

Abstract
DNA polymerase δ (pol δ) is a high fidelity eukaryotic enzyme that participates in DNA repair and is essential for DNA replication. Toward the goal of dissecting its multiple biological functions, here we describe the biochemical properties of Saccharomyces cerevisiae pol δ with a methionine replacing conserved leucine 612 at the polymerase active site. Compared with wild type pol δ, L612M pol δ has normal processivity and slightly higher polymerase specific activity. L612M pol δ also has normal 3′ exonuclease activity, yet it is impaired in partitioning mismatches to the exonuclease active site, thereby reducing DNA synthesis fidelity. Error rates in vitro for L612M pol δ are elevated for both base substitutions and single base deletions but in a highly biased manner. For each of the six possible pairs of reciprocal mismatches that could arise during replication of complementary DNA strands to account for any particular base substitution in vivo (e.g. T-dGMP or A-dCMP for T to C transitions), L612M pol δ error rates are substantially higher for one mismatch than the other. These results provide a biochemical explanation for our observation, which confirms earlier genetic studies, that a haploid pol3-L612M S. cerevisiae strain has an elevated spontaneous mutation rate that is likely due to reduced replication fidelity in vivo.
DNA polymerase δ (pol δ),2 one of four eukaryotic B family polymerases, is required for replicating the nuclear genome and also synthesizes DNA during recombination, mismatch repair, and excision repair of DNA damage (1). Although complete disruption of the Saccharomyces cerevisiae POL3 gene encoding pol δ is lethal, conditional pol3 alleles have effectively been used to probe several functions of pol δ in vivo. For example, studies using pol3 alleles that inactivate the intrinsic 3′ exonuclease activity of pol δ but leave the polymerase activity intact have provided important insights into the contribution of the 3′ exonuclease of pol δ to proofreading of replication errors (2, 3), suppression of cancer in mice (4), and generation of ligatable nicks at the 5′ ends of Okazaki fragments during lagging strand replication (5). These studies add to growing evidence (for review, see Ref. 6) that pol δ participates in lagging strand replication.
To better understand where and when pol δ and other eukaryotic B family DNA polymerases operate during replication of large and complex nuclear genomes, we have been searching for new alleles of DNA polymerases that retain robust replicative capacity while also generating a mutator phenotype in cells due to reduced DNA synthesis fidelity. Identifying polymerases with reduced fidelity has been facilitated by elegant structural information. For example, numerous studies (e.g. for review, see Ref. 7) have shown that non-conservative replacements of conserved amino acids in the nascent base pair binding pockets of many DNA polymerases can reduce their fidelity, albeit often with concomitant reduction in catalytic efficiency. In addition to simply having lower fidelity, an ideal property for assigning a polymerase to a particular DNA transaction in vivo would be a distinctive error specificity that generates a specific mutational signature. We were encouraged that mutator polymerases with distinctive error signatures could be constructed by the observation that an amino acid substitution in the nascent base pair binding pocket of the large Klenow fragment of Escherichia coli DNA polymerase I (E710A) not only reduced fidelity but did so only for a subset of the 12 possible single base-base mismatches (8, 9). For example, E710A Klenow polymerase was 75-fold less accurate than the wild type polymerase for errors involving A-dCMP mismatches, but it retained normal fidelity for errors involving T-dGMP mismatches (9).
Glu-710 of Klenow polymerase is located in the highly conserved sequence motif A, which along with motifs B and C form the polymerase active site. Motif A is conserved in multiple polymerases, including B family enzymes like pol δ, and its homologs, pol α, pol ε, and pol ζ and bacteriophage T4 and RB69 pols. Interestingly, the crystal structure of RB69 pol (10) revealed that Tyr-416 in motif A occupies a position in the polymerase active site (Fig. 1A) that is structurally similar to that of Glu-710 of Klenow polymerase, which when mutated reduced fidelity for some but not all mismatches, as described above. On that basis, in a previous study we replaced the homologous tyrosine residues in S. cerevisiae pol α, pol δ, pol ε, and pol ζ (Fig. 1B) with alanine and constructed haploid yeast strains with each of these four mutant polymerase alleles. The phenotypes of these strains (11) strongly suggested that these mutant polymerases all had significantly reduced catalytic activity. The strongest phenotype was for the Y613A mutant of yeast pol δ, which resulted in lethality. This is consistent with the major role that pol δ plays in replicating the nuclear genome, and the inference that Tyr-416 in RB69 pol is critical for efficient catalysis because it directly interacts with and properly positions the correct incoming dNTP (Fig. 1A).
FIGURE 1. Conservation of Leu-612 in pol δ and putative contribution to the nascent base pair binding pocket.
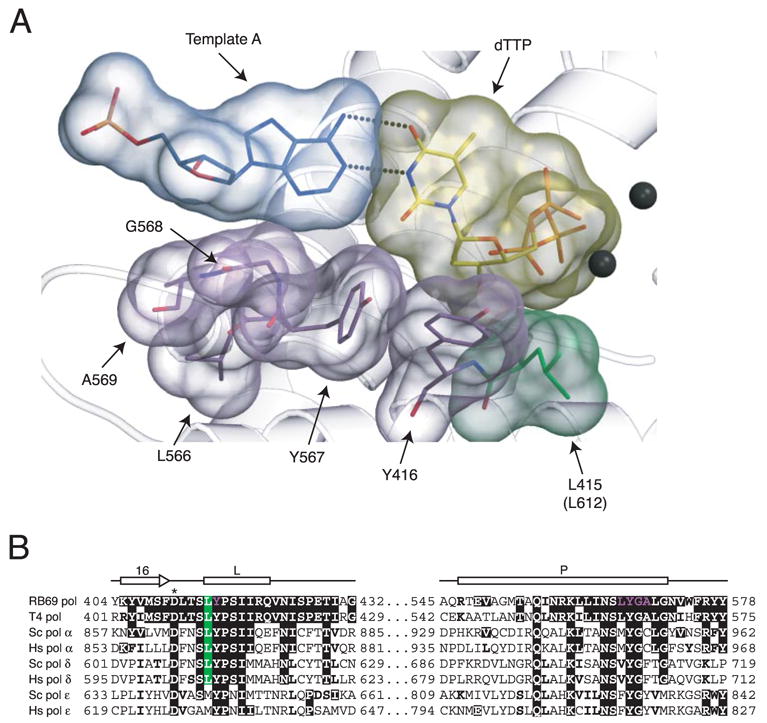
A, surface representation of the nascent base pair and several amino acids in RB69 pol that form the DNA minor groove edge of the binding pocket at the polymerase active site (adapted after Fig. 3B in Ref. 10). Leu-612 in yeast pol δ (in parentheses) aligns with Leu-415 in RB69 pol (green). The adjacent Tyr-416 in RB69 pol aligns with Tyr-869 in yeast pol α, which when substituted with alanine results in a mutator phenotype (11). The two divalent metal ions at the polymerase active site are depicted as black spheres. The figure was made with Molscript (34), Grasp (35), and Raster3D (36). B, alignment showing conservation of Leu-612 in yeast pol δ. Identical and conserved residues with respect to RB69 pol are shown in bold white letters against a black background and bold black letters, respectively. The conserved leucine (residue 415 in RB69 pol) is shown in bold white letters against a green background. Residues Tyr-416, Leu-566, Tyr-567, Gly-568, and Ala-569 in RB69 pol (purple residues in panel A) are shown in bold purple letters against a black background. Sc, S. cerevisiae; Hs, Homo sapiens. Secondary structure elements (depicted as arrows and rectangles) are as described in Wang et al. (37). One of the three catalytic aspartic acid residues is labeled with an asterisk (*).
More recently, we have turned our attention to the immediately adjacent leucine residue. In RB69 pol (Fig. 1A), this leucine (Leu-415) does not directly interact with the dNTP, but it does interact with Tyr-416. We reasoned that if the homologous residue in yeast pol δ (Leu-612) was replaced with a somewhat conservative amino acid, this might indirectly reduce fidelity without strongly reducing polymerase activity. This prediction was partly based on seminal studies of B family polymerases from three other laboratories. The first studies were of the B family homolog T4 pol, where substituting conserved Leu-412 with methionine resulted in phage with a normal DNA synthesis rate but a modest mutator phenotype (12). The purified L412M enzyme had normal polymerase activity except that it was more sensitive than wild type T4 pol to inhibition by phosphonoacetic acid, a pyrophosphate analog (12). Interestingly, although L412M T4 pol retains normal 3′ exonuclease activity, this mutant was less efficient in proofreading due to a defect in partitioning between the polymerase and exonuclease active sites (12–14). The second studies were of yeast pol α, where an L868M mutant was found to have normal polymerase specific activity (3, 15) but enhanced mismatch extension capacity (3) and reduced DNA synthesis fidelity in vitro (3, 15). Yeast strains harboring this pol α mutant allele had elevated spontaneous mutation rates that were strongly enhanced by inactivating mismatch repair (15), indicating reduced replication fidelity in vivo. The mutator effect of the pol α L868M mutant allele was also strongly elevated by inactivating the 3′ exonuclease activity of pol δ (3), suggesting that the 3′ exonuclease activity of pol δ may excise errors made by pol α, a type of extrinsic proofreading (16, 17). The third, and most recent studies were published after we initiated this work and reported the phenotypes of yeast strains harboring mutant alleles of pol δ containing replacements for Leu-612. One study (18) showed that the pol3-L612M strain is very sensitive to phosphonoacetic acid, is non-viable in the absence of RAD27, and has an elevated spontaneous mutation rate that is strongly enhanced when mismatch repair is inactivated. Another very extensive study (19) reported the phenotypes conferred by different substitutions for Leu-612. Of the 19 possible amino acid replacements for Leu-612, 8 pol3-L612X strains were viable, but their sensitivities to treatment with hydroxyurea and methylmethane sulfonate, their cell cycle behavior, their cellular morphology, and their effect on spontaneous mutagenesis varied over a wide range. Among the eight alleles examined, the phenotypes of the pol3-L612M strain were most similar to the wild type strain, with the exception that the spontaneous mutation rate was elevated 7-fold, suggesting reduced replication fidelity in vivo (19).
Collectively, the studies described above predict that yeast pol δ containing the L612M amino acid replacement should have several biochemical properties: it should retain robust polymerization activity and normal 3′ exonuclease activity but have reduced DNA synthesis fidelity and impaired partitioning of mismatched termini between the polymerase and 3′ exonuclease active sites. One objective of the present study was to test these four predictions, all of which are fulfilled. Given its high catalytic efficiency and reduced fidelity, a second objective was to determine the error specificity of L612M pol δ in detail toward the goal of identifying an informative mutator polymerase for in vivo studies. Fortuitously, the L612M mutant of pol δ not only retains high catalytic activity, it also has a distinctive error signature.
EXPERIMENTAL PROCEDURES
Materials
Yeast replication protein A, proliferating cell nuclear antigen, and replication factor C were purified from E. coli overproducing strains as described (20, 21). The three forms of pol δ (wild type, L612M, and 5-DV) were overexpressed and purified as previously described (22). All materials for the fidelity assay were from previously described sources (23, 24).
Construction of the Pol δ L612M Mutant
The pol3-L612M mutation was introduced into plasmid pBL335 (Bluescript, 2 μm ori, TRP1, M13 ori, GAL1–10 GST-POL3) by site-directed mutagenesis. The identity of the entire pol3-L612M gene was verified by sequencing. L612M pol δ was overproduced in yeast and purified exactly as described for the wild type enzyme (22). Because of the presence of a glutathione S-transferase fusion to the N terminus of L612M pol δ encoded on plasmid pBL335-L612M, the mutant enzyme could be purified free from chromosomally encoded wild type pol δ through glutathione-affinity chromatography. After affinity purification, the tag was removed proteolytically, and L612M pol δ was further purified by Mono S chromatography as described (22).
Measurements of Polymerase-specific Activity
The reaction mixture (60 μl) contained 20 mM Tris (pH 7.8), 200 μg/ml bovine serum albumin, 1 mM dithiothreitol, 90 mM NaCl, 16.5 nM [α-32P]dCTP, 100 μM concentrations of each cold dNTP, 8 mM magnesium acetate, and 1.5 μg of activated calf thymus DNA (GE Healthcare). Reactions were initiated by adding 5 nM wild type or L612M pol δ. Reaction mixtures were incubated at 30 °C, and time points were stopped after 1, 2, 5, and 10 min by adding EDTA to 20 mM. Duplicate samples were processed for incorporation by adding 0.5 ml of 10% trichloroacetic acid. After a 10-min incubation on ice, acid-precipitable material was collected by filtration through Whatman GF/C filters. The filters were rinsed 3 times with 5 ml of 5% trichloroacetic acid, 1% sodium pyrophosphate and once with 5 ml of ethanol, dried, and counted in scintillation fluid.
Measurements of Polymerase and Exonuclease Activities
The substrate used for primer extension analysis (Fig. 2A) was generated by annealing a 32P-labeled primer strand (5′-GTAACGCCAGGGTTTTCTCA-3′) to a template strand (5′-ACGTCGTGACTGAGAAAACCCTGGCGTTACCCA-3′). Reactions (10 μl) were performed with 1000 fmol (100 nM) of DNA substrate in a buffer containing 20 mM Tris (pH 7.8), 200 μg/ml bovine serum albumin, 1 mM dithiothreitol, 90 mM NaCl, 8 mM magnesium acetate, and 100 μM concentrations of each dNTP. Reactions were initiated by the addition of 2 fmol (0.2 nM) of polymerase and transfer to 30 °C. Time points were stopped after 1, 3, and 5 min by the addition of an equal volume of formamide loading dye and were analyzed by electrophoresis on a denaturing 12% polyacrylamide gel. Products were detected and quantified using a PhosphorImager and ImageQuant software (GE Healthcare). The substrate used for analysis of single-stranded exonuclease activity (Fig. 2B) was a 32P-labeled primer strand (5′-GTAACGCCAGGGTTTTCTCG-3′). Reactions were performed as described above, except that dNTPs were omitted.
FIGURE 2. Catalytic activities of L612M pol δ.
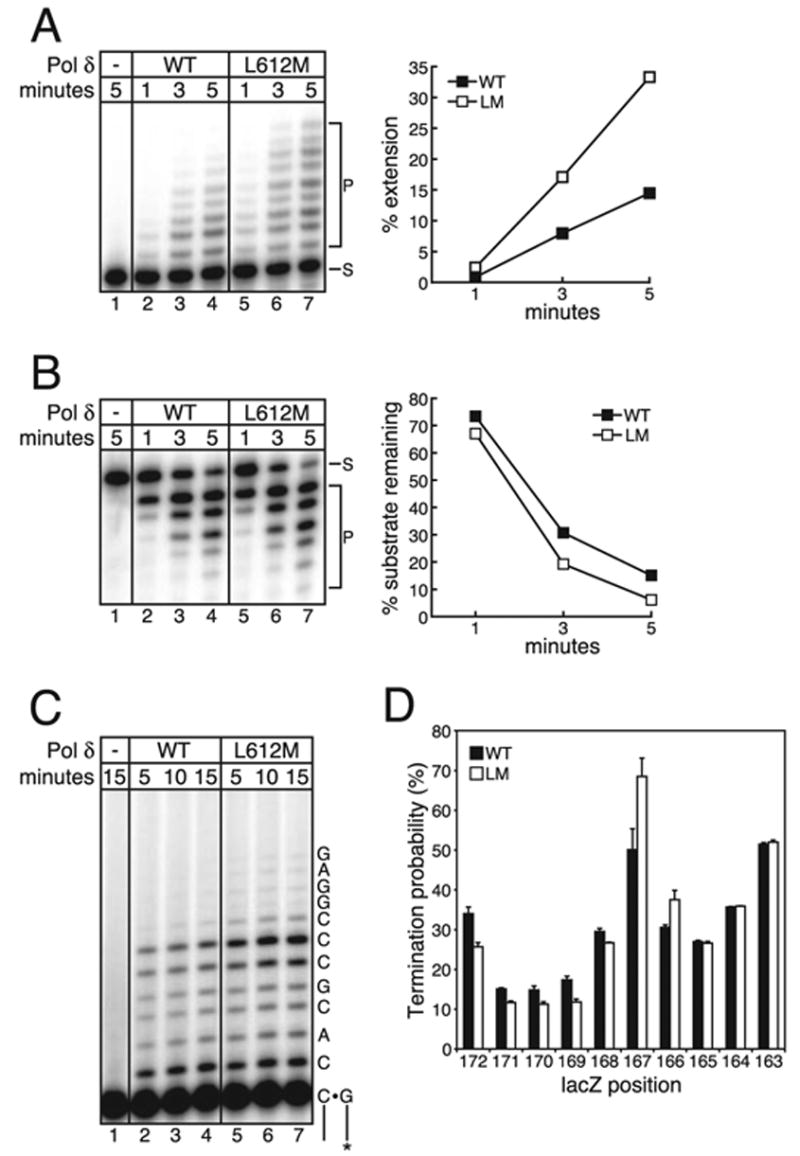
A, primer extension activity. A 32P-labeled primed template was present (100 nM) in all reactions. 0.2 nM wild type (WT) or L612M (LM) pol δ was added as noted. Reaction time was 1, 3, or 5 min. S, substrate; P, primer extension products. The % extension is plotted at the right. B, single-stranded exonuclease activity. A 32P-labeled single-stranded substrate was present (100 nM) in all reactions. 0.2 nM wild type or L612M pol δ was added as noted. Reaction time was 1, 3, or 5 min. The % substrate remaining is plotted at the right. C, processivity of wild type and L612M pol δ. A primed single-stranded M13mp2 DNA substrate was present (5 nM) in all reactions. 0.1 nM wild type or L612M pol δ was added as noted. Reaction times were 5, 10, or 15 min. A portion of the template sequence is shown on the right. D, termination probabilities at each template position, expressed in percentage as the ratio of products at each site to the products at that site plus all greater-length products. The termination probability shown is the average of the three time points shown in panel C. Error bars denote the S.D. of the three averaged time points.
Measurements of Polymerase-Exonuclease Partitioning
Extension of a T-dGMP mismatch (see Fig. 4, B and C) was analyzed using a substrate made by annealing a 32P-labeled primer strand (5′-GTAACGCCAGGGTTTTCTCG-3′) to a template strand (5′-ACGTCGTGACTGAGAAAACCCTGGCGTTACCCA-3′) generating a terminal T-dGMP mismatch. Extension of a T-dTMP mismatch (see Fig. 4, D and E) was performed using a substrate made by annealing a 32P-labeled primer strand (5′-GTAACGCCAGGGTTTTCTCT-3′) to a template strand (5′-ACGTCGTGAATGAGAAAACCCTGGCGTTACCCA-3′), generating a terminal T-dTMP mismatch. Reactions were performed as described above except that 100 μM concentrations of only the next correct dNTP was added; for the T-dGMP mismatch, dGTP was added, and for the T-dTMP mismatch, dTTP was added.
FIGURE 4. Mismatch extension by L612M pol δ.
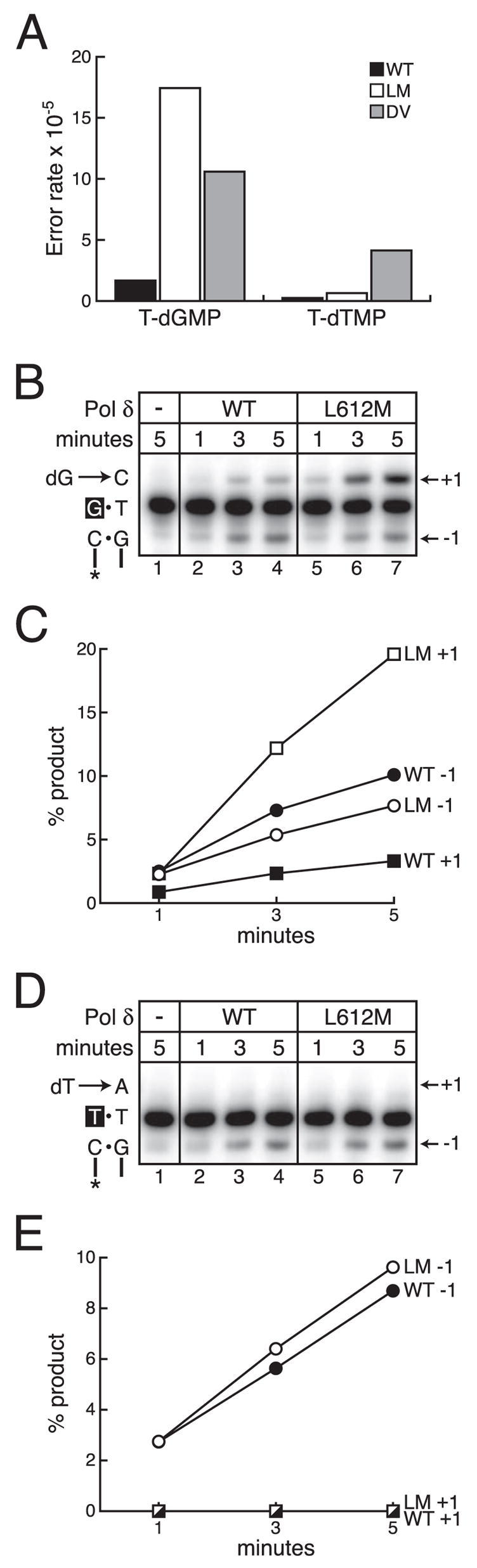
A, average error rates from the (+) and (−) strand gap substrates of wild type (WT), L612M (LM), and 5-DV (DV) pol δ for T-dGMP and T-dTMP mispairs. B, mismatch extension of a T-dGMP mismatch by wild type and L612M pol δ. A 32P-labeled primed template with a terminal T-dGMP mismatch was present (100 nM) in all reactions. The sequence of a portion of the substrate is shown on the left. The mismatched dGMP primer terminus is shown in white on a black background, and the incoming dGTP (dG) is shown. 0.2 nM wild type or L612M pol δ was added as noted. Reaction times were 1, 3, or 5 min. +1, mismatch extension product; −1, mismatch excision product. C, the percent of mismatch extension (+1; squares) and mismatch excision (−1; circles) product is plotted versus reaction time for wild type (filled symbols) and L612M (open symbols) pol δ for the T-dGMP mismatch. D, mismatch extension of a T-dTMP mismatch by wild type and L612M pol δ. A 32P-labeled primed template with a terminal T-dTMP mismatch was present (100 nM) in all reactions. The sequence of a portion of the substrate is shown on the left. The mismatched dTMP primer terminus is shown in white on a black background, and the incoming dTTP (dT) is shown. 0.2 nM wild type or L612M pol δ was added as noted. Reaction times were 1, 3, or 5 min. +1, mismatch extension product; −1, mismatch excision product. E, the percent of mismatch extension (+1; squares) and mismatch excision (−1; circles) product is plotted versus reaction time for wild type (closed symbols) and L612M (open symbols) pol δ for the T-dTMP mismatch. No extension of the T-dTMP mismatch was detected for either wild type or L612M pol δ.
Measurements of Polymerase Processivity
The substrate used was primed single-stranded M13mp2 DNA with the primer strand annealing to nucleotides +173 to +192 of the lacZ target sequence. Reactions (30 μl) were performed in the standard reaction buffer described above and contained 150 fmol (5 nM) of primed single-stranded M13mp2 substrate, 1 mM concentrations of each dNTP, and 3 fmol (0.1 nM) wild type or L612M pol δ. Reactions were incubated at 30 °C, and time points were stopped by the addition of an equal volume of formamide loading dye after 5, 10, and 15 min. Reactions were performed in triplicate, and the termination probability at each template position was determined as described previously (25). The termination probabilities for each of the three replicate reactions were averaged for each time point, and then the three averaged time points were averaged to obtain a final termination probability for each template position. Error bars in Fig. 2D denote the S.D. of the three averaged time points.
Gap-filling DNA Synthesis Reactions and Product Analysis for Determining Fidelity
Reactions (25 μl) contained 20 mM Tris (pH 7.7), 8 mM magnesium acetate, 90 mM NaCl, 0.5 mM ATP, 100 μM concentrations of each dNTP, 1 mM dithiothreitol, 100 mg/ml bovine serum albumin, 500 fmol of proliferating cell nuclear antigen, 200 fmol of replication factor C, 5 pmol of replication protein A, 150 fmol of pol δ and 25 fmol (1 nM) of gapped DNA that was prepared as described previously (26). Polymerization reactions were incubated at 30 °C for 3 min. Under these conditions, when DNA products were analyzed by agarose gel electrophoresis as described (23), all reactions filled the gap without obvious strand displacement (data not shown, but for a typical result, see Fig. 3 in Ref. 23). DNA products of gap-filling reactions were introduced into E. coli cells and plated as described (23) to score blue M13 plaques (correct synthesis) and light blue and colorless plaques (containing errors). The types of errors were determined by sequencing the lacZα complementation gene in single-stranded DNA isolated from independent mutant M13 plaques, allowing calculation of error rates as previously described (27).
FIGURE 3. Fidelity of L612M pol δ.
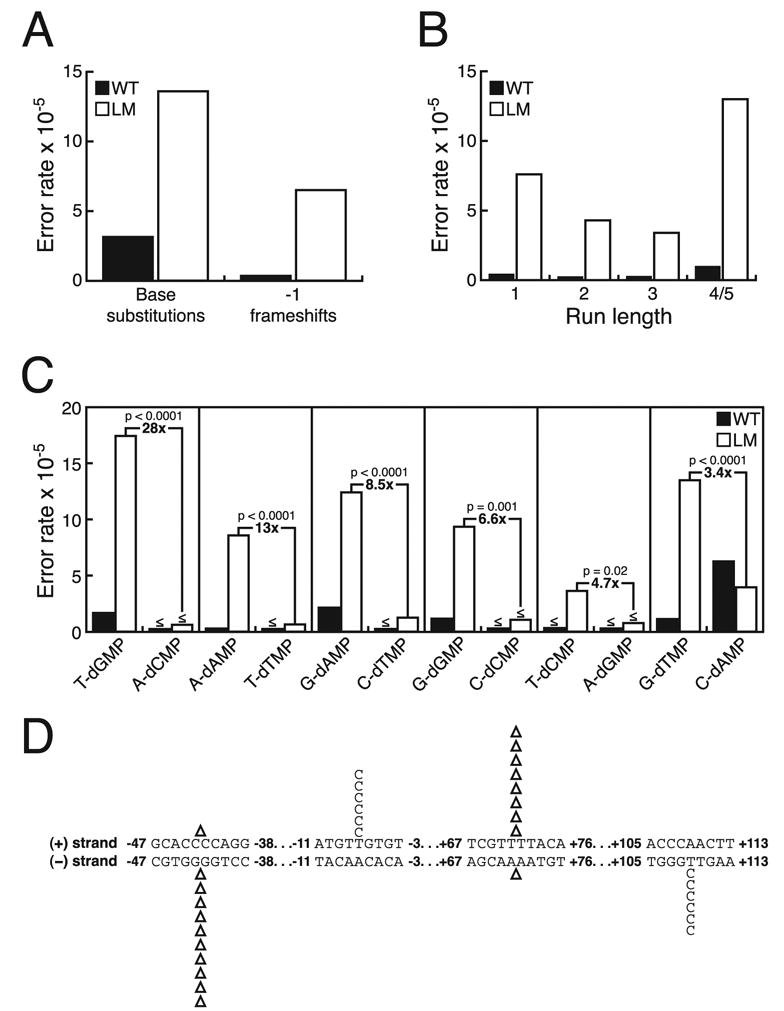
A, average error rates from the (+) and (−) strand gap substrates for wild type (WT) and L612M (LM) pol δ for single base substitutions and −1 frameshift errors. B, average error rates from the (+) and (−) strand gap substrates for wild type and L612M pol δ for −1 frameshift errors as a function of the length of the repeat element. C, average error rates from the (+) and (−) strand gap substrates for wild type and L612M pol δ for each of the 12 possible base-base mispairs. The mispairs are grouped into reciprocal mispairs. The average background error rate for C to T base substitutions inferred to be due to template deamination in the unfilled gap substrates (3.55 × 10−5) was subtracted from the error rates for C-dAMP mispairs. The -fold difference between the error rates of L612M pol δ for each set of reciprocal mispairs is shown with the p value from the Fisher’s exact test for statistical significance. D, selected regions of the spectra of point mutations made by L612M pol δ on the (+) strand and (−) strand gap substrates. Base substitutions and single nucleotide deletions (Δ) are shown above the (+) strand sequence and below the (−) strand sequence. Numbers denote the position in the lacZ target sequence.
Measurements of Mutation Rates in Yeast
Forward mutation rates at CAN1 were determined by fluctuation analysis using 12 independent cultures as described (28, 29).
RESULTS
Specific Activity and Processivity of L612M Pol δ
We first measured the polymerase activity of the L612M pol δ mutant relative to wild type pol δ. Using activated calf thymus DNA as a substrate, wild type pol δ incorporated 0.8 pmol of dCTP/nmol of pol δ/min, whereas L612M pol δ incorporated 1.6 pmol of dCTP/nmol of pol δ/min. L612M pol δ was also ~2-fold more active than wild type pol δ in a simple primer extension assay (Fig. 2A). In a reaction containing a high DNA to enzyme ratio such that products result from a single encounter between pol δ and the primer-template (Fig. 2C), L612M pol δ extended 18% of the available primer and cycled 9 times, whereas wild type pol δ extended 10% of the available primer and cycled 5 times. Despite this 2-fold difference, the probability of termination of processive synthesis at each template position was similar for wild type and L612M pol δ (Fig. 2D). To determine whether the increase in activity was specific to polymerase activity or also included the 3′ exonuclease activity of pol δ, we compared the ability of L612M pol δ and wild type pol δ to digest single-stranded and double-stranded DNA. The 3′ exonuclease activity of L612M pol δ was slightly increased relative to wild type pol δ when either single-stranded DNA (Fig. 2B) or double-stranded DNA (data not shown) was used as a substrate. However, the differences are slight, such that the L612M mutation appears to selectively increase polymerase activity relative to exonuclease activity. At a minimum, replacing Leu-612 with methionine does not reduce polymerase activity or processivity nor does it reduce the intrinsic exonuclease activity of pol δ.
Fidelity of L612M Pol δ
Next we determined if L612M pol δ synthesizes DNA with reduced fidelity during synthesis to copy the lacZα complementation sequence in gapped circular M13 DNA substrates (23). For comparison, parallel analyses were performed with wild type pol δ and an exonuclease-deficient mutant of pol δ (D520V(5-DV)) (attempts to construct and purify exonuclease-deficient L612M pol δ were unsuccessful, possibly because the combination of the L612M and 5-DV mutations in POL3 causes lethality in yeast due to error catastrophe). All reactions were performed in the presence of proliferating cell nuclear antigen, replication factor C, and replication protein A because these accessory proteins operate with pol δ as it fulfills its in vivo functions. To define error specificity when replicating complementary strand sequences, error rates for all three forms of pol δ were determined with two different gapped DNA substrates, one containing the (+) strand lacZ template and another containing the complementary (−) strand lacZ template (26).
Wild type, L612M, and 5-DV pol δ all filled the gaps in both lacZ substrates (see “Experimental Procedures”). The DNA products of these reactions were introduced into a lacZα-complementation E. coli strain, and the cells were plated to score DNA synthesis errors as light blue and colorless M13 plaques among much greater numbers of dark blue plaques resulting from correct synthesis (Table 1, top three rows). With both templates, the lacZ mutant frequencies generated by L612M pol δ were about 6-fold higher than those for wild type pol δ, clearly indicating that L612M pol δ has reduced fidelity. Interestingly, the lacZ mutant frequencies were similar for L612M and 5-DV pol δ despite the fact that the L612M enzyme retains robust 3′ exonuclease activity (Fig. 2B), whereas the 5-DV polymerase is exonuclease-deficient.
TABLE 1.
Fidelity of wild type, L612M, and 5-DV pol δ
| (+) strand | (−) strand | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT pol δ | L612Mpol δ | 5-DVpol δ | WTpol δ | L612Mpol δ | 5-DVpol δ | |||||||
| Total plaques | 39208 | 12025 | 12321 | 16116 | 7929 | 9613 | ||||||
| lacZ mutants | 116 | 235 | 263 | 66 | 194 | 250 | ||||||
| Mutants sequenced | 116 | 235 | 236 | 66 | 166 | 250 | ||||||
| Mutant frequency | 0.0030 | 0.020 | 0.021 | 0.0041 | 0.024 | 0.026 | ||||||
| Total # mutations | 116 | 244 | 240 | 67 | 170 | 255 | ||||||
|
|
||||||||||||
| Number Observed | Error Rate (× 10−5) | Number Observed | Error Rate (× 10−5) | Number Observed | Error Rate (× 10−5) | Number Observed | Error Rate (× 10−5) | Number Observed | Error Rate (× 10−5) | Number Observed | Error Rate (× 10−5) | |
|
|
||||||||||||
| Base Substitutions | 100 | 2.6 | 161 | 13 | 193 | 17 | 59 | 3.7 | 95 | 14 | 201 | 21 |
| Transitions | 79 | 2.6 | 98 | 11 | 73 | 8.4 | 46 | 3.7 | 48 | 9.0 | 73 | 9.8 |
| A to G (A-dCMP) | 0 | ≤0.15 | 0 | ≤0.48 | 1 | 0.51 | 1 | 0.32 | 1 | 0.75 | 1 | 0.54 |
| G to A (G-dTMP) | 8 | 1.2 | 29 | 14 | 21 | 11 | 4 | 1.0 | 21 | 13 | 26 | 11 |
| C to T (C-dAMP) | 62 | 6.7 | 21 | 7.3 | 31 | 11 | 35 | 13 | 9 | 7.7 | 26 | 16 |
| T to C (T-dGMP) | 9 | 1.2 | 48 | 21 | 20 | 9.3 | 6 | 2.1 | 17 | 14 | 20 | 12 |
| Transversions | 21 | 0.65 | 63 | 6.2 | 120 | 13 | 13 | 0.96 | 47 | 8.1 | 128 | 16 |
| A to C (A-dGMP) | 0 | ≤0.21 | 0 | ≤0.66 | 0 | ≤0.71 | 0 | ≤0.38 | 0 | ≤0.89 | 2 | 1.3 |
| A to T (A-dAMP) | 1 | 0.13 | 17 | 7.1 | 25 | 11 | 1 | 0.43 | 10 | 10 | 34 | 24 |
| C to A (C-dTMP) | 0 | ≤0.13 | 2 | 0.84 | 11 | 4.9 | 0 | ≤0.36 | 2 | 1.7 | 11 | 6.5 |
| C to G (C-dCMP) | 1 | 0.18 | 2 | 1.2 | 3 | 1.9 | 0 | ≤0.41 | 0 | ≤0.96 | 1 | 0.69 |
| G to C (G-dGMP) | 6 | 1.0 | 12 | 6.6 | 15 | 8.9 | 3 | 1.3 | 12 | 12 | 9 | 6.5 |
| G to T (G-dAMP) | 12 | 1.8 | 26 | 12 | 41 | 21 | 8 | 2.5 | 17 | 12 | 59 | 31 |
| T to A (T-dTMP) | 1 | 0.18 | 1 | 0.58 | 10 | 6.2 | 0 | ≤0.31 | 1 | 0.73 | 4 | 2.1 |
| T to G (T-dCMP) | 0 | ≤0.16 | 3 | 1.5 | 15 | 8.2 | 1 | 0.49 | 5 | 5.7 | 8 | 6.6 |
| Single base frameshifts | 16 | 0.32 | 81 | 5.1 | 46 | 3.1 | 8 | 0.38 | 73 | 8.1 | 51 | 4.0 |
| −1 | 16 | 0.32 | 80 | 5.1 | 45 | 3.1 | 8 | 0.38 | 72 | 8.0 | 51 | 4.0 |
| run | 9 | 0.35 | 29 | 3.6 | 21 | 2.8 | 2 | 0.19 | 33 | 7.2 | 27 | 4.2 |
| non-run | 7 | 0.28 | 51 | 6.5 | 24 | 3.3 | 6 | 0.58 | 39 | 8.7 | 24 | 3.8 |
| +1 | 0 | ≤ 0.020 | 1 | 0.063 | 1 | 0.068 | 0 | ≤ 0.047 | 1 | 0.11 | 0 | ≤ 0.079 |
The total number of specific mutations reported is sometimes greater than the number of lacZ mutants sequenced due to the presence of two detectable errors in a single mutant. A new detectable mutation is confirmed by observation as the sole mutation causing a detectable phenotype in at least two independent mutant plaques. However, 36 mutations that were observed only once were considered detectable sites, as they were the only mutations observed in plaques with a detectable phenotype. Background error rates for unfilled (+) strand gap substrate are 1.8 × 10−5 for base substitutions and 0.087 × 10−5 for single base frameshifts. Background error rates for unfilled (−) strand gap substrate are 1.8 × 10−5 for base substitutions and 0.24 × 10−5 for single base frameshifts. There were a small number of more complex events for L612M pol δ (CG to T at +168 to +169 of (+) strand gap, CGC to TTT at +177 to +179 of (+) strand gap, GGGG to GTG at (−41) to (−44) of (−) strand gap, deletion of TA or AT at +13 to +14 or +14 to +15 of (−) strand gap) and 5-DV pol δ (CCCC to TCC at (−41) to (−44) of (−) strand gap, deletion of GT at +74 to −75 of (−) strand gap, deletion of GG at (−41) to (−44) of (−) strand gap, deletion of TT at +91–+94 of (−) strand gap). WT, wild type.
To determine the types and locations of errors and to calculate error rates, we sequenced the lacZα complementation gene in DNA isolated from collections of independent mutant plaques (Table 1, the fourth and fifth rows). As expected based on earlier results with wild type and exonuclease-deficient pol δ (22), no large deletions were observed due to the presence of the accessory proteins proliferating cell nuclear antigen, replication protein A, and replication factor C. Although a small number of mutations involved more than a single base change (see legend to Table 1), the vast majority of errors were single base substitutions and single base deletions (Table 1). From the mutant frequencies and the proportion of each type of error, we calculated the rates (see “Experimental Procedures”) at which wild type, L612M, and 5-DVpol δ generated various errors when copying the (+) and (−) strand templates (Table 1). The results with wild type and 5-DV pol δ are similar to those recently published (22), illustrating the reproducibility of the fidelity analysis and providing a basis for comparison to the error rates of L612M pol δ. For all three polymerases, mutant frequencies and error rates were similar when results from the (+) and (−) strand substrates were compared (Table 1); therefore, error rates for the two substrates were averaged for all subsequent comparisons between wild type, L612M, and 5-DV pol δ (Figs. 3, A–C, and 4A).
Single Base Deletions
The average single base deletion error rate of L612M pol δ is 19-fold higher than that of wild type pol δ (Fig. 3A). The highest error rates are for single base deletions within the longest homopolymeric runs (4–5 bases) in the lacZ target (Fig. 3B), consistent with involvement of a misaligned intermediate containing an extra template strand nucleotide stabilized by the correct base-pairing possible in a repetitive sequence (for review, see Ref. 30). Interestingly, the deletion rate differs when L612M pol δ copies the same run in the two complementary strands (Fig. 3D). For example, when copying the (+) strand template, L612M pol δ deletes a T from a TTTT run at positions +70 to +73 at a rate that is 4.7-fold higher than the rate at which it deletes an A from the complementary AAAA run when copying the (−) strand template. Similarly, when copying the (−) strand template, L612M pol δ deletes a G from a GGGG run at positions −41 to −44 at a rate that is 17-fold higher than the rate at which it deletes a C from the complementary CCCC run when copying the (+) strand template. L612M pol δ is also less accurate than wild type pol δ when copying shorter homonucleotide runs and when copying non-iterated bases (Fig. 3B). The high rate of single base deletions in non-iterated sequences is consistent with L612M pol δ-mediated misinsertion followed by primer relocation to generate a misaligned intermediate and/or with misalignment in the active site (for review, see Ref. 30).
Single Base Substitutions
The majority of errors made by L612M pol δ were single base substitutions (Table 1). The overall average base substitution error rate of L612M pol δ is 4.5-fold higher than that of wild type pol δ (Fig. 3A). Notably, the error rate of L612M pol δ is significantly higher than that of wild type pol δ for only 6 of the 12 possible single base mismatches (Fig. 3C). Remarkably, the six base substitution error rates that increase and the six that do not can be paired as “reciprocal” mispairs (Fig. 3C), i.e. those that would result in the same base substitution in vivo if the mistake was made when replicating either the (+) or the (−) strand. This behavior is illustrated with two examples in Fig. 3D. When copying the (+) strand template, synthesis by L612M pol δ led to six T to C substitution errors via stable misincorporation of dGMP opposite template T at position −7 in the lacZ sequence. In contrast, the same base pair substitution (i.e. T-A to C-G) would result from misincorporating dCMP opposite template A at position −7 on the complementary (−) strand, yet that error was not detected in the spectrum of errors made by L612M pol δ. A second example was observed for the two reciprocal mispairs at position +109. In this instance stable misincorporation of dGMP opposite template T was observed when the (−) strand template was being copied, but its complementary (+) strand mismatch was again not observed. Thus, the asymmetry in error rates of reciprocal mispairs is intrinsic to L612M pol δ rather than the strand being copied. The asymmetry is also not unique to the two particular sites used to illustrate this point (Fig. 3D). Rather, the differences in error rates for reciprocal mispairs shown in Fig. 3C reflect average rates when copying a large number of phenotypically detectable positions in both the (+) and (−) strand templates.
Impaired Partitioning between the Polymerase and Exonuclease Active Sites
The error rate of L612M pol δ for certain mispairs is elevated relative to wild type pol δ (Figs. 4A and 3C) despite the fact that L612M pol δ retains 3′ exonuclease activity. This could result from defective partitioning of a mismatched primer terminus between the polymerase and exonuclease active sites, as has been demonstrated for L412M T4 pol (12–14) and suggested for L612M yeast pol δ (18). Such a partitioning defect could result from promiscuous mismatch extension by L612M pol δ, as has been observed with naturally exonuclease-deficient L868M pol α (3). Therefore, we designed an assay to concomitantly monitor both mismatch extension and mismatch excision by exonuclease-proficient pol δ. The first substrate we used contained a primer terminal G mismatched with a template T whose 5′ neighbor is a template C (Fig. 4B). In a reaction containing onlyd GTP, the 3′ exonuclease can remove the terminal G to generate a −1 product (Fig. 4B, lanes 2–7). After doing so, subsequent polymerization from the −1 product can only regenerate the initial mismatched substrate becaused GTP is the only nucleotide present. In this way, correct incorporation of dGMP opposite template C to generate a +1 product must necessarily represent extension of the mismatched primer.
We first examined extension versus excision of a T-dGMP mismatch because the error rate for the corresponding base substitution in the gap-filling assay, which requires both misinsertion and mismatch extension, is elevated for the L612M mutant relative to wild type pol δ (Fig. 4A). Both wild type and L612M pol δ were able to extend the T-dGMP mismatch to generate a +1 product, and both were able to excise the mismatched primer terminal G to generate a −1 product (Fig. 4B). However, the ratio of the two products demonstrates that L612M pol δ favors mismatch extension by a factor of ~2.5-fold, whereas wild type pol δ favors mismatch excision by ~3-fold (Fig. 4C). These data are consistent with the interpretation that L612M pol δ extends the T-dGMP mismatch more efficiently than does wild type pol δ and, thus, proofreads this mismatch less efficiently despite retaining normal 3′ exonuclease activity.
In contrast to results with the T-dGMP mismatch, the error rates for the base substitution resulting from a T-dTMP mismatch in the gap-filling assay are similar for L612M and wild type pol δ (Fig. 4A). However, 5-DV pol δ is a mutator for T-dTMP mispairs (Fig. 4A), suggesting that wild type and L612M pol δ are capable of misinserting dTMP opposite template T, but both favor excision over extension of the T-dTMP mismatch to a similar extent. Indeed, when this was examined, L612M and wild type pol δ both excised the mismatch with similar efficiency, and neither generated detectable +1 extension products with a T-dTMP mismatch (Fig. 4, D and E). These data are consistent with the low error rate for T to A substitutions by both wild type and L612M pol δ in the gap-filling assay (Fig. 4A and Table 1).
Spontaneous Mutator Phenotype of a pol3-L612M Yeast Strain
Consistent with two recent reports (18, 19), a yeast strain harboring the L612M pol δ allele has a spontaneous mutation rate at the CAN1 locus that is elevated 9-fold compared with a wild type strain (Table 2).
TABLE 2.
Forward mutation rates at CAN1 in wild type and pol3-L612M S. cerevisiae strains
| Strain | Canr mutation rate (×10−7) (95% CI) | -Fold increase |
|---|---|---|
| Wild type | 1.9 (1.5–3.2) | 1 |
| pol3-L612M | 17.4 (10.8–24.7) | 9 |
CI, confidence intervals.
DISCUSSION
The properties of yeast L612M pol δ described here are interesting in comparison to earlier studies of three other B family polymerases; they are unanticipated regarding error specificity, they are informative regarding several phenotypes of a yeast pol3-L612M strain, and they may be useful for dissecting the roles of pol δ during DNA replication in vivo.
Comparison to Earlier Biochemical Studies
L612M pol δ has wild type processivity, and its polymerase activity is ~2-fold higher than wild type pol δ. This 2-fold increase in polymerase-specific activity is likely to be significant in light of the observation that both L612M and wild type pol δ have similar 3′ exonuclease-specific activities. This collection of three biochemical properties differs slightly from results with the analogous L412M mutant of T4 pol, which has wild type polymerase activity but increased processivity (12). Nonetheless, L412M T4 pol and L612M pol δ share a defect in proofreading even though both polymerases retain wild type levels of 3′ exonuclease activity. Biochemical studies of L412M T4 pol indicate that the proofreading defect is due to inefficient transfer of the DNA strand from the polymerase active site to the exonuclease active site (12–14). Our own partitioning results with L612M pol δ (Fig. 4) and the observation that the homologous L868M pol α mutant is promiscuous in extending mismatches (3), suggest that replacing Leu-612 at the polymerase active site of pol δ with methionine enhances mismatch extension efficiency, thus decreasing the probability that the mismatched primer terminus will partition to the exonuclease active site, thereby suppressing proofreading.
Error Specificity
The ratio of extension versus excision by L612M pol δ differs with the T-dGMP and T-dTMP mismatches (Fig. 4), suggesting that the probability of partitioning between the polymerase and exonuclease active sites differs for different mismatches. This is one possible explanation for the observation that L612M pol δ error rates for the 12 possible single base mismatches differ by more than 20-fold (Table 1, Fig. 3C). An additional, non-exclusive possibility is that the methionine replacement also increases misinsertion probabilities in a mismatch-specific manner. This is consistent with and supported by kinetic studies demonstrating that an L868F replacement in pol α strongly increases misinsertion efficiency (3, 15). Regardless of the relative contributions of misinsertion, mismatch extension, and/or partitioning, the ultimate result is an error specificity of L612M pol δ that is truly remarkable. In comparing the two possible mismatches that could arise during replication of complementary strands to account for any particular base substitution in vivo, the error rate of L612M pol δ is consistently and substantially higher for one mismatch than the other (Fig. 3C). One possible molecular contribution to this asymmetric pattern comes from the study of the E710A mutant of Klenow fragment DNA polymerase (9). When Glu-710 was changed to alanine, the E710A Klenow mutant behaved similarly to what is observed here with the L612M pol δ mutant, in two ways. First, E710A was much less accurate than the wild type polymerase for errors involving an A-dCMP mismatch but retained normal fidelity for errors involving the reciprocal T-dGMP mismatch. Second, the E710A mutant was also much less accurate than the wild type polymerase for errors involving a G-dTMP mismatch, but again retained normal fidelity for errors involving the reciprocal C-dAMP mismatch. To explain these asymmetries, kinetic analysis of correct and incorrect dNTP insertion by the wild type and E710A polymerase were combined with information on the structure of the polymerase active site and the known asymmetry of wobble base pairs. The results led to the suggestion that the side chain of glutamate 710 excludes wobble base pairs between template pyrimidines and purine triphosphates by steric clash. They further suggested that the glutamate side chain enhances the stability of incoming correct dNTPs such that loss of this interaction upon replacement with alanine leads to lower selectivity against mismatches involving incoming pyrimidines. Similar explanations could be relevant to the L612M pol δ base substitution error rate pattern in Fig. 3C, especially since Leu-612 in pol δ is inferred to be immediately adjacent to Tyr-613, which (from Ref. 10) is inferred to occupy a position in the polymerase active site similar to that of Glu-710 in Klenow polymerase. Regardless of the molecular explanation, the biased error rates of L612M pol δ may be useful for in vivo studies, as described below.
Using pol3-L612M to Probe Pol δ Function during Replication in Vivo
A major objective of this study was to determine the biochemical properties of L612M pol δ toward the goal of using the pol3-L612M allele to probe pol δ functions during DNA replication in vivo. The pol3-L612M allele appears to be ideal for this purpose. The specific activity and processivity of L612M pol δ is similar to that of wild type pol δ, which is consistent with a recent study (19) reporting that a haploid yeast strain harboring the pol3-L612M allele was very similar to the wild type strain with respect to cell cycle behavior, cellular morphology, and sensitivity to treatment with hydroxyurea and methylmethane sulfate. These facts all indicate that the pol3-L612M strain has relatively normal replicative capacity. The observation that L612M pol δ has reduced DNA synthesis fidelity is consistent with two studies (18, 19) showing that the pol3-L612M strain has an elevated spontaneous mutation rate, a result that we have confirmed here. This fact and the observation that the spontaneous mutation rate of a pol3-L612M strain is substantially enhanced when mismatch repair is inactivated (18) strongly suggest that the spontaneous mutagenesis observed in the pol3-L612M strain results from inaccurate replication by L612M pol δ.
With these observations in mind, it may be possible to use the asymmetry in error rates between reciprocal mispairs that is depicted in Fig. 3, C and D, to assign L612M pol δ to replication of the lagging strand, for which there is already considerable evidence (6), and/or to the leading strand (31). For example, the observation that L612M pol δ has a 28-fold higher rate for T-dGMP errors than for A-dCMP errors (Fig. 3C) potentially allows one to infer which strand is being replicated by L612M pol δ when an A-T to G-C mutation is generated in a pol3-L612M mutator strain. The error rate bias of L612M pol δ implies that >95% of A-T to G-C errors would result from dGMP misinsertion opposite the template T of the A-T base pair rather than from dCMP misinsertion opposite the template A. Whether the template T is replicated by the leading strand replication machinery or the lagging strand replication machinery can be controlled by placing a reporter gene for mutagenesis close to a known origin of replication either in opposite orientations on the same side of the origin or in the same orientation on opposite sides of the origin (32). By comparing the distribution of A-T to G-C mutations observed in a pol3-L612M mutator strain under these circumstances, it may be possible to infer whether pol δ replicates the leading strand, the lagging strand, or both. Experiments to address these possibilities are currently in progress. Based on the asymmetric single base deletion error rates of L612M pol δ (Fig. 3D), it may also be possible to use the same strategy to determine whether microsatellite instability results from lagging strand replication, leading strand replication, or both and to determine whether pol δ is the enzyme responsible for these errors. Finally, by manipulating the genetic background of the yeast strains, it may be possible to use the pol3-L612M strain to examine the efficiency of repair of mismatches generated specifically by pol δ on one or both strands (33) and/or as a function of location in the genome.
Acknowledgments
We thank Dmitry Gordenin and Zachary Pursell for thoughtful suggestions on this manuscript and Dinh Nguyen for assistance in DNA substrate preparation and sequence analysis of lacZ mutants.
The abbreviation used is
- pol δ
polymerase δ
Footnotes
This work was funded in part by the Intramural Research Program of the NIEHS, National Institutes of Health (to T. A. K.) and in part by National Institutes of Health Grant GM32431 (to P. M. J. B.).
References
- 1.Bebenek K, Kunkel TA. Adv Protein Chem. 2004;69:137–165. doi: 10.1016/S0065-3233(04)69005-X. [DOI] [PubMed] [Google Scholar]
- 2.Morrison A, Johnson AL, Johnston LH, Sugino A. EMBO J. 1993;12:1467–1473. doi: 10.1002/j.1460-2075.1993.tb05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pavlov YI, Frahm C, Nick McElhinny SA, Niimi A, Suzuki M, Kunkel TA. Curr Biol. 2006;16:202–207. doi: 10.1016/j.cub.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Goldsby RE, Hays LE, Chen X, Olmsted EA, Slayton WB, Spangrude GJ, Preston BD. Proc Natl Acad Sci U S A. 2002;99:15560–15565. doi: 10.1073/pnas.232340999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin YH, Ayyagari R, Resnick MA, Gordenin DA, Burgers PM. J Biol Chem. 2003;278:1626–1633. doi: 10.1074/jbc.M209803200. [DOI] [PubMed] [Google Scholar]
- 6.Garg P, Burgers P. Crit Rev Biochem Mol Biol. 2005;40:115–128. doi: 10.1080/10409230590935433. [DOI] [PubMed] [Google Scholar]
- 7.Kunkel TA, Bebenek K. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 8.Minnick DT, Bebenek K, Osheroff WP, Turner RM, Jr, Astatke M, Liu L, Kunkel TA, Joyce CM. J Biol Chem. 1999;274:3067–3075. doi: 10.1074/jbc.274.5.3067. [DOI] [PubMed] [Google Scholar]
- 9.Minnick DT, Liu L, Grindley ND, Kunkel TA, Joyce CM. Proc Natl Acad Sci U S A. 2002;99:1194–1199. doi: 10.1073/pnas.032457899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franklin MC, Wang J, Steitz TA. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 11.Pavlov YI, Shcherbakova PV, Kunkel TA. Genetics. 2001;159:47–64. doi: 10.1093/genetics/159.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reha-Krantz LJ, Nonay RL. J Biol Chem. 1994;269:5635–5643. [PubMed] [Google Scholar]
- 13.Beechem JM, Otto MR, Bloom LB, Eritja R, Reha-Krantz LJ, Goodman MF. Biochemistry. 1998;37:10144–10155. doi: 10.1021/bi980074b. [DOI] [PubMed] [Google Scholar]
- 14.Fidalgo da Silva E, Mandal SS, Reha-Krantz LJ. J Biol Chem. 2002;277:40640–40649. doi: 10.1074/jbc.M203315200. [DOI] [PubMed] [Google Scholar]
- 15.Niimi A, Limsirichaikul S, Yoshida S, Iwai S, Masutani C, Hanaoka F, Kool ET, Nishiyama Y, Suzuki M. Mol Cell Biol. 2004;24:2734–2746. doi: 10.1128/MCB.24.7.2734-2746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nick McElhinny SA, Pavlov YI, Kunkel TA. Cell Cycle. 2006;5:958–962. doi: 10.4161/cc.5.9.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Albertson TM, Preston BD. Curr Biol. 2006;16:209–211. doi: 10.1016/j.cub.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 18.Li L, Murphy KM, Kanevets U, Reha-Krantz LJ. Genetics. 2005;170:569–580. doi: 10.1534/genetics.104.040295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venkatesan RN, Hsu JJ, Lawrence NA, Preston BD, Loeb LA. J Biol Chem. 2006;281:4486–4494. doi: 10.1074/jbc.M510245200. [DOI] [PubMed] [Google Scholar]
- 20.Henricksen LA, Umbricht CB, Wold MS. J Biol Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- 21.Ayyagari R, Gomes XV, Gordenin DA, Burgers PM. J Biol Chem. 2003;278:1618–1625. doi: 10.1074/jbc.M209801200. [DOI] [PubMed] [Google Scholar]
- 22.Fortune JM, Stith CM, Kissling GE, Burgers PM, Kunkel TA. Nucleic Acids Res. 2006;34:4335–4341. doi: 10.1093/nar/gkl403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bebenek K, Kunkel TA. Methods Enzymol. 1995;262:217–232. doi: 10.1016/0076-6879(95)62020-6. [DOI] [PubMed] [Google Scholar]
- 24.Shcherbakova PV, Pavlov YI, Chilkova O, Rogozin IB, Johansson E, Kunkel TA. J Biol Chem. 2003;278:43770–43780. doi: 10.1074/jbc.M306893200. [DOI] [PubMed] [Google Scholar]
- 25.Ohashi E, Bebenek K, Matsuda T, Feaver WJ, Gerlach VL, Friedberg EC, Ohmori H, Kunkel TA. J Biol Chem. 2000;275:39678–39684. doi: 10.1074/jbc.M005309200. [DOI] [PubMed] [Google Scholar]
- 26.Zhong X, Garg P, Stith CM, Nick McElhinny SA, Kissling GE, Burgers PM, Kunkel TA. Nucleic Acids Res. 2006;34:4731–4742. doi: 10.1093/nar/gkl465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fortune JM, Pavlov YI, Welch CM, Johansson E, Burgers PM, Kunkel TA. J Biol Chem. 2005;280:29980–29987. doi: 10.1074/jbc.M505236200. [DOI] [PubMed] [Google Scholar]
- 28.Tran HT, Degtyareva NP, Koloteva NN, Sugino A, Masumoto H, Gordenin DA, Resnick MA. Mol Cell Biol. 1995;15:5607–5617. doi: 10.1128/mcb.15.10.5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. Mol Cell Biol. 1997;17:2859–2865. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Diaz M, Kunkel TA. Trends Biochem Sci. 2006;31:206–214. doi: 10.1016/j.tibs.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Johnson A, O’Donnell M. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 32.Pavlov YI, Newlon CS, Kunkel TA. Mol Cell. 2002;10:207–213. doi: 10.1016/s1097-2765(02)00567-1. [DOI] [PubMed] [Google Scholar]
- 33.Pavlov YI, Mian IM, Kunkel TA. Curr Biol. 2003;13:744–748. doi: 10.1016/s0960-9822(03)00284-7. [DOI] [PubMed] [Google Scholar]
- 34.Kraulis PJ. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 35.Nicholls A, Sharp KA, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 36.Merritt EA, Bacon DJ. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]