

Abstract
Protein based cellular therapeutics have been limited by getting the molecules into cells and the fact that many proteins require post-translational modifications for activation. Protein transduction domains (PTDs), including that from the HIV TAT protein (TAT), are small arginine rich peptides that carry molecules across the cell membrane. We have shown that the heat shock-related protein, HSP20 is a downstream mediator of cyclic nucleotide-dependent relaxation of vascular smooth muscle and is activated by phosphorylation. In this study, we co-expressed in E. coli the cDNAs encoding the catalytic subunit of protein kinase G and a TAT-HSP20 fusion protein composed of the TAT PTD (-YGRKKRRQRRR-) fused to the N-terminus of human HSP20. Immunoblot and HPLC-ESI-MS/MS analysis of the purified TAT-HSP20 demonstrated that it was phosphorylated at serine 40 (equivalent to serine 16 in wild-type human HSP20). This phosphorylated TAT-HSP20 was physiologically active in intact smooth muscles in that it inhibited 5-hydroxytryptamine-induced contractions by 57% ± 4.5. The recombinant phosphorylated protein also led to changes in actin cytoskeletal morphology in 3T3 cells. These results delineate strategies for the expression and activation of therapeutic molecules for intracellular protein based therapeutics.
Keywords: Protein kinase G (PKG), HSP20, phosphorylation, co-expression, protein transduction
Introduction
Smooth muscle relaxation can be induced by a variety of endogenous and pharmacologic agents such as nitric oxide (NO) through cyclic nucleotide signaling cascades. NO released from stimulated endothelial cells or by NO donors such as sodium nitroprusside (SNP), diffuses across cell membranes activating guanylyl cyclase, increasing intracellular cGMP concentrations, and activating cGMP-dependent protein kinase [1]. An alternate pathway for smooth muscle relaxation involves activation of adenylyl cyclase (by agonists such as forskolin), increasing intracellular cAMP concentrations, and activation of cAMP-dependent protein kinase [2].
One downstream target of both cyclic nucleotide signaling cascades is the heat shock-related protein 20, HSP20. HSP20 is highly and constitutively expressed in skeletal, cardiac, and smooth muscle and increases in the phosphorylation of HSP20 are associated with cyclic nucleotide-dependent relaxation of vascular smooth muscle [3,4]. Like other members of the heat shock protein family, HSP20 requires phosphorylation at a serine residue (S16) to become functionally active [5,6]. HSP20 is a specific substrate protein of PKA and PKG and accumulating evidence suggests HSP20 has several physiological and biochemical roles including controlling muscle tone, regulating cell motility, modulating actin filament dynamics and protection against intimal hyperplasia and ischemia/reperfusion injury [7–14]. Although the molecular mechanisms of action for HSP20 have not been determined fully, it is an actin-associated protein and we have shown that HSP20 peptide treatment leads to loss of actin stress fibers and disruption of focal adhesion complexes [9–11]. One possible mechanism for the changes in the actin cytoskeleton is that phosphorylated HSP20 contains a 14-3-3 binding motif. The actin depolymerizing protein, cofilin, has also been shown to bind to 14-3-3. Treatment with phosphopeptide analogues of HSP20 leads to a dissociation and activation of cofilin resulting in depolymerization of the actin cytoskeleton [10]. Collectively, these data suggest HSP20 may modulate cytokinetic processes via alterations in actin filament dynamics.
Recently, we used a direct approach to establish the role of phosphoHSP20 in smooth muscle relaxation. We transduced phosphoproteins and peptides into tissues using protein transduction domains (PTDs) and in doing so prevented smooth muscle contraction [12,13]. The use of PTDs as tools for intracellular delivery is a relatively new approach that is receiving increased attention as new methods for its implementation are developed and evidence for its efficacy is established. PTDs, first discovered independently by the Green and Frankel laboratories, are positively charged amino acid sequences facilitating the entry of proteins into cells through macropinocytotic mechanisms [15–17]. Our laboratory has successfully implemented the use of these sequences to transduce different proteins and peptides into numerous tissues and cells [8–14].
One limitation of previous approaches was the requirement for in vitro phosphorylation of expressed, purified protein prior to physiological testing. In view of the potential shortcomings of this method for large-scale preparations, an alternate phosphorylation scheme was undertaken. The bacterial co-expression of the catalytic subunit of cGMP-dependent protein kinase (cPKG) and a fusion protein composed of a transduction domain (TAT) and the PKG substrate, HSP20 (TAT-HSP20) was performed to determine if biologically active recombinant phospho-proteins could be produced. The results delineate possible strategies for the production of other proteins through co-expression of the enzyme responsible for post-translational modification.
Materials and methods
cDNA cloning and co-expression
A 1600 bp cDNA encoding the type I catalytic subunit of cGMP-dependent protein kinase from Bos taurus (encoded residues 334–670) was liberated from pBKS+ cPKG (gift from Dr. Thomas Lincoln, University of South Alabama) by NcoI-XhoI digestion and cloned into a NcoI-XhoI-digested shuttle vector pET29a yielding pET29a PKG. pACYC Duet PKG was constructed by ligating the BglII-XhoI insert of pET29a PKG and the backbone of BglII-XhoI-digested pACYC Duet (Stratagene). Construction of pET14b TAT-HSP20 has been described previously [13].
pACYC Duet PKG and pET14b TAT-HSP20 were co-transformed into the E. coli expression strain BL21(DE3) and a single colony resistant to 50 mg/L ampicillin and 30 mg/L chloramphenicol used to inoculate 400 mL of Luria Broth (LB) seed culture and grown overnight on a platform shaker (220 rpm at 37ºC). The following morning, 4 liters of LB was inoculated with the seed culture and the culture was induced with 2 mM isopropyl-β-D-thiopyranogalactoside (IPTG - Research Organics, Cleveland, OH) at an OD of 0.5. Induced cells were grown overnight (14hr) and the next morning harvested from media by centrifugation at 6000 × g for 15 min at 4ºC.
TAT-HSP20 purification and refolding
Frozen cells were thawed and thoroughly resuspended in 100 mL 1X TNE buffer (50 mM NaCl, 1 mM EDTA, and 500 mM Tris-HCl, pH 8.0, 10nM Caliculin A, 50μl Sigma Phosphatase Inhibitor Cocktail II, 50μl Sigma Protease Inhibitor Cocktail and 200 μg deoxyribonuclease) and incubated at 4°C for 30 min. After sonication on ice, the inclusion bodies containing recombinant protein were harvested by centrifugation (19,000 g, 10 min). Metal chelation affinity chromatography was used to purify recombinant TAT-pHSP20. Briefly, inclusion bodies were first resuspended in binding buffer (20 mM Na2HPO4, 0.5 M NaCl, 50 mM imidazole, pH 7.4, 8 M urea). The sample was then added to Ni2+-charged Chelating Sepharose Fast Flow (Pharmacia Biotech, Peapack, NJ) and incubated for 30 min at room temperature. The resin was then loaded in a water-chilled XK-26 column and washed extensively with binding buffer on an AKTÄ fast-performance liquid chromatography system (Pharmacia Biotech). Protein was refolded by using an overnight linear gradient of urea from 8 to 0 M and eluted with binding buffer containing 500 mM imidazole. The eluate was concentrated and the imidazole removed by repeatedly reducing eluate volume 50% using a stirred ultrafiltration cell (8200, Millipore, Bedford, MA) with a 10,000-nominal molecular weight limit filter under 75 p.s.i. nitrogen and bringing the volume back to 100% again with PSS (physiological salt solution – 140 mM NaCl, 5 mM KCl, 1.6 mM CaCl2, 1.2 mM MgCl2, 1.2 mM Na2HPO4, 5.6 mM glucose, 2 mM MOPS, and 0.02 mM EDTA, pH 7.4).
In vitro analysis of phosphorylation
One and two dimensional SDS-PAGE, immunoblot analyses and quantification were performed as described previously [13].
Immunoprecipitation
A 300 mg bacterial pellet from an induced culture expressing pACYC Duet PKG and pET14b TAT-HSP20 was frozen in liquid nitrogen then added to 300 μl of a lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM EGTA, 0.3% NP-40, 0.5% sodium deoxycholate and 0.01% Triton X-100) containing 0.3 μM aprotinin, 100 mM sodium fluoride, 10 mM benzamadine, 2 mM phenylmethylsulfonyl fluoride (PMSF), 100 uL Sigma Phosphatase Inhibitor Cocktail 1 (Sigma Chemical Co., St. Louis, MO; per 10 mLs), 100 uL Sigma Phosphatase Inhibitor Cocktail 2 (per 10 mLs), and 100 uL Sigma Protease Inhibitor Cocktail (per 10 mLs lysis buffer) added just before use. The sample was placed in a Disruptor Genie, (Scientific Industries, Bohemia, NY) for 15 min at 4 °C, centrifuged at 15,000 × g for 5 min and the supernatant removed. The supernatant was pre-cleared of non-specific cross-reacting antigens by batch (noncolumn) incubation with ant-rabbit IgGs conjugated to agarose (Invitrogen, Carlsbad, CA) for 1 h at 4 °C using end-over-end rotation. After removal of anti-rabbit IgG-agarose at 15,000 × g, the lysate was added to 700 μl of immunoprecipitation buffer and 5 μl of anti-HSP20 monoclonal antibodies (Upstate, Charlottesville, VA). The mixture was incubated overnight at 4°C with end-over-end rotation to purify HSP20 by immunoprecipitation (IP). Anti-HSP20 IgG complexes were captured using 40 μl of Protein A-conjugated agarose for 2h at 4°C using end-over-end rotation. Agarose beads were pelleted and washed three times with IP buffer. Immunoprecipitated protein were eluted from the agarose with 100 uL of UDC buffer (8M urea, 2% CHAPS, 10 mM DTT) and subjected to SDS-PAGE. A dominant band of approximately 20 kDa was used for mass spectrometric identification.
In-gel digestion
The gel portion containing HSP20 were excised, placed in a 0.6-ml polypropylene tube, washed with 400 μl of 40 mM NH4HCO3, destained twice with 300 μl of 50% acetonitrile (ACN) in 40 mM NH4HCO3 and dehydrated with 100% ACN for 5 minutes. After removal of ACN by aspiration, the gel pieces were dried in a vacuum centrifuge at 60 °C for 5 minutes. The gel pieces were then reduced in 100 μL of 10 mM dithiothreitol (DTT) for 1 hr at 55°C and alkylated in 100 μL of 50 mM of iodoacetamide (IDA) at room temperature for 45 min in the dark. After two wash steps with ammonium bicarbonate/ACN, the gel was dehydrated with ACN and dried in a vacuum centrifuge at 60°C for 20 minutes. Trypsin (250 ng; Sigma Chemical Co.) in 20 μl of 40 mM H4HCO3 was added and the samples were maintained at 4°C for 15 minutes prior to the addition of 50 μl of 40 mM NH4HCO3. The digestion was allowed to proceed at 37ºC overnight and was terminated by addition of 10 μl 5% formic acid (FA). After further incubation at 37°C for 30 minutes and centrifugation for 1 minute, each supernatant was transferred to a clean polypropylene tube. The extraction procedure was repeated using 40 μl of 5% FA, and the two extracts were combined. The resulting peptide mixtures were purified by solid-phase extraction (C18 ZipTip, Millipore, Billerica, MA) after sample loading in 0.05% heptafluorobutyric acid:5% FA (v/v) and elution with 50 % ACN:1% FA (v/v). The samples were dried by vacuum centrifugation and 4 μl of 0.1% FA:2%ACN (v/v) was added.
Mass spectrometry
HPLC-ESI-MS/MSn was performed on a Thermo Finnigan (San Jose, CA) LTQ-FT fitted with a PicoView™ nanospray source (New Objective, Woburn, MA). On-line HPLC was performed using a Michrom BioResources Paradigm MS4 micro HPLC (Alburn, CA) with a PicoFrit™ column (New Objective, Woburn, MA, 75 μm i.d., packed with ProteoPep™ II C18 material, 300 Å); mobile phase, linear gradient of 2 to 27% ACN in 0.1 % FA in 45 minutes, a hold of 5 minutes at 27% ACN, followed by a step to 50% ACN, hold 5 minutes and then a step to 80%; flow rate, 250 nl/min.
A “top-10” data-dependent MS/MS analysis was performed (acquisition of a full scan spectrum followed by collision-induced dissociation (CID) mass spectra of the 10 most abundant ions in the survey scan) to identify HSP20 peptides. The survey scan was acquired using the FT mass analyzer, which offers high mass accuracy and resolution. For localization of sites of phosphorylation, we used a scan protocol of 1 survey scan (FT), followed by 5 targeted MS/MS and MS/MS/MS scans (CID spectra of specified m/z values were acquired using the LTQ mass analyzer).
All uninterpreted tandem MS data were searched using Mascot (Matrix Science, London, UK) with 5 parts per million (ppm) error tolerance for precursor ion masses (measured in FT) and 0.5 Da error tolerance for product ion masses (measured in LTQ). Search parameters allowed for oxidation of methionine, N-terminal acetylation, carobyamidomethylation of cysteine, and phosphorylation of serine, threonine, and tyrosine. Assignments of the phosphopeptides were confirmed by manual comparison of the tandem mass spectra with the predicted fragmentation generated in silico by the MS-Product component of ProteinProspector (http://prospector.ucsf.edu).
Physiological activity in intact tissues
All animal procedures were performed under the guidance of the Institutional Animal Care and Use Committee (IACUC) using an approved protocol. Briefly, 400g Sprague-Dawley rats were sacrificed via CO2 asphyxiation and the aorta of each rat was dissected out, cleaned of excess fat and connective tissue, stored in PSS buffer and 1 hr later cut transversely into rings that were 1 mm in width. Arterial tissues were tied to 4.0 silk, fixed at one end to a stainless steel wire, and attached to a Kent Scientific (Litchfield, CT) force transducer (TRN001) interfaced with a Data Translation analog-to-digital board, DT2801 (Data Translation, Marlboro, MA). Data were acquired with Powerlab software (AD Instruments, Colorado Springs, CO). The rings were repeatedly contracted with 110 mM KCl (with equimolar replacement of NaCl in buffer), and the length was progressively adjusted until maximal tension was obtained. The tissue was washed with PSS buffer and equilibrated for at least 30 min between contractions. Contractile responses to serotonin (5-hydroxytryptamine, 5-HT) were subsequently tested by treating the tissue with increasing doses (0.01, 0.1, and 1μM) of 5-HT (not shown). Tissues were rinsed and treated with 0.5 μM 5-HT (submaximal dose) to determine pretreatment contraction. Any tissue failing to contract was considered nonviable and was not used in further experiments. After washing tissues and allowing a return to baseline tension, TAT-pHSP20 (10μM) protein was added to the bath and the ring incubated for 10 min. The effectiveness of the pretreatment was assessed by addition of 0.5 μM 5-HT, and the contractile response recorded. Percent contraction was calculated relative to force generated with 5-HT prior to pretreatment with TAT-pHSP20. Protein extracts of vascular rings were obtained by flash freezing tissues still connected to the transducer, then vortexing frozen vascular rings in UDC (9M urea, 2% CHAPS, 10 mM DTT) sample buffer for 1 hr. For treatments with synthesized PTD-HSP20 peptides, tissues were contracted with 0.5 μM 5-HT (submaximal dose) followed by increasing doses of peptide (100 μM, 500 μM and 1 mM, respectively).
NIH 3T3 cell culture and actin staining
NIH 3T3 cells (ATCC CRL 1658) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) BCS, penicillin (100 U/ml), and 100μg/ml streptomycin (Gibco/BRL), L-glutamine (4 mM) in a CO2 incubator (5% CO2) at 37 °C. Serum free media supplemented with insulin-transferrin sodium selenite (5 ng/ml) and HEPES (pH 7.55 - 25mM) was used to quiesce and potentiate cells for stress fiber formation.
To visualize the effect of the TAT-pHSP20 on the actin cytoskeleton, serum-starved cells seeded on coverslips in 6-well dishes (50-70% confluent) were rinsed briefly with phosphate buffered saline (PBS) then treated with recombinant TAT-pHSP20 (10 μM) in PSS for 1 h at 37°C. Following this incubation, cells were washed in PBS, fixed in 4% formaldehyde (w/v) for 1hr at RT and permeabilized with 0.1% Triton X-100 for 20 min. Washed cells were subsequently incubated with Alexa 568-conjugated phalloidin (0.2 U/mL) and DAPI (1 μg/mL) for 1 hr at RT, washed with PBS then viewed using a Zeiss Axiovert 200M fluorescent microscope (Carl Zeiss, Inc., Thornwood, NY, USA) equipped with DAPI and Alexa 568 excitation/emission filter sets. Digital micrographs were acquired using an Axiocam MRM high-resolution camera then adjusted for contrast and assembled into figures using Adobe Photoshop (Mountain View, CA).
Results
Co-expression of cPKG and TAT-HSP20
The plasmids pET14b-TAT-HSP20 and pACYC-Duet cPKG were constructed as described in Materials and Methods, sequenced and the proteins expressed in the E. coli strain BL21(DE3), predominantly in insoluble inclusion body forms. Both proteins were expressed at approximately 20 mg/L in four liter induction experiments. Fig. 1 shows aligned regions of interest from immunoblots generated by probing bacterial lysates before (lanes 1, 3 and 5) or after protein induction with 2 mM IPTG (lanes 2, 4 and 6). Blots were probed with monoclonal antibodies raised against human HSP20 (top), anti-PKG polyclonal IgGs (middle), and anti-phospho-HSP20 specific IgGs (bottom). As judged by SDS-PAGE, TAT-HSP20 migrated with a molecular weight of ~25 kDa while cPKG migrated at approximately 39 kDa. Antibody detections of bacterial lysates expressing the TAT-HSP20 cDNA alone with or without IPTG induction (lanes 1 and 2, respectively) revealed an immunoreactive polypeptide that was of the molecular weight predicted for TAT-HSP20 [13]. Anti-PKG antibodies or anti-phospho-HSP20 specific IgGs were not reactive. Bacterial lysates containing the cPKG cDNA alone (lanes 3 and 4) revealed an anti-PKG reactive band only in the sample induced with IPTG (lane 4). Immunoblots performed on bacterial samples transformed with both pET14b-TAT-HSP20 and pACYC-Duet cPKG plasmids before and after IPTG induction (lanes 5 and 6, respectively) showed immunoreactive patterns with both anti-HSP20 and anti-PKG antibodies similar to those observed in lanes 1–4. In addition, a prominent polypeptide band immunoreactive with anti-phospho-HSP20 specific IgGs (lane 6 – bottom panel) appeared that was absent in lanes 1–5. Pellets from four liter expression experiments where TAT-HSP20 cDNA was expressed alone or co-expressed with cPKG in the presence of IPTG (~20 g each) were used subsequently for TAT-HSP20 protein purification and physiological testing. Recombinant proteins from both E. coli strains were purified independently by urea-solubilization and metal chelation affinity binding then refolded by the gradual removal of urea during a linear urea-physiological salt solution (PSS) gradient. Proteins were dialyzed against PSS to remove imidazole and concentrated to ~ 10 μM.
Figure 1.
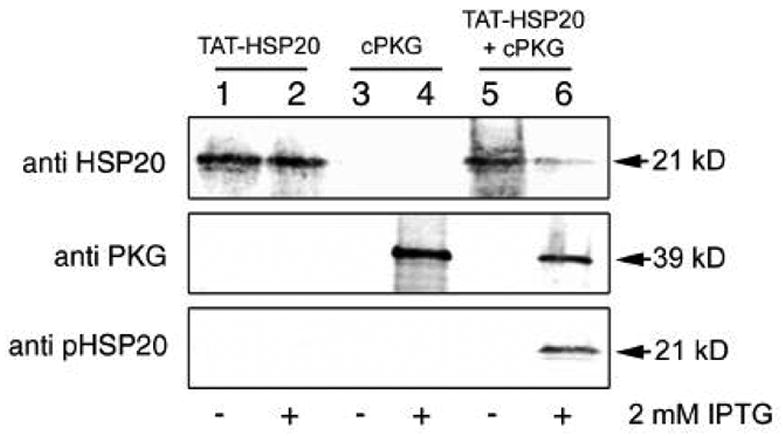
TAT-pHSP20 is expressed and phosphorylated in E. coli. Representative immunoblot analyses of BL21(DE3) bacterial extracts taken from a large scale (4L) co-expression experiment. Extracts were generated before (-) or after (+) induction with 2 mM IPTG and 20 μg protein was loaded per well. Lanes 1 and 2 - pET14b TAT-HSP20 vector only; lanes 3 and 4 - pACYC Duet PKG C subunit vector only; lanes 5 and 6 - pET14b TAT-HSP20 and pACYC Duet PKG C subunit vectors together. Blots were probed with the antibodies indicated on the left.
Determination of TAT-HSP20 phosphorylation
To determine the extent to which TAT-pHSP20 was phosphorylated by co-expression with cPKG, purified phosphorylated and non-phosphorylated protein samples were separated by isoelectric focusing followed by SDS-PAGE, blotted to nitrocellulose and probed with anti-HSP20 antibodies (Fig. 2). A control protein of weight and composition similar to TAT-HSP20 (PTD-HSP27) did not react with anti-HSP20 antibodies demonstrating antibody specificity (Fig. 2A). Fig. 2B, immunoblots of TAT-HSP20 purified from E. coli revealed a single spot at a pI of ~9.0 and an electrophoretic mobility of ~ 25 kD. Sequences comprising the amino-terminal PTD are basic in nature, can alter electrophoretic mobility and contribute to a higher pI (~9.0) compared to HSP20 without a PTD (pI ~ 5.9, mass ~17kD) [6,13]. In contrast, immunoblots of TAT-pHSP20 purified from E. coli co-expressing cPKG revealed two immunoreactive proteins. One spot exhibited a pI of ~9.0 and a mass of 25 kD while the second spot was more acidic by about 0.4 pI units. Sensitive fluorescence-based quantitative analyses of these spots revealed that the more acidic form had a relative intensity of 3.46 (arbitrary units) while the intensity of the more basic spot was 3.70 indicating that the more acidic or phosphorylated spot represented 48% of the total anti-HSP20 immunoreactive protein population isolated from E. coli. The relative differences between the pIs of the two proteins observed in Fig. 2C are consistent with reports published previously on HSP20 phosphorylation [6, 13]. These results coupled with the phospho-HSP20-specific detections in Fig. 1 suggest that a distinct portion of TAT-HSP20 is phosphorylated in E. coli when co-expressed with cPKG.
Figure 2.

TAT-HSP20 co-expressed with cPKG yields two anti-HSP20 immunoreactive polypeptides. Two-dimensional gel electrophoresis and Western blot analyses of purified recombinant proteins, PTD-HSP27 (A), TAT-HSP20 (B), TAT-pHSP20 (C). 75 μg of recombinant protein were subjected to isoelectric focusing (linear pH gradient range 3–10, 7 cm length) then separated in the second dimension (4-20% polyacrylamide). Proteins were transferred onto PVDF membranes and immunoblots were incubated with mouse anti-HSP20 and IRDye 700DX conjugated affinity purified goat anti-mouse secondary antibodies. Arrow indicates the phosphorylated TAT-HSP20 isoform.
We employed high-performance liquid chromatography electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS) to determine the site within TAT-HSP20 phosphorylated by cPKG. This valuable tool is widely recognized as a gold-standard in characterizing post-translational modifications [18,19]. From the tryptic digests of TAT-HSP20 immunoprecipitates, we were able to obtain 100% sequence coverage of TAT-HSP20 using HPLC-ESI-MS/MS followed by MASCOT database searching. Moreover, one phosphopeptide was identified. Fig. 3 shows the CID spectrum of TAT-HSP2038–51 (RApSAPLPGLSAPGR). The presence of unique ions b9, y5, y8, y10 and y11 clearly indicates that Ser40 (corresponding to Ser16 in wild-type human HSP20) is the phosphorylation site. Furthermore, when [M+2H-98]2+ (m/z 666.4) was used as a precursor for an MS3 experiment, unique ions interpreted as y7, y10 and y11 were detected, appropriate for phosphorylation at Ser40 and not Ser47.
Figure 3.
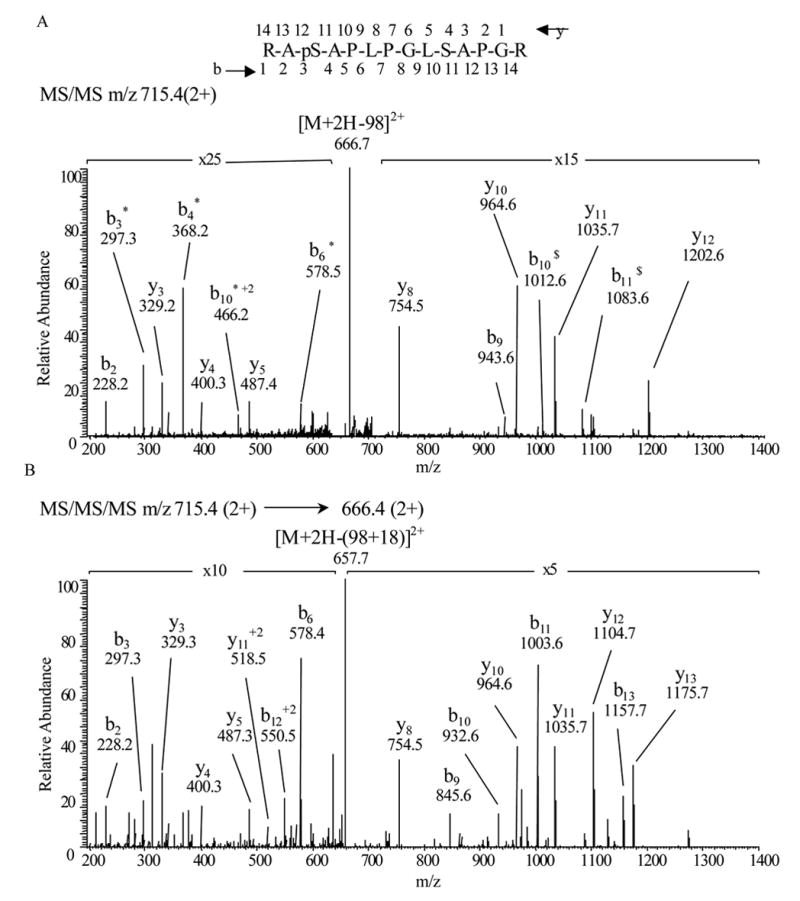
(A) Collision-ionization-disassociation mass spectra of RApSAPLPGLSAPGR (residues 38–51 of TAT- HSP20, corresponding to residues 14–27 in wild-type human HSP20) observed in a tryptic digest of TAT-HSP20 isolated from bacterially-derived human TAT-HSP20 co-expressed with cPKG. MS/MS m/z 715.4, 2+. * indicates loss of H3PO4 (98 u) and $ indicates loss of H20 from the indicated fragment (18u). (B) MS/MS/MS, m/z spectrum 715.4 (2+) → 666.4(2+).
Physiological activity of TAT-pHSP20 analogs
The physiological activity of TAT-pHSP20 from bacterial co-expression experiments was assessed in muscle bath experiments. Rings of male rat aorta were washed, equilibrated with PSS and treated with sub-maximal doses of 5-HT to determine tissue responsiveness. After washing in PSS and letting tension return to baseline levels, rings were incubated in PSS buffer alone (control – Fig. 4A) or in PSS buffer containing TAT-pHSP20 (10μM – Fig. 4B) for 10 min. Tissues were subsequently challenged with the same sub-maximal doses of 5-HT (0.5μM) and the contractile responses recorded. There were no significant differences in the magnitude of contraction in response to serial treatments with 5HT (Fig. 4A). In contrast, pre-incubation with TAT-pHSP20 inhibited 5-HT-induced contraction by 57 ± 4.5%. Similar results were observed with pig coronary artery where pre-incubation with TAT-pHSP20 (10 μM) inhibited 5-HT contractions by 60% (data not shown). These results are similar to the inhibition observed using TAT-HSP20 that was phosphorylated in vitro where pre-incubation with TAT-pHSP20 inhibited contraction of human umbilical artery by 81.99 ± 6.83% [13].
Figure 4.
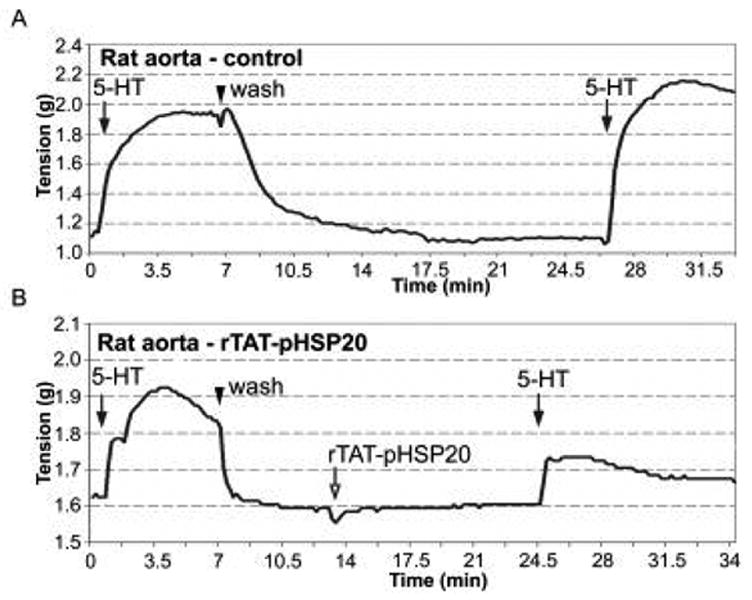
Purified recombinant TAT-pHSP20 (phosphorylated in bacteria) inhibits smooth muscle contraction. Representative force tracing are shown for contractile responses generated by male rat aorta rings. Tissues were pre-contracted with 5 HT (0.5 μM) followed by treatment with PSS buffer alone (A) or in vivo phosphorylated TAT-pHSP20 protein (10 μM) (open arrow) (B). Following pre-treatment with TAT-pHSP20, rings were challenged with 0.5 μM 5HT and responses recorded. Quantified contractile responses (% contraction) revealed a 57% inhibition of 5HT-induced contraction. Similar results were obtained using pig coronary artery where a 60% inhibition of contraction was observed (data not shown).
The in vivo efficacy of molecules with putative therapeutic application is often established using peptide mimetics. Peptide mimetics are advantageous in determining full-length protein activity as they have fewer domains available for binding/interaction, are usually easier to synthesize in large quantities, and less susceptible to proteolytic activity. A synthesized HSP20 analog, PTD-pHSP20 composed of an optimized protein transduction domain, fused to sequences encoding the active portion (phosphorylation site, serine 16 of HSP20) [6] has been shown to relax a wide array of smooth muscles including bovine carotid artery, porcine coronary artery, human saphenous vein and human umbilical artery [8,10,12,13]. To compare the magnitude of the physiological responses of the entire recombinant molecule to the peptide mimetics, rings of rat aorta contracted with 5-HT (0.5 μM) displayed steady contractions that were sustained for long periods of time (> 20 min), whereas treatment of aorta rings with increasing amounts of PTD-pHSP20 (100 μM, 500 μM and 1 mM respectively) led to a dose-dependent decrease in contraction that was maximal at a PTD-pHSP20 concentration of 1 mM (data not shown). Collectively, the data from physiological experiments indicate that the full-length, transducible, recombinant, and phosphorylated HSP20 (TAT-pHSP20) obtained by co-expressing TAT-HSP20 together with cPKG in E. coli, diminished rat aortic contractions at concentrations 100 times less than a peptide control (10 μM vs. 1 mM).
Effect of TAT-pHSP20 on actin stress fibers
NIH 3T3 cells were used to further delineate physiological activities of the bacterially-derived HSP20 protein. 3T3 cells represent a model widely used for studies of actin reorganization and there is a rich history available in this regard. Serum starved cells were treated with either physiological salt solution (PSS) or TAT-pHPS20 (10 μM) in PSS for 1 hr. Cells were then washed with PBS and processed for fluorescence microscopy. Fig. 5A shows fixed and permeabilized control cells with the actin cytoskeleton (red – Alexa 568-phalloidin) and nuclei (blue – DAPI) labeled. Actin filaments are organized evenly throughout each cell and prominent stress fibers are visible. In contrast, the micrograph in Fig. 5B shows TAT-pHSP20-treated cells exhibiting actin reorganizations, i.e., cells clearly lack central actin stress fibers and exhibit cortical actin staining at the cell periphery. Considering that cytoskeletal rearrangements are involved in many cytokinetic processes, it is possible that the physiology results observed in Fig.2 are mediated by thin filament altering mechanisms.
Figure 5.
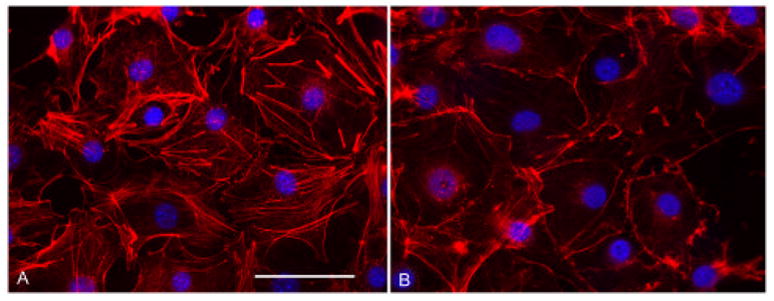
Purified recombinant TAT-pHSP20 induces actin rearrangements in 3T3 cells. Immunofluorescence microscopic images of NIH 3T3 cells serum starved for 24 hrs – treated with physiological salt solution (PSS) (A) or treated with 10 μM recombinant TAT-pHSP20 recombinant protein in PSS (B). Cells were washed, fixed, detergent permeabilized, then probed with Alexa568-phalloidin and DAPI. Cells treated with TAT-pHSP20 lack stress fibers and central actin filaments. Scale bar = 50 μM.
Discussion
Several protein kinases and their subunits have been expressed in E. coli in both active and inactive forms, including cGMP- and cAMP dependent protein kinases, myosin light chain kinase, and casein kinase [20–29]. Full length bovine cGMP-dependent protein kinase has been expressed previously in E. coli using both T7 RNA polymerase and tac+ promoters [21]. Both experiments resulted in predominantly insoluble and catalytically inactive kinases with a small fraction of soluble PKG able to bind cGMP but not phosphorylate a GRTGRRNSI peptide substrate. Despite these findings, the catalytic subunit of bovine PKG (cPKG) was selected as the kinase of choice for this study as it is well able to autophosphorylate itself, it has been shown to phosphorylate HSP20 in vitro and in vivo, and it has been expressed successfully in E. coli before [5,13,30,31]. Furthermore, this sequence expressed in and partially purified from baculovirus-infected Sf9 cells generated a protein capable of phosphorylating IP3 receptor peptide and histone substrates [32].
In these studies, at least partial activation of this cPKG was achieved in E. coli, when another cDNA (HSP20) was coordinately expressed. The identity of the coordinately expressed substrate protein with kinase activity was established by multiple criteria. First, bacterial extracts from cells co-expressing TAT-HSP20 and cPKG as well as refolded TAT-pHSP20 purified under denaturing conditions revealed proteins that were immunoreactive to anti-phospho-HSP20 antibodies (Fig. 1). Secondly, two-dimensional gel electrophoresis of TAT-HSP20 protein obtained from cells co-expressing cPKG, but not TAT-HSP20 alone, resulted in the appearance of a second, more acidic, anti-HSP20-immunoreactive spot consistent with TAT-HSP20 phosphorylation (Fig. 2). Third, HPLC-ESI-MS/MSn analyses revealed the presence of phosphorylation at Ser40 within a peptide (amino acids 38-51, corresponding to Ser16 in wild-type human HSP20) from TAT-pHSP20 (Fig. 3). Fourth, biological assays wherein TAT-pHSP20 was examined for its effects in inhibiting smooth muscle contraction revealed tissue responses indicative of phosphorylation (Fig. 4 and 5). Finally, cultured cells pretreated with TAT-pHSP20 displayed actin cytoskeleton rearrangements consistent with the activity reported for pHSP20 (Fig. 5) [33]. Collectively, these results support the notion that at least a portion of cPKG is catalytically active when expressed in E. coli and can phosphorylate a co-expressed substrate protein, HSP20. In addition, with the use of a transduction domain fused to the substrate protein, these studies are the first to demonstrate that an active (phosphorylated) protein can be generated which has both morphologic and physioloigic activity in cultured cells and intact tissues.
Our findings suggesting that a functionally active PKG catalytic subunit can be expressed in E. coli contrast previous reports [21,30]. In the former report [19], the full length PKG encoding both catalytic and regulatory subunits was expressed and in the latter [30] a cPKG (residues 335-670)-maltose binding protein (MBP) fusion was expressed. Although the fusion protein was soluble no activity could be detected. Proteolytic cleavage of the MBP fragment with Factor Xa yielded a spontaneously aggregating catalytic subunit for which activity was not reported. It seems likely that the secondary structures imparted by the attached MBP or the presence of a regulatory subunit abrogated detectable activity using the assays described. Our encouraging results suggest that bacteria may possess a rudimentary chaperone system potentiating activation and productive folding of at least a portion of cPKG.
Substitution of a negatively charged amino acid can mimic the conformational effects of phosphorylation on some proteins, particularly when the major function of phosphorylation is to add negative charge [13]. The functionality of phosphatase-resistant mimics is diminished however, when residue substitutions lead to protein instability or when the geometric positioning of the aspartyl or glutamyl sidechains is incompatible with the interacting protein counterpart. In the case of HSP20, the molecular structure has not yet been elucidated but a transducible phosphomimetic of HSP20 peptide bearing an S16E mutation inhibited 5-HT induced contractions of umbilical artery smooth muscle by 50% as opposed to the 80% inhibition observed with the S16 peptide counterpart [13]. The biological activity of constitutively active full-length mutants (S16E and S16D) mutants should lend further insight into the role of HSP20 phosphorylation.
The observation that treatment of serum-starved cultured cells with TAT-pHSP20 resulted in the destruction of actin stress fibers is congruent with previously published studies on heat shock proteins and the actin cytoskeleton [8–10,33–35]. In general, the presence of stress fibers is associated with a proliferative response whereas the absence of stress fibers is associated with a quiescent phenotype [36]. Recent data from our laboratory and others suggests that thin filament stability and dynamics are regulated by competition between molecules stabilizing actin filaments (HSP27) and the severing/depolymerizing activities of proteins such as cofilin. Interestingly, cofilin, HSP27 and HSP20 all possess binding motifs for the scaffolding protein 14-3-3. 14-3-3 proteins comprise a highly conserved family of dimeric and cytosolic proteins that bind other proteins in a phosphorylation-dependent manner and thereby modulate their activity or prevent interaction with other proteins. Recent observations from our group have shown that the intracellular receptor for the peptide analog of HSP20 (PTD-pHSP20) is the scaffolding protein 14-3-3 [10]. Consequently, we expected similarities in the cellular responses to full-length TAT-pHSP20 (described here) and the peptide mimics of HSP20. After either treatment, stress fibers were markedly reduced with most actin redistributed to the cell periphery [Fig. 5B; [10]. These data suggest that phosphorylated HSP20 may modulate actin cytoskeletal dynamics by competing with phosphorylated cofilin for binding to 14-3-3. Once released, cofilin is then dephosphorylated, activated and available to sever actin filaments. Mass spectrometry-based proteomic analyses of 14-3-3 and HSP20 protein complexes from tissues in different physiological conditions should greatly increase our understanding of the functional consequences of phosphorylation in cytoskeletal reorganization.
The substantial differences in effective concentration required to induce relaxation for transducible recombinant HSP20 fusion proteins compared to transducible HSP20 peptides (1μM vs. 10μM, respectively) has been observed previously [13]. This observation is not altogether surprising given the greater conformational structure and additional interactive binding sites present in the entire molecule. The approach of generating recombinant molecules linked to a transduction domain allows for assessment of the biological function of the protein, with or without modifications, prior to generating and optimizing peptide mimetics. The large scale expression and purification of proteins in bacteria is a cost effective and desirable means of protein production primarily when post-translational modifications of the resultant product are not required. The results reported here lend credence to the approach of activating proteins in bacteria through co-expression of kinase(s) when alternate means such as in vitro phosphorylation or site-directed mutagenesis introducing acidic residues mimicking the effects of phosphorylation are not feasible. These studies also confirm that the addition of a transduction domain to the candidate protein allow entry of the active protein into tissues. This approach of co-expressing a substrate protein fused to a transduction domain with the relevant kinase provides a unique opportunity to generate protein based therapeutics and to better elucidate the biologic functions of specific proteins.
Table I.
Summary of purification scheme used to purify phosphorylated TAT-HSP20
| Step 1 | Inclusion body isolation |
| Step 2 | Inclusion body solubilization in buffer containing 8M urea |
| Step 3 | Immobilized metal affinity chromatography |
| Step 4 | Overnight resin wash using 8M to 0M urea gradient |
| Step 5 | Elution using imidazole |
| Step 6 | Imidazole removal via stepwise dialysis against physiological saline solution |
Acknowledgments
We thank Dr. Thomas Lincoln and the members of his group at the University of South Alabama for the PKG catalytic subunit cDNA clone and technical expertise. This work was supported by the National Institute of Health Grants RO1HL58027-01 to C. Brophy and R01DK47936-11 to L. Mandarino.
Abbreviations
- cPKG
catalytic subunit of cGMP dependent protein kinase
- HSP20
heat shock related protein 20 kD
- PTD
protein transduction domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lincoln TM, Komalavilas P, Boerth NJ, MacMillan-Crow LA, Cornwell TL. cGMP signaling through cAMP- and cGMP-dependent protein kinases. Adv Pharmacol. 1995;34:305–322. doi: 10.1016/s1054-3589(08)61094-7. [DOI] [PubMed] [Google Scholar]
- 2.Murray KJ. Cyclic AMP and mechanisms of vasodilation. Pharmacol Ther. 1990;47:329–45. doi: 10.1016/0163-7258(90)90060-f. [DOI] [PubMed] [Google Scholar]
- 3.Rembold CM, Foster DB, Strauss JD, Wingard CJ, Eyk JE. cGMP-mediated phosphorylation of heat shock protein 20 may cause smooth muscle relaxation without myosin light chain dephosphorylation in swine carotid artery. J Physiol. 2000;524(Pt 3):865–78. doi: 10.1111/j.1469-7793.2000.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kato K, Goto G, Inaguma Y, Hasegawa K, Morishita R, Asano T. Purification and characterization of a 20-kDa protein that is highly homologous to alphaB-crystallin. J Biol Chem. 1994;269:15302–15309. [PubMed] [Google Scholar]
- 5.Beall AC, Kato K, Goldenring JR, Rasmussen H, Brophy CM. Cyclic nucleotide-dependent vasorelaxation is associated with the phosphorylation of a small heat shock-related protein. J Biol Chem. 1997;272:11283–11287. doi: 10.1074/jbc.272.17.11283. [DOI] [PubMed] [Google Scholar]
- 6.Beall A, Bagwell D, Woodrum D, Stoming TA, Kato K, Suzuki A, Rasmussen H, Brophy CM. The small heat shock-related protein, HSP20, is phosphorylated on serine 16 during cyclic nucleotide-dependent relaxation. J Biol Chem. 1999;274:11344–51. doi: 10.1074/jbc.274.16.11344. [DOI] [PubMed] [Google Scholar]
- 7.Fan G, Ren X, Qian J, Nicolaou QYP, Wang Y, Jones W, Chu G, Kranias E. A Novel Cardioprotective Role of a Small Heat-Shock Protein, Hsp20, Against Ischemia/Reperfusion Injury. Circulation. 2005;111:1792–9. doi: 10.1161/01.CIR.0000160851.41872.C6. [DOI] [PubMed] [Google Scholar]
- 8.Tessier DJ, et al. Transduction of peptide analogs of the small heat shock-related protein HSP20 inhibits intimal hyperplasia. J Vasc Surg. 2004;40:106–114. doi: 10.1016/j.jvs.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 9.Tessier DJ, Komalavilas P, Panitch A, Joshi L, Brophy CM. The small heat shock protein (HSP) 20 is dynamically associated with the actin cross-linking protein actinin. J Surg Res. 2003;111:152–7. doi: 10.1016/s0022-4804(03)00113-6. [DOI] [PubMed] [Google Scholar]
- 10.Dreiza CM, et al. Transducible heat shock protein 20 (HSP20) phosphopeptide alters cytoskeletal dynamics. FASEB J. 2005;19:261–3. doi: 10.1096/fj.04-2911fje. [DOI] [PubMed] [Google Scholar]
- 11.McLemore EC, et al. Transducible recombinant small heat shock-related protein, HSP20, inhibits vasospasm and platelet aggregation. Surgery. 2004;136:573–578. doi: 10.1016/j.surg.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 12.Flynn CR, et al. Transduction of biologically active motifs of the small heat shock- related protein, HSP20, leads to relaxation of vascular smooth muscle. FASEB J. 2003;10:1358–60. doi: 10.1096/fj.02-1028fje. [DOI] [PubMed] [Google Scholar]
- 13.Flynn CR, Brophy CM, Furnish EJ, Komalavilas P, Tessier D, Thresher J, Joshi L. Transduction of phosphorylated heat shock-related protein 20, HSP20, prevents vasospasm of human umbilical artery smooth muscle. J App Phys. 2005;98:1836–45. doi: 10.1152/japplphysiol.01043.2004. [DOI] [PubMed] [Google Scholar]
- 14.McLemore EC, Tessier DJ, Thresher J, Komalavilas P, Brophy CM. Role of the small heat shock proteins in regulating vascular smooth muscle tone. J Am Coll Surg. 2005;201:30–6. doi: 10.1016/j.jamcollsurg.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 15.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–93. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 16.Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–88. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 17.Ho A, Schwarze SR, Mermelstein SJ, Waksman G, Dowdy SF. Synthetic protein transduction domains: enhanced transduction potential in vitro and in vivo. Cancer Res. 2001;61:474–7. [PubMed] [Google Scholar]
- 18.Mann M, Ong SE, Gronborg M, Steen H, Jensen ON, Pandey A. Analysis of Protein Phosphorylation Using Mass Spectrometry Deciphering the phosphoproteome. Trends Biotechnol. 2002;20:261–268. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- 19.Jensen ON. Modification-Specific Proteomics Characterization of Post-Translational Modifications by Mass Spectrometry. Curr Opin Chem Biol. 2004;8:33–41. doi: 10.1016/j.cbpa.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 20.Bagchi D, Kemp BE, Means A. Myosin light chain kinase structure function analysis using bacterial expression. J Biol Chem. 1989;264:15843–9. [PubMed] [Google Scholar]
- 21.Feil R, Bigl M, Ruth P, Hormann F. Expression of cGMP-dependent protein kinase in Escherichia coli. Mol Cell Biochem. 1993;127:71–80. doi: 10.1007/978-1-4615-2600-1_6. [DOI] [PubMed] [Google Scholar]
- 22.Seeliger M, Young M, Henderson MN, Pellicena P, King D, Falick A, Kuriyan J. High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein Sci. 2005;14:3135–39. doi: 10.1110/ps.051750905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsui T, Tanihara K, Date T. Expression of unphosphorylated form of human double-stranded RNA-activated protein kinase in Escherichia coli. Biochem Biophys Res Commun. 2001;284:798–807. doi: 10.1006/bbrc.2001.5039. [DOI] [PubMed] [Google Scholar]
- 24.Shi Y, Brown E, Walsh C. Expression of recombinant human casein kinase II and recombinant heat shock protein 90 in Escherichia coli and characterization of their interactions. Proc Natl Acad Sci USA. 1994;91:2767–2771. doi: 10.1073/pnas.91.7.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wild N, Herberg F, Hofmann F, Dostmann W. Expression of a chimeric cGMP-sensitive regulatory subunit of the cAMP-dependent protein kinase type I. FEBS Lett. 1995;374:356–62. doi: 10.1016/0014-5793(95)01146-6. [DOI] [PubMed] [Google Scholar]
- 26.Girod A, Kinzel V, Bossemeyer D. In vivo activation of recombinant cAPK catalytic subunit active site mutants by coexpression of the wild-type enzyme, evidence for intermolecular cotranslational phosphorylation. FEBS Lett. 1996;391:121–25. doi: 10.1016/0014-5793(96)00717-x. [DOI] [PubMed] [Google Scholar]
- 27.Slice L, Taylor S. Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli. J Biol Chem. 1989;264:20940–20946. [PubMed] [Google Scholar]
- 28.Gosse ME, Padmanabhan A, Fleishmann RD, Gottesman MM. Expression of Chinese hamster cAMP-dependent protein kinase in Escherichia coli results in growth inhibition of bacterial cells: A model system for the rapid screening of mutant type I regulatory subunits. Proc Natl Acad Sci USA. 1993;90:8159–8163. doi: 10.1073/pnas.90.17.8159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herberg FW, Bell SM, Taylor SS. Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli: multiple isozymes reflect different phosphorylation states. Protein Eng. 1993;6:771–777. doi: 10.1093/protein/6.7.771. [DOI] [PubMed] [Google Scholar]
- 30.Dostmann W, Endres R, Hofmann F. Cloning and bacterial expression of the catalytic and regulatory subunits of cGMP-dependent protein kinast I alpha (cGK) Naunyn Schmiedebergs Arch Pharmacol. 1993;347:66. [Google Scholar]
- 31.Heil W, Landgraf W, Hofmann F. A catalytically active fragment of cGMP-dependent protein kinase. Occupation of its cGMP-binding sites does not affect its phosphotransferase activity. Eur J Biochem. 1987;168:117–21. doi: 10.1111/j.1432-1033.1987.tb13395.x. [DOI] [PubMed] [Google Scholar]
- 32.Boerth NJ, Lincoln TM. Expression of the catalytic domain of cyclic GMP-dependent protein kinase in a baculovirus system. FEBS Lett. 1994;342:255–260. doi: 10.1016/0014-5793(94)80512-1. [DOI] [PubMed] [Google Scholar]
- 33.Woodrum DA, Brophy CM, Wingard CJ, Beall A, Rasmussen H. Phosphorylation events associated with cyclic nucleotide-dependent inhibition of smooth muscle contraction. Am J Physiol. 1999;277:H931–9. doi: 10.1152/ajpheart.1999.277.3.H931. [DOI] [PubMed] [Google Scholar]
- 34.Woodrum D, Pipkin W, Tessier D, Komalavilas P, Brophy CM. Phosphorylation of the heat shock-related protein, HSP20, mediates cyclic nucleotide-dependent relaxation. J Vasc Surg. 2003;37:874–81. doi: 10.1067/mva.2003.153. [DOI] [PubMed] [Google Scholar]
- 35.Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol. 1995;15:505–516. doi: 10.1128/mcb.15.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramakers GJ, Moolenaar WH. Regulation of astrocyte morphology by RhoA and lysophosphatidic acid. Exp Cell Res. 1998;245:252–62. doi: 10.1006/excr.1998.4224. [DOI] [PubMed] [Google Scholar]