal DA on the contralateral (right) sides showed no differences with any of the treatments. MPP+-I infusion reduced DA on the ipsilateral (left) side. GEE co-infusion with MPP+ provided partial protection. The n for each group is given in parenthesis. aDifferent from saline control; bdifferent from MPP+ alone. (C) Striatal glutathione (GSH) levels were elevated following GEE infusion, but were significantly lower with MPP+ or MPP+ plus GEE. aDifferent from saline control; cdifferent from GEE alone.
To determine if GEE could protect against striatal dopamine loss caused by chronic central MPP+ infusion, rats were given either saline-I (0.108 mg/kg/d), GEE (10 mg/kg/d), MPP+ (0.142 mg/kg/d) or GEE plus MPP+ for 28d into the left cerebral ventricle (icv) via osmotic pump (Figs. 8B & C). After 28d, MPP+ reduced striatal dopamine levels to 30% of saline-I treated controls ipsilateral to infusion. Concurrent administration of GEE with MPP+ provided partial protection, with dopamine levels 51% of control and 68% higher than with MPP+ alone. Dopamine levels on the contralateral side were unaffected (Fig. 8B). GSH levels were decreased by 25% on the left side with MPP+ (Fig. 8C). GEE alone enhanced GSH levels by 30% on the ipsilateral side. In animals treated concurrently with GEE and MPP+, GSH levels were 88% of controls. GSH levels in this group were not significantly different from either saline or MPP+ suggesting that GEE may have partially preserved GSH levels in MPP+ treated animals.
Discussion
The present work had 2 objectives. The first was to characterize the ability of GSH or its ethyl ester to elevate intracellular GSH, measure the half-life for clearance of preloaded GSH and determine the ability of peripherally delivered GSH to elevate brain levels. The second objective was to assess whether elevation of intracellular GSH in vitro and in vivo would protect dopamine neurons from damage due to oxidative stress or mitochondrial impairment. These issues are important to consider as GSH is currently in use by some clinicians as a therapeutic treatment for PD and there is little specific information on its uptake or clearance by neurons, although there is evidence in animal models to support a neuroprotective role.
With respect to the first objective, our findings show that the ethyl ester of GSH, but not GSH per se significantly elevated intracellular GSH in mesencephalic cultures. In a variety of non-neuronal cell types, GSH is not elevated by exogenous addition (Dolphin et al. 1989) and our work in mesencephalic mixed cultures indicates that this is also true for neurons and glia. In contrast, the monoethyl ester of GSH readily increases intracellular GSH levels several fold. Some toxicity was noted when loading cultures with GEE. Toxicity was not found with equivalent amounts of GSH suggesting that toxicity was related to the ethyl ester moiety. Toxicity with the ethyl esters of GSH has been noted previously (Anderson et al. 1985; Scaduto et al. 1988; Tsan et al. 1989; Gale et al. 1990) and may be due to the ethanol formed as GSH is released. One report attributes toxicity to impurities in the ester compound rather than to ethanol formation (Meister 1991). The finding of toxicity with the ethyl ester of GSH indicates caution with its use in vivo.
Despite the rise in intracellular GSH levels with GEE, the issue of BBB permeability is also a potential constraint to elevation of brain levels of GSH in vivo. There is a carrier mediated transport of GSH across the BBB (Kannan et al. 1990), but it is found to be of low capacity (Cornford et al. 1978). Indeed, brain extraction of radiolabeled GSH after intracarotid injection was only 0.5% (Cornford et al. 1978). The issue of BBB permeability to GSH is somewhat moot given that extracellular GSH is incapable of elevating intracellular levels. The ability of peripherally administered GEE to elevate brain levels of GSH has not been examined previously. Findings reported here indicate that subcutaneous administration of up to 50 mg/kg/d for 28d is not capable of increasing brain tissue levels of GSH. Only central delivery of GEE into the lateral ventricle was found to significantly elevate GSH levels. Elevation of GSH was only observed on the infused side and may be due to the anterior placement of the cannula into the ventricle (Yazdani et al. 2006).
The half-life for clearance of preloaded GSH was 10.14 hr. This has implications for the timing of GSH administration and suggests that daily administration would be required to maintain an elevation in intracellular GSH. The half-life for clearance was not significantly different from cells that had undergone prior depletion of GSH with BSO. Basal levels of GSH in BSO treated cultures were approximately half that of control cultures throughout the experiment. These findings suggest that clearance of exogenously loaded GSH is not regulated by intracellular GSH concentrations unlike regulation of GSH synthesis by feedback inhibition of gamma-glutamycysteine synthetase (Anderson & Lou 1998).
Given our findings that GSH per se does not elevate brain or neuronal intracellular levels, what would account for the reported positive benefits of intravenous GSH in PD patients (Sechi et al. 1996), and see Official Journal of the National Parkinson Foundation vol. XVI issue 4, Fall 2005. Serum contains the GSH catabolizing enzyme, gamma glutamyltranspeptidase (Magnani et al. 1984). This would suggest that GSH is metabolized to cysteinylglycine and glutamate in the bloodstream. Cysteineylglycine may serve as a substrate for GSH biosynthesis (Anderson et al. 1985; Boyd-Kimball et al. 2005), but whether it crosses the BBB is unknown. The restriction of the BBB to small peptides, however, makes the uptake of cysteinylglycine unlikely (Cornford et al. 1978). Cysteinylglycine could be further metabolized by dipeptidases to cysteine and glycine. Cysteine is rate limiting in GSH biosynthesis (Wang & Ballatori 1998). Cysteine may be transported across the BBB by the L-carrier system (Wade & Brady 1981). A caveat to this is that elevation of cysteine in brain could pose a potential problem as cysteine can bind to glutamate receptors to produce an excitotoxicity (Olney et al. 1990; Puka-Sundvall et al. 1995). Additionally, it has been reported that exogenous GSH added to human plasma is rapidly oxidized to glutathione disulfide by the side oxidase activity of gamma glutamyltranspeptidase (Magnani et al. 1984), although it is unclear how this would relate to protection. Assuming that future evidence supports a protective role for peripherally administered GSH in PD, the fate of GSH and the mechanism underlying any benefit remains to be clarified.
A second objective of this study was to determine whether intracellular elevation of GSH would protect dopamine neurons both in vitro and in vivo from oxidative stress or mitochondrial impairment. In vitro, GEE preloading of mesencephalic cultures provided near complete protection of both the mesencephalic dopamine and GABA populations from oxidative stress due to H2O2 exposure. Significant protection was observed with 2.5 and 5mM GEE. Mitochondrial impairment with the complex II inhibitor, malonate, or complex I inhibitor, MPP+ was toxic to dopamine, but not GABA neurons. GEE preloading completely protected dopamine neurons against malonate toxicity and MPP+ toxicity with 1mM GEE and 2.5 mM GEE, respectively. There was less protection with 5 mM GEE preloading and this could be due to toxicity from GEE per se at the higher concentration.
Acute in vivo models of mitochondrial impairment have been used extensively to model PD. Chronic, generalized mitochondrial deficits, however, are found in sporadic PD (Parker et al. 1989; Schapira et al. 1989). Chronic mitochondrial impairment may, therefore, better replicate the disease process and importantly, may better predict clinical efficacy. Our laboratory has been developing a chronic model of central delivery of MPP+ in rats (Yazdani et al. 2006). Using this model, we examined whether co-administration of MPP+ with GEE provided protection. MPP+ delivered into the left lateral ventricle for 28d at 0.142mg/kg/d produced selective striatal loss of dopamine and as we have reported previously, loss of tyrosine hydroxylase positive and Nissl positive neurons in the substantia nigra pars compacta (Yazdani et al. 2006). GEE at 10mg/kg/d, a concentration found to significantly elevate GSH in the striatum provided parital protection from loss of striatal dopamine. Protection in this chronic model of mitochondrial impairment, although significant, was not as robust as that observed in the acute in vitro treatments with an oxidative or mitochondrial insult. This likely reflects the greater challenge of a chronic mitochondrial perturbation. Alternatively, it is possible that the higher levels of intracellular GSH achieved in vitro (Fig. 1) as compared with in vivo, (Fig. 6) provided more complete protection. Consistent with our findings, others have reported protection of dopamine terminals from acute MPTP toxicity with brain GSH elevation using N-acetylcysteine (NAC) (Munoz et al. 2004; Park et al. 2004). NAC, however, can also produce acidification (Gillissen et al. 2006) and damage to non-dopamine neurons (Munoz et al. 2004) and may, therefore, be of mixed benefit for enhancing GSH levels in cells.
Since GSH is a major antioxidant in cells, its depletion very likely contributes to the initial oxidative damage as well as the progressive damage during the latter stages of PD. The striking accumulation of oxidative damage in the SN of PD brain indicates a brain region under free radical attack. Enhancing antioxidant capabilities in the nigra, therefore, constitutes a rationale approach to preventing ongoing damage. These efforts for the most part have met with only modest success. High dose ascorbate plus α-tocopherol supplementation in one clinical trial delayed the need for L-Dopa therapy by 2–4 years in 75% of PD patients (Fahn 1992). Treatment with α-tocopherol alone has been shown to have no effect (Shoulson 1998). There have been some promising results with coenzyme Q10. In PD patients treated for 16 mo with 1200 mg per day of CoQ10, there was a 44% slowing of disability between the CoQ10 group and the placebo group. CoQ10 has known antioxidant properties, but may also enhance mitochondrial function, serve as a cofactor for uncoupling proteins (Echtay et al. 2000) or inhibit the activation of the mitochondrial permeability transition (Echtay et al. 2002). Which of these properties of CoQ10 contributes to protection is at present unclear.
Increasing GSH levels in brain would also be a rationale approach to antioxidant supplementation. This is compelling for 2 reasons. Firstly, there is a known deficit of GSH in the nigra in PD and secondly, GSH serves many different roles in the cell in addition to it being an antioxidant. It is possible that loss of a number of functions of GSH in addition to its antioxidant capabilities contributes to dopamine cell susceptibility. Supplementation with an antioxidant other than GSH may overcome some, but not all of the consequences produced by loss of GSH. Thus, a strong case can be made for specifically enhancing GSH in brain. The best approach to doing this, however is more problematic. GEE, although able to elevate intracellular levels of GSH required central delivery and in vitro demonstrated some toxicity. Cysteine is taken up by the BBB and can be transported into neurons for GSH biosynthesis, but potentially can interact with glutamate receptors to produce excitotoxicity (Olney et al. 1990; Puka-Sundvall et al. 1995). NAC can elevate intracellular GSH levels but, its ability to cross the BBB is controversial (McLellan et al. 1995). In addition, NAC can cause acidification (Gillissen et al. 2006) and is also reported to cause neuronal toxicity (Munoz et al. 2004). Enhancing γ-glutamylcysteine synthetase expression and activity through the use of viral vectors may be one possible approach. The issue of how to best restore GSH levels requires further investigation.
In summary, similar to other cell types, GSH was not able to elevate intracellular levels in neuronal cells, whereas the ethyl ester of GSH resulted in significant increases in GSH concentration. Use of the ethyl ester to elevate GSH, however, was problematic in that it produced toxicity at the higher concentration used, needed to be delivered centrally in order to elevate brain GSH and had a half-life for clearance that would require daily supplementation in order to maintain elevated levels. Elevation of GSH was found to provide significant protection from dopamine cell damage due to oxidative stress or mitochondrial impairment both in vitro and in vivo. Therefore, despite the difficulty in optimizing the best approach to supplementation of GSH levels in neuronal cells, given the multifunctional roles served by GSH, this remains an avenue of significant therapeutic potential.
Acknowledgments
This work was supported by grants from the Michael J. Fox Foundation for Parkinson’s Research and from The National Institutes of Health (NS36157).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alam ZI, Daniel SE, Lees AJ, Marsden CD, Jenner P, Halliwell B. A generalized increase in protein carbonyls in the brain in Parkinson’s disease but not incidental Lewy body disease. Journal of Neurochemistry. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Lou JL. Glutathione therapy: from prodrugs to genes. Semin Liver Dis. 1998;18:415–424. doi: 10.1055/s-2007-1007174. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Powrie F, Puri RN, Meister A. Glutathione monoethyl ester: preparation, uptake by tissues and conversion to glutathione. Arch Biochem & Biophsy. 1985;239:538–548. doi: 10.1016/0003-9861(85)90723-4. [DOI] [PubMed] [Google Scholar]
- Boyd-Kimball D, Sultana R, Abdul HA, Butterfield DA. Gamma-glutamylcysteine ethyl ester-induced up-regulation of glutathione protects neurons against AB(1–42)-mediated oxidative stress and neurotoxicity: implications for Alzheimer’s disease. Journal of Neuroscience Research. 2005;79:700–706. doi: 10.1002/jnr.20394. [DOI] [PubMed] [Google Scholar]
- Cornford EM, Braun LD, Crane PD, Oldendorf WH. Blood-brain barrier restriction of peptides and the low uptake of enkephalins. Endocrinology. 1978;103:1297–1303. doi: 10.1210/endo-103-4-1297. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Holley AE, Flitter WD, Slater TF, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994a;9:92–97. doi: 10.1002/mds.870090115. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Sian J, Rose S, Hindmarsh JG, Mann VM, Cooper JM, Wells FR, Daniel SE, Lees AJ, Schapira AHV, Jenner P, Marsen CD. Indexes of Oxidative Stress and Mitochondrial-Function in Individuals with Incidental Lewy Body Disease. Annals of Neurology. 1994b;35:38–44. doi: 10.1002/ana.410350107. [DOI] [PubMed] [Google Scholar]
- Dolphin D, Poulson R, Avramovic O. Glutathione: Chemical, Biochemical and Medical Aspects. Wiley; New York: 1989. pp. 22–23. [Google Scholar]
- Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand RM. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Winkler E, Klingenberg M. Coenzyme Q is an oligatory cofactor for uncoupling protein function. Nature. 2000;408:609–613. doi: 10.1038/35046114. [DOI] [PubMed] [Google Scholar]
- Fahn S. A pilot trial of high-dose alpha-tocopherol and ascorbate in early Parkinson’s disease. Ann Neurol. 1992;32:S128–132. doi: 10.1002/ana.410320722. [DOI] [PubMed] [Google Scholar]
- Gale GR, Smith AB, Atkins LM, Jones MM, Singh PK, Meredith MJ. Glutathione status, glutathione monoisopropyl ester, and cadmium metabolism in mice. Res Commun Chem Pathol Pharmacol. 1990;70:71–91. [PubMed] [Google Scholar]
- Gillissen A, Joworska M, Orth M, Coffiner M, Maes P, App EM, Cantin AM, Schultze-Werninghaus G. Nacystelyn, a novel lysine salt of N-acetylcysteine, to augment cellular antioxidant defence in vitro. Respiratory Medicine. 2006;91:159–168. doi: 10.1016/s0954-6111(97)90052-4. [DOI] [PubMed] [Google Scholar]
- Jenner P, Dexter DT, Sian J, Schapira AHV, Marsden CD. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental lewy body disease. Ann Neurol. 1992;32:S82–S87. doi: 10.1002/ana.410320714. [DOI] [PubMed] [Google Scholar]
- Kannan R, Kuhlenkamp JF, Jeandidler E, Trinh H, Ookhtens M, Kkaplowitz N. Evidence for carrier-mediated transport of glutathione across the blood brain barrier in the rat. J Clin Invest. 1990;85:2009–2013. doi: 10.1172/JCI114666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf J, Venema K. Amino acids in the substantia nigra of rats with striatal lesions produced by kainic acid. Journal of Neurochemistry. 1983;40:1171–1173. doi: 10.1111/j.1471-4159.1983.tb08109.x. [DOI] [PubMed] [Google Scholar]
- Lomaestro BM, Malone M. Glutathione in health and disease: pharmacotherapeutic issues. Ann Pharmacother. 1995;29:1263–1273. doi: 10.1177/106002809502901213. [DOI] [PubMed] [Google Scholar]
- Magnani M, Novelli G, Palloni R. Human plasma glutathione oxidation in normal and pathological conditions. Clin Physiol Biochem. 1984;2:287–290. [PubMed] [Google Scholar]
- McLellan L, Lewis AD, Hall DJ, Ansell JD, Wolf CR. Uptake and distribution of N-acetylcysteine in mice: tissue specific effects on glutathione concentrations. Carcinogenesis. 1995;16:2099–2166. doi: 10.1093/carcin/16.9.2099. [DOI] [PubMed] [Google Scholar]
- Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol & Therap. 1991;51:155–194. doi: 10.1016/0163-7258(91)90076-x. [DOI] [PubMed] [Google Scholar]
- Munoz AM, Rey P, Soto-Otero R, Guerra MJ, Labandeira-Garcia JL. Systemic administration of N-acetylcysteine protects dopaminergic neurons against 6-hydroxydopamine-induced degeneration. Journal of Neuroscience Research. 2004;76:551–562. doi: 10.1002/jnr.20107. [DOI] [PubMed] [Google Scholar]
- Ogita K, Enomoto R, Nakahara F, Ishitsubo N, Yoneda Y. A possible role of glutathione as an endogenous agonist at the N-methyl-D-aspartate recognition domain in rat brain. J Neurochem. 1995;64:1088–1096. doi: 10.1046/j.1471-4159.1995.64031088.x. [DOI] [PubMed] [Google Scholar]
- Olney JW, Zorumski C, Price MT, Labruyere J. L-cysteine, a bicarbonate-sensitive endogenous excitotoxin. Science. 1990;248:596–599. doi: 10.1126/science.2185543. [DOI] [PubMed] [Google Scholar]
- Papucci L, Schiavone N, Witort E, Donnini M, Lapucci A, Tempestini A, Formigli L, Zecchi-Orlandini S, Oriandini G, Carella G, Brancato R, Capaccioli S. Coenzyme Q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J Biol Chem. 2003;278:28220–28228. doi: 10.1074/jbc.M302297200. [DOI] [PubMed] [Google Scholar]
- Park SW, Kim SH, Park KH, Kim SD, Kim JY, Baek SY, Chung BS, Kang CD. Preventive effect of antioxidants in MPTP-induced mouse model of Parkinson’s disease. Neuroscience Letters. 2004;3633:243–246. doi: 10.1016/j.neulet.2004.03.072. [DOI] [PubMed] [Google Scholar]
- Parker WD, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol. 1989;26:719–733. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in stereotaxic coordinates. Academic Press; Orlando: 1986. [Google Scholar]
- Perry TL, Yong VW. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neuroscience Letters. 1986;67:269–274. doi: 10.1016/0304-3940(86)90320-4. [DOI] [PubMed] [Google Scholar]
- Puka-Sundvall M, Eriksson P, Nilsson M, Sandberg M, Lehmann A. Neurotoxicity of cysteine: interaction with glutamate. Brain Research. 1995;705:65–70. doi: 10.1016/0006-8993(95)01139-0. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ramos J, Overvik E, Ames BN. A marker of oxyradical-mediated DNA damage (8-hydroxy-2′-deoxyguanosine) is increased in nigrostriatum of Parkinson’s disease brain. Neurodegeneration. 1994;3:197–204. [Google Scholar]
- Scaduto RC, Jr, Gattone VH, Grotyohann LW, Wertz J, Martin LF. Effect of an altered glutathione content on renal ischemic injury. Am J Physiol. 1988;255:F911–F921. doi: 10.1152/ajprenal.1988.255.5.F911. [DOI] [PubMed] [Google Scholar]
- Schapira AHV, Cooper JM, Dexter D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989 June;3:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- Sechi G, Deledda MG, Bua G, Satta WM, Deiana GA, Pes GM, Rosati G. Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog Neuro-Psychopharmacol Biol Psychiat. 1996;20:1159–1170. doi: 10.1016/s0278-5846(96)00103-0. [DOI] [PubMed] [Google Scholar]
- Shoulson I. DATATOP: a decade of neuroprotective inquiry. Parkinson study group. Deprenyl and tocopherol antioxidative therapy of Parkinsonism. Ann Neurol. 1998;44:S160–S166. [PubMed] [Google Scholar]
- Shults CW, Oakes D, Kieburtz K, et al. Effects of coenzyme Q10 in early Parkinson’s disease. Arch Neurol. 2002;59:1541–1550. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting the basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- Tsan MF, White JE, Rosano CL. Modulation of endothelial GSH concentrations: effect of exogenous GSH and GSH monoethyl ester. J Appl Physiol. 1989;66:1029–1034. doi: 10.1152/jappl.1989.66.3.1029. [DOI] [PubMed] [Google Scholar]
- Wade LA, Brady HM. Cysteine and cystine transport at the blood-brain barrier. J Neurochem. 1981;37:730–734. doi: 10.1111/j.1471-4159.1982.tb12548.x. [DOI] [PubMed] [Google Scholar]
- Wang W, Ballatori N. Endogenous glutathione conjugates: occurrence and biological functions. Pharmacol Rev. 1998;50:335–355. [PubMed] [Google Scholar]
- Yazdani U, German DC, Liang CL, Manzino L, Sonsalla PK, Zeevalk GD. Rat model of Parkinson’s disease: Chronic central delivery of 1-methyl-4-phenylpyridinium (MPP+) Exp Neurol. 2006;200 (1):172–183. doi: 10.1016/j.expneurol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno K. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson’s disease. Proc Natl Acad Sci. 1996;93:2713. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevalk GD, Bernard LP. Energy status, oxidative stress and ubiquitin proteasomal function during chronic and acute complex I inhibition with rotenone in mesencephalic cultures. Antiox Redox Signal. 2005;7:662–672. doi: 10.1089/ars.2005.7.662. [DOI] [PubMed] [Google Scholar]
- Zeevalk GD, Bernard LP, Nicklas WJ. Oxidative stress during energy impairment in mesencephalic cultures is not a downstream consequence of a secondary excitotoxicity. Neuroscience. 2000;96:309–316. doi: 10.1016/s0306-4522(99)00567-9. [DOI] [PubMed] [Google Scholar]