

Abstract
The S checkpoint response to ultraviolet radiation (UVC) that inhibits replicon initiation is dependent on the ATR and Chk1 kinases. Downstream effectors of this response, however, are not well characterized. Data reported here eliminated Cdc25A degradation as intrinsic components of the UVC-induced pathway of inhibition of replicon initiation in human cells. A sublethal dose of UVC (1 J/m2), which selectively inhibits replicon initiation by 50%, failed to reduce the amount of Cdc25A protein or decrease Cdk2/cyclin E kinase activity. Cdc25A degradation was observed after irradiation with cytotoxic fluences of UVC, suggesting that severe inhibition of DNA chain elongation and activation of the replication checkpoint might be responsible for the UVC-induced degradation of Cdc25A. Another proposed effector of the S checkpoint is the Cdc7/Dbf4 complex. Dbf4 interacted with Chk1 in vivo and was recognized as a substrate for Chk1-dependent phosphorylation in vitro. Flag-Dbf4 formed complexes with endogenous Cdc7 and this interaction was stable in UVC-irradiated HeLa cells. Over-expression of Flag- or Myc-tagged Dbf4 abrogated the S checkpoint response to UVC. These findings implicate a Dbf4-dependent kinase as a possible target of the ATR- and Chk1-dependent S checkpoint response to UVC.
Human cells continuously experience damage to DNA from both reactive cellular metabolites and environmental sources. If damaged DNA is not repaired before DNA replication, mutations and chromosomal aberrations might ensue. To maintain high fidelity replication of the genome, human cells have evolved a number of biochemical pathways that respond to perturbations in DNA structure and facilitate error-free replication of damaged DNA (1). The S checkpoint maintains genomic stability by transiently inhibiting the initiation of new replicons and thereby limiting the number of active replication forks that encounter DNA lesions. This transient inhibition of replicon initiation should provide more time for DNA repair pathways to recognize and eliminate DNA damage prior to replication.
Replicon initiation is a biochemically and genetically conserved process across eukaryotic phyla that requires the stepwise formation of pre-replication complexes (Pre-RC) at origins of DNA replication (2). The six-subunit origin recognition complex (ORC) binds to replication origins throughout the cell cycle and acts as a landing pad for Cdc6. ORC and Cdc6 collaborate with the chaperone protein Cdt1 to load the six subunit MCM complex into the developing Pre-RC. Mcm2–7 forms a hexameric structure, which is hypothesized to be the mammalian DNA helicase that unwinds duplex DNA to allow DNA polymerases to synthesize daughter strands. The ORC-Cdc6-Cdt-MCM pre-replication complex is regulated by two S phase kinase complexes, a cyclin-dependent kinase (CDK) and a Dbf4-dependent kinase (DDK). S phase CDKs phosphorylate Cdc6, which results in its degradation (yeast) or nuclear export (mammals) (3–8). Cdc7/Dbf4 phosphorylates several members of the MCM complex including Mcm2 (9–11). DDK-dependent phosphorylation is thought to activate the MCM helicase causing local unwinding of origin-specific DNA and the binding of the single-stranded DNA binding protein, RPA. CDK and DDK-dependent phosphorylations are required for the loading of Cdc45 (12–15). Cdc45 acts as a chaperone protein, which recruits the DNA polymerase α/DNA primase complex to origin DNA (13). TopBP1 and Mcm10 are also required for Cdc45 loading following DDK-dependent activation of the MCM helicase (16,17). Loading of the DNA polymerase α/DNA primase complex on RPA-coated template strands initiates bi-directional, semi-conservative DNA replication.
The S checkpoint pathway that responds to IR-induced DNA damage and inhibits replicon initiation in S phase cells has been intensively investigated (18). Upon recognition of IR-induced DNA strand breaks by the Mre11/Rad50/Nbs1 (MRN) complex, the sensor kinase ATM is activated by intramolecular autophosphorylation (19). Activated ATM inhibits the initiation of new replicons through a complex signaling pathway that involves a number of mediators of checkpoint signaling including 53BP1, MDC1, and Brca1. Inhibition of the function of any of these mediator proteins results in a radioresistant DNA synthesis phenotype (RDS) in which cells are less able or unable to inhibit DNA synthesis after IR treatment (20–23). It was previously demonstrated that ATM regulates two parallel pathways to inhibit replicon initiation following IR-induced DNA damage (24). One involves ATM-mediated phosphorylation of Chk2 on threonine 68. Chk2 in concert with Chk1 phosphorylates Cdc25A phosphatase on a number of serine residues, resulting in the accelerated ubiquitin-mediated proteolysis of Cdc25A (25,26). Degradation of Cdc25A, an activator of CDKs, inhibits Cdk2/cyclin E and binding of Cdc45 to origins of DNA replication (24). The second ATM-dependent pathway that is activated in response to IR-induced strand breaks involves ATM-dependent phosphorylation of SMC1 (24), but it remains to be determined how this pathway regulates replication origin firing.
We have shown previously that S checkpoint activation in human cells exposed to low, sublethal doses of UV was independent of ATM and the MRN complex, but dependent on the ATR and Chk1 kinases (27). Claspin and Timeless, two mediators of ATR-dependent checkpoint signaling, also appear to be required for the response to UV-induced DNA damage (28,29). Little is known about the mechanism by which ATR- and Chk1-dependent signaling inhibits replicon initiation. As mentioned above, Cdc25A is the downstream target of the ATM-dependent, IR-induced S checkpoint. Cdc25A degradation was also observed in cells treated with the antimetabolites hydroxyurea (HU) (30) and 5-fluoro-uracil (31), the topoisomerase inhibitors etoposide and camptothecin (32), as well as lethal fluences of UV (33–35). However, it remains to be determined whether Cdc25A is a required target for the UV-induced inhibition of replicon initiation.
There is evidence that the Cdc7/Dbf4 complex may be a target of the S checkpoint response to genotoxic stress (36). In HU-treated S. pombe Cdc7/Him1/Dfp1 (spDbf4) undergoes Cds1-dependent phosphorylation (37,38). In HU-treated S. cerevisiae, Rad53 physically interacts with scCdc7 and phosphorylates scDbf4. Rad53-dependent phosphorylation of scDbf4 has been shown to result in the removal of the kinase complex from chromatin and inhibition of kinase activity (39,40). Recently, evidence for the involvement of vertebrate DDK as a target of the S checkpoint has been reported (41). In Xenopus egg extracts, treatment with etoposide or exonuclease III activated an ATR-dependent checkpoint that inhibited the initiation of DNA replication; this response was accompanied by the inhibition of Dbf4 binding to chromatin and its interaction with Cdc7, ultimately resulting in the down-regulation of Cdc7/Dbf4 kinase activity and the inhibition of Cdc45 binding to origins (41). Further, the ATR-dependent S checkpoint response to single stranded DNA regions (daughter-strand gaps) did not inhibit Cdk2/cyclin E kinase activity. However, inhibition of Cdk2/cyclin E was observed in Xenopus egg extracts in the presence of double-stranded DNA breaks. This S checkpoint response was shown to be ATM-dependent and inhibited the initiation of DNA synthesis through inactivation of Cdk2/cyclin E (42). Thus, at least two DNA damage checkpoint mechanisms regulate origin firing in Xenopus.
The goal of the present study was to characterize further the downstream signaling targets of the ATR- and Chk1-dependent S checkpoint response to UV-induced DNA damage. Our data indicate that, in contrast to the IR-induced S checkpoint, the UVC-induced response is independent of Chk1-mediated proteolysis of Cdc25A and does not involve inhibition of Cdk2/Cyclin E. This is consistent with different signaling intermediates being phosphorylated and activated when normal human fibroblasts are treated with UVC or IR. Furthermore, data presented herein suggest that Chk1, when activated by UVC irradiation, may regulate the Cdc7/Dbf4 kinase complex. Taken together, the data reveal several mechanisms for inhibition of replicon initiation following DNA damage in human cells.
EXPERIMENTAL PROCEDURES
Cell Lines and Culture Conditions
hTERT-immortalized normal human fibroblasts (NHF1), HeLa, and HEK-293T cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 2 mM glutamine (Invitrogen), and 10% fetal bovine serum (Hyclone). All cell lines were maintained at 37°C in a humidified atmosphere of 5% CO2. Cells were routinely established to be free of contamination with mycoplasma using a commercial kit (Gen-Probe Inc.)
Cell Treatments
Prior to treatment with UVC, culture medium was removed and reserved. Cultures were washed once with warm PBS, then placed uncovered under a UV lamp emitting primarily 254 nm radiation at a fluence rate of 0.7 J/m2/s. Following irradiation, the reserved medium was replaced and the cultures incubated for the indicated periods of time. Sham-treated cultures were handled exactly the same way, except that they were not exposed to UVC. Cells treated with ionizing radiation (IR) were maintained in culture medium and exposed to 137Cs γ-rays at a dose rate of 0.84 Gy/min. Cells were treated with HU by adding the appropriate amount of a 1 M stock solution in PBS to the culture medium for a final concentration of 2 mM. Where indicated, cells were treated with 25 μg/ml N-acetyl-Leu-Leu-norleucinal (LLnL, Sigma) 30 min prior to UVC or IR exposure and for the duration of the experiment.
Western Immunoblot Analyses
Logarithmically growing cells were seeded at 106 per 100-mm dish and incubated for 40 h. Cultures were treated with UVC or IR as described above and incubated in reserved medium for 45 min at 37° C. Cells were harvested by trypsinization, washed once in PBS, and resuspended in MCLB [50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5 mM EDTA, 2 mM DTT, 1% NP40, 10 mM β–glycerophosphate, 1 mM Na3VO4] supplemented with 1X protease inhibitor cocktail (Sigma). After clarifying the extract by centrifugation, protein concentration was determined using the Bio-Rad DC Protein Assay (Bio-Rad Laboratories). Samples containing equal amounts of protein were mixed with equal volumes of 2X Laemmli sample buffer [125 mM Tris-HCl (pH 6.8), 4% SDS, 20% glycerol] containing 5% β-mercaptoethanol, boiled, and separated by SDS-PAGE. Proteins were transferred to nitrocellulose and probed with antibodies against Chk1 (Santa Cruz, G4), Chk2 (Oncogene), phospho-serine 317 Chk1, phospho-serine 345 Chk1, and phospho-threonine 68 Chk2 (Cell Signaling), Flag epitope (Anti-flag M2, Sigma), Cdc7 (Abcam), Myc (Santa Cruz, E10), Cdc25A (Labvision), Nbs1, phospho-serine 343 Nbs1, Smc1, phosphor-serine 655 Smc1, Rad17, and phospho-serine 645 Rad17 (Bethyl Laboratories).
Cdk2/cyclin E Kinase Assay
Cdk2/cyclin E kinase assays were performed as previously described (43). Briefly, cyclin E was immunoprecipitated from 500 μg of whole cell extract with anti-human cyclin E Powerclonal antibody (Upstate Biotechnology, Inc., Lake Placid, NY). Kinase reactions were carried out in histone H1 kinase buffer [20 mM HEPES (pH 7.3), 80 mM β-glycerophosphate, 20 mM EGTA, 50 mM MgCl2, 5 mM MnCl2, 1 mM dithiothreitol, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μM cyclic AMP-dependent protein kinase-inhibitory peptide], with 8 μg of histone H1 and 10 μCi of [gamma-32P]ATP (3,000 Ci/mmol, Amersham Pharmacia Biotech) for 30 min at 37 °C. The kinase reactions were stopped by addition of 2X Laemmli sample buffer and proteins resolved by 12% SDS-PAGE. Gels were stained with Coomassie Blue to verify equal histone protein loading, dried, and subjected to autoradiography with Hyperfilm MP (Amersham Pharmacia Biotech). The radiolabeled protein substrates in the dried gels were then quantified using a Molecular Dynamics PhosphorImager and ImageQuant software.
RNA Interference
siRNA duplexes were synthesized by Dharmacon against the following target sequences: ATR, AACCTCCGTGATGTTGCTTGA; and Chk1, AAGCGTGCCGTAGACTGTCCA. Transfections of 200 nM siRNA oligonucleotide duplexes were performed with Oligofectamine (Invitrogen) per manufacturer’s instructions.
Immunoprecipitation of Flag-Dbf4
HeLa cells were transfected with 10 μg of pME18S-2XFlag-Dbf4 plasmid (per 100-mm plate containing approximately 1×106 cells) using Fugene 6 reagent (Roche Molecular Biochemicals), according to manufacturer’s instructions. Forty-eight hours after transfection, cells were harvested and lysed in MCLB. Pre-cleared lysates (1 mg protein) were incubated with 40 μl of anti-Flag M2 affinity gel (Sigma) at 4°C overnight. Proteins were separated on 8% SDS-PAGE and probed with the indicated antibodies.
Affinity Purification of Flag-Dbf4 and Flag-Chk1
HEK-293T cells were transfected with 25 μg of either pME18S-2XFlag-Dbf4, pcDNA3-Flag-Chk1WT, or pcDNA3-Flag-Chk1KD plasmids by using a calcium phosphate transfection method (44). Forty-eight hours after transfection, cells were washed with PBS and lysed in 1.5 ml of lysis buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM β-glycerophosphate, 10% glycerol, 1% Tween 20, 0.1% NP-40, 1 mM Na3VO4, 1 mM NaF, and protease inhibitors (Roche Molecular Biochemicals)] for 30 min on ice. After centrifugation at 14,000 rpm for 30 min, the supernatants were incubated with 40 μl of anti-Flag M2 affinity gel (Sigma) at 4°C overnight. For Flag-Dbf4-transfected cells, the agarose beads were washed three times with 1.5 ml of buffer containing 50 mM Tris-HCl (pH 7.5) and 1 M NaCl; the proteins were eluted in 100 μl of elution buffer that contained 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% glycerol, 1 mM dithiothreitol (DTT), and 200 μg/ml of 3XFlag-peptide (Sigma). For Flag-Chk1 transfected cells, bead washing was performed only under low salt conditions (150 mM NaCl). For in vivo interaction studies, pME18S-Myc-Dbf4, pcDNA3-Flag-Chk1, or pcDNA3-ATR plasmids were transfected alone or in combination by using a calcium phosphate transfection method. Forty-eight hours after transfection, cells were harvested, lysed, and Flag-tagged proteins immunoprecipitated as described. Flag-beads were washed with low-salt buffer and proteins separated on 8% SDS-PAGE.
Radioresistant DNA Synthesis (RDS) Assay
Logarithmically growing cells were plated at a density of 2.5×105 cells per 60-mm dish and grown at 37° C for 30–40 h in medium containing 10 nCi/ml of [14C]thymidine (ICN Radiochemicals) to uniformly label DNA. Radioactive medium was replaced with fresh medium to chase 14C-labeled precursors into DNA for at least three hours. To determine the ability of cells to repress DNA synthesis in the presence of DNA damage, cells were either sham-treated or exposed to UVC. Cells were incubated at 37° C for 30 min and then 15 min with 20 μCi/ml [3H]thymidine. Radioactive medium was removed and the cells were washed twice in cold PBS. Cells were harvested by scraping from the plate with a rubber policeman into 0.5 ml 0.1 M NaCl containing 0.01 M EDTA (pH 8.0) and passed 5X through a 25-gauge needle. Aliquots (200 μl) of the cell suspension were added to separate tubes containing 200 μl of lysis buffer (1 M NaOH, 0.02 M EDTA) and acid-insoluble material collected on GF/C microfibre glass filters (45). Net 3H radioactivity corrected for 14C spillover was normalized for cell number (total 14C radioactivity).
Velocity Sedimentation Analysis
The methodology used to determine the steady-state distribution of sizes of nascent DNA 30–45 min after irradiation of log-phase cultures with either 1 J/m2 UVC or 5 Gy IR was described previously (46,47).
RESULTS
UVC and IR activate different signaling intermediates in human cells
Human S phase cells inhibit replicon initiation by 50% when exposed to low doses of UVC and IR. However, it was previously shown that the upstream sensor and transducer kinases involved in the responses to these radiations were different (27). ATR and Chk1 regulate the S checkpoint response to UVC, while ATM is required for the response to IR. Since the ATR- and ATM-dependent S checkpoints inhibit replicon initiation to the same extent, it was hypothesized that two independent signaling pathways converged downstream, ultimately regulating the same origin-proximal targets. To test this hypothesis, signaling intermediates were compared after the two checkpoint pathways were activated. First, the doses of UVC and IR that induced an equivalent inhibition of replicon initiation were determined. Velocity sedimentation analysis reveals the steady-state distribution of pulse-labeled nascent DNA molecules in S phase cells. This technique was used to quantify DNA damage-induced inhibition of DNA synthesis in replicons that were scheduled to initiate after irradiation. Treatment of NHF1 cells with eitherm 1 J/m2 UVC or 5 Gy IR induced an equivalent biological response, mainly the same selective inhibition of incorporation of radiolabeled precursor into low-molecular-weight nascent DNA intermediates (Fig 1A).
Fig. 1. UVC and IR activate different signaling intermediates in human cells.
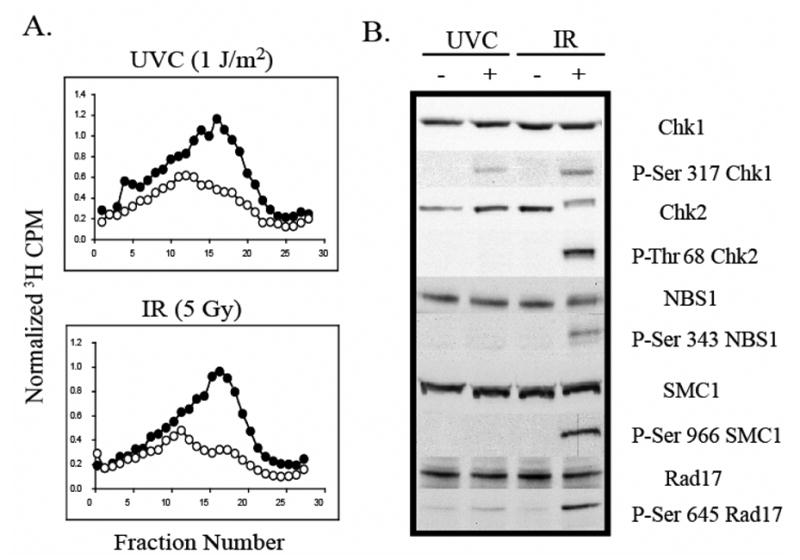
A.) NHF1 cells were grown in the presence of [14C]thymidine for ~ 40 h to label DNA uniformly, and then in non-radioactive medium overnight. Cells were sham-treated or exposed to UVC (1 J/m2) or IR (5 Gy), incubated at 37 °C for 30 min, and then labeled for 15 min in medium containing [3H]thymidine. Cells were harvested and nascent DNA separated by velocity sedimentation. Net 3H radioactivity corrected for 14C spillover was normalized to cell number (total 14C radioactivity). Closed circles (●) represent profiles from sham-treated cells while grey circles (○) represent those from irradiated cultures. B.) Normal human fibroblasts were sham treated or irradiated with either 1 J/m2 UVC or 5 Gy IR. Cells were harvested 1 h after irradiation and cell extracts prepared for western immunoblot analysis.
To determine if downstream signaling targets were shared between the two S checkpoint pathways, NHF1 fibroblasts were harvested 45 min post-irradiation, when inhibition of replicon initiation was demonstrated. Consistent with previous reports, treatment with IR induced the phosphorylation of Chk1, Chk2, Nbs1, Smc1, and Rad17, (48–55) (Fig. 1B). In contrast, UVC failed to induce phosphorylation of Chk2, Nbs1, or Smc1, and induced Rad17 phosphorylation only modestly over the level seen in the sham-treated control. These data are consistent with previously published observations that the activation of the S checkpoint by UVC required the phosphorylation of Chk1 by ATR and was independent of ATM, Nbs1, and Mre11 (27).
Cdc25A degradation was not associated with activation of the S checkpoint by UVC
The ATM-dependent, IR-induced S checkpoint inhibits replicon initiation by regulating the stability of Cdc25A. Checkpoint-induced degradation of this phosphatase results in persistent inhibition of Cdk2/Cyclin E and attenuation of replicon initiation. Since checkpoint-dependent regulation of Cdc25A stability was also observed in cells treated with lethal doses of UVC (33,34,56), we hypothesized that Cdc25A was a downstream target of the ATR-dependent S checkpoint. To test this hypothesis, NHF1 and Hela cells were treated with doses of UVC and IR that induced equivalent inhibitions of replicon initiation (Fig. 1A). Cdc25A degradation, and Chk1 and Chk2 phosphorylation were observed after treating cells with IR (Fig. 2A). However, under conditions in which low-dose UVC activated ATR-dependent Chk1 phosphorylation, Cdc25A degradation was not observed (Fig. 2A).
Fig. 2. Cdc25A degradation is not required for the UVC-induced S checkpoint response.
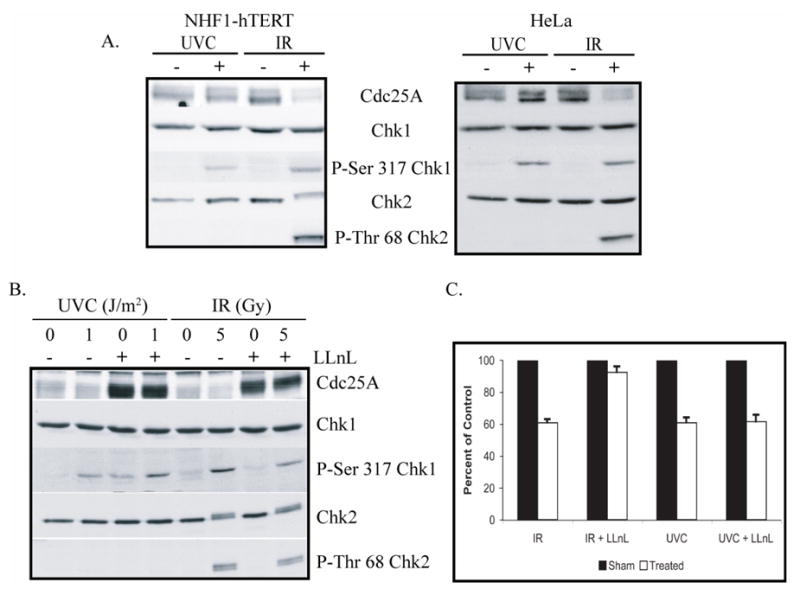
A.) NHF1 and HeLa cells were sham treated or irradiated with either 1 J/m2 UVC or 5 Gy IR. Cells were harvested 1 h after irradiation and cell extracts prepared for western immunoblot analysis. B.) NHF1 cells were pretreated with either DMSO or 25 μg/ml LLnL for 30 min. Cells were then sham treated or irradiated with either 1 J/m2 UVC or 5 Gy IR. Cells were harvested 1 h after irradiation and cell extracts prepared for western immunoblot analysis. C.) NHF1 cells were grown in the presence of [14C]thymidine to label DNA uniformly, then in non-radioactive medium overnight, and pretreated with DMSO or LLnL (as indicated in B). After sham treatment or irradiation with either 1 J/m2 UVC or 5 Gy IR, cells were incubated at 37 °C for 30 min, and then labeled for 15 min in medium containing [3H]thymidine. DNA synthesis activity was measured by the ratios of 3H/14C radioactivity in acid-precipitable material from cell lysates. Net 3H radioactivity was corrected for 14C spillover before normalized to cell number (total 14C radioactivity). The 3H/14C values were expressed as percentages of the paired, sham-treated controls (n=3; black bars, sham-treated controls; white bars, cell exposed to IR or UVC, as indicated; error bars correspond to one standard deviation of the mean.).
It has been shown previously that DNA damage-induced degradation of Cdc25A is mediated by the proteosome (25,35), and this mechanism has recently been elucidated (57,58). If Cdc25A degradation was required for the UVC-induced inhibition of replicon initiation, one would predict that inhibition of the proteasome with N-acetyl-Leu-Leu-norleucinal (LLnL) would override the UVC-induced S checkpoint response. To test this hypothesis, NHF1 cells were pretreated with LLnL for 30 min prior to being irradiated with either UVC or IR. Cells were harvested 45 min later and extracts prepared. When cells were pretreated with LLnL, the basal level of Cdc25A was dramatically increased (Fig. 2B). Further, ATR- and ATM-dependent phosphorylations of Chk1 and Chk2 were observed in LLnL-treated cells. In separate experiments, cells were pulse-labeled with [3H]thymidine for 30 min after irradiation to measure the effect of LLnL on DNA damage-induced inhibition of DNA synthesis. Consistent with previously published data (25), increasing the concentration of Cdc25A above basal levels almost fully reversed the IR-induced S checkpoint response (Fig. 2C). However, LLnL pretreatment had no effect on the UVC-induced inhibition of DNA synthesis, suggesting that proteasome-mediated degradation of Cdc25A was not associated with the ATR-dependent S checkpoint response to UVC.
Low-dose UVC does not inhibit Cdk2/cyclin E kinase activity
Cdk2/cyclin E is essential for the initiation of DNA replication and is inhibited through ATM-dependent Cdc25A degradation following treatment of cells with IR (25). To further characterize signaling within the UV-induced S checkpoint, the activity of Cdk2/cyclin E was measured in cells treated with either UVC or IR (Fig. 3A). In NHF1 cells treated with 5 Gy as a positive control, there was a 44% inhibition of Cdk2/cyclin E kinase activity toward histone H1, relative to sham-treated controls (Fig. 3B). This degree of inhibition is comparable to the degree of inhibition of replicon initiation quantified by velocity sedimentation analysis (Fig. 1A). In contrast, treatment with a UVC fluence that induced a ~40% inhibition of DNA synthesis had no effect on Cdk2/cyclin E kinase activity. Together, these results indicate that the UVC-induced S checkpoint inhibits replicon initiation without regulating Cdc25A stability or Cdk2/cyclin E activity.
Fig. 3. Cdk2/cyclinE inhibition is not associated with activation of the UVC-induced S checkpoint.
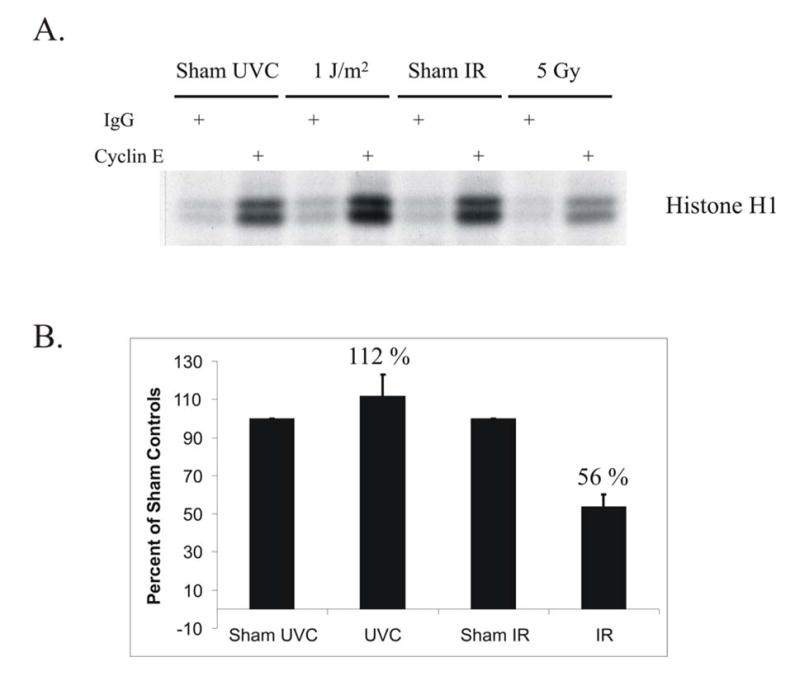
A.) NHF1 cells were sham treated or irradiated with either 1 J/m2 UVC or 5 Gy IR. Cells were harvested 1 h after irradiation and cell extracts prepared. Cdk2/cyclin E complexes were immunoprecipitated and kinase activity measured in vitro against histone H1. In control reactions, the same extracts were incubated with non-specific IgG. B.) Compilation data of 4 independent experiments were graphed as percent of sham-treated controls. Error bars represent one standard deviation of the mean.
UVC-induced Cdc25A degradation is a high-dose effect
Several studies have described Chk1-dependent, UV-induced degradation of Cdc25A and inhibition of Cdk2/cyclin E (33–35). These observations, however, were made after treating cells with much higher doses than the 1 J/m2 that selectively inhibits replicon initiation. UV doses above 1 J/m2 incrementally inhibit DNA chain elongation in active replicons, with doses above 10 J/m2 (producing >7 cyclobutane pyrimidine dimers in an average 20 μm replicon) inhibiting chain elongation by >90%. Cdc25A degradation has also been observed in cells treated with HU for an extended period of time, conditions known also to inhibit DNA chain elongation severely (30). To reconcile these results with those shown in Figures 2 and 3, it was hypothesized that Chk1-dependent UV-induced Cdc25A degradation is secondary to severe inhibition of chain elongation. To test this hypothesis, the effect of increasing doses of UVC on the steady-state distribution of sizes of nascent DNA molecules in S phase cells was measured (Fig. 4A). Treatment of cells with 1 J/m2 UVC had only a small effect on DNA chain elongation in active replicons (gradient fractions 7–15), but selectively inhibited synthesis of low molecular weight nascent DNA by ~50% (fractions 16–24), i.e. inhibited replicon initiation. When the dose of UVC was increased to deposit incrementally more lesions on template strands, there was a dose-dependent inhibition of DNA chain elongation in active replicons. After treating cells with 8 J/m2, DNA chain elongation in active replicons was inhibited severely, causing the predominant accumulation of abnormally small nascent strands. The mechanisms of this dose-dependent inhibition of DNA chain elongation have been thoroughly discussed (45).
Fig. 4. UVC-induced Cdc25A degradation is a high-dose effect.
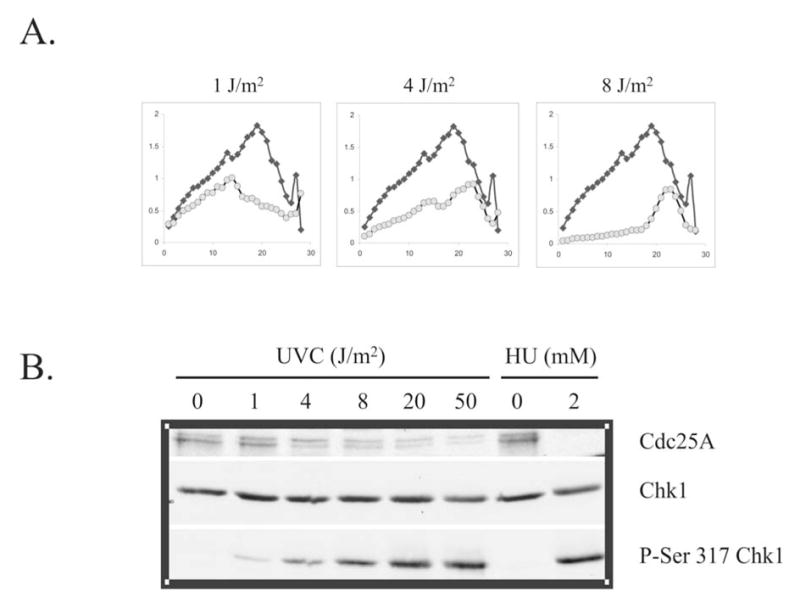
A.) NHF1 cells were treated with 0, 1, or 8 J/m2 UVC and inhibition of DNA synthesis determined by velocity sedimentation as described in the legend to Fig. 1 (27). Closed diamonds (♦) represent sham treated cultures, while grey circles (○) represent UVC-irradiated cultures. B.) NHF1 cells were sham treated or irradiated with increasing fluences of UVC (0–50 J/m2) and harvested 1 h later. Parallel cultures were incubated for 24 h in medium containing 2 mM HU. Cells were harvested and extracts prepared for western immunoblot analysis.
To determine if UV-induced Cdc25A degradation was observed under conditions of severe inhibition of DNA chain elongation, NHF1 cells were irradiated with increasing fluences of UVC (0–50 J/m2) and then harvested after 1-h incubation. Other cells were incubated with HU for 24 h, a treatment that has been shown to activate the replication checkpoint and induce Chk1-dependent Cdc25A degradation (30) (Fig. 4B). In the positive control (24-h incubation with HU), Cdc25A was virtually undetectable. In contrast, Cdc25A degradation was not observed in NHF1 cells that had been treated with 1 J/m2 UVC. Higher fluences of UVC were progressively more effective in triggering Cdc25A degradation. Within 1 h after treatment with 50 J/m2 UVC, the steady-state amount of Cdc25A was reduced to <20% of control levels. UVC induced a dose-dependent increase in Chk1 phosphorylation. Under conditions in which there was only a selective and transient inhibition of replicon initiation (1 J/m2), Chk1 was modestly phosphorylated above basal levels. When cells were treated with HU or high doses of UVC that inhibit chain elongation severely, Chk1 was more extensively phosphorylated. Taken together, these data indicate that UVC-induced Cdc25A degradation is a high-dose phenomenon associated with severe inhibition of DNA chain elongation in active replicons.
Chk1 phosphorylates Dbf4 in vitro and interacts with Dbf4 in vivo
Cdc7/Dbf4 is a conserved kinase complex that has been shown to be essential for the initiation of DNA synthesis in S phase eukaryotic cells (2,59). In yeasts, it was reported that ScCdc7-ScDbf4 and SpCdc7-SpDbf4 may play important roles in S phase checkpoint control and recovery from DNA damage (36). scDbf4 was shown to physically interact with Rad53 following exposure of cells to HU (39). This interaction led to Rad53-dependent phosphorylation of scDbf4, inhibition of DDK kinase activity, and inhibition of late origin firing. To determine if checkpoint-dependent regulation of Dbf4 was conserved in humans, the ability of human Chk1 to phosphorylate human Dbf4 was tested. Flag-Dbf4, Flag-Chk1WT, and Flag-Chk1KD were expressed independently in HEK-293T cells and affinity purified using anti-Flag agarose. Flag-Dbf4 was incubated with either Flag-Chk1WT or Flag-Chk1KD in the presence of [32P]ATP. Dbf4 was efficiently phosphorylated by wild-type Chk1, but not when incubated with kinase-dead Chk1, indicating that Dbf4 was a substrate for Chk1 phosphorylation in vitro (Fig. 5A). We attempted to use mass spectrometry to determine the Chk1 phosphorylation sites on Dbf4, but several experiments were unsuccessful. Sequence analysis predicted two conserved Chk1 target sites within the Dbf4 coding region. We mutated serines 84 and 251 in Dbf4 by site-directed mutagenesis. The mutant proteins were expressed and purified as described above. However, when mutant Dbf4 proteins were incubated with Chk1 in the presence of [32P]ATP we observed no measurable difference in phosphorylation as compared to wild-type Dbf4 (data not shown).
Fig. 5. Chk1 phosphorylates Dbf4 in vitro and interacts with Dbf4 in vivo.
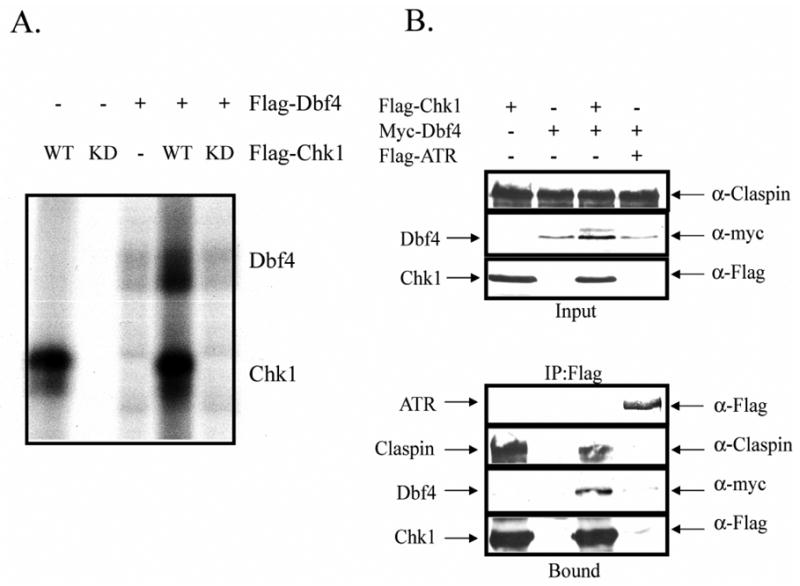
A.) HEK-293T cells were transfected with either Flag-Chk1WT, Flag-Chk1KD, or Flag-Dbf4 expression vectors. Forty-eight hours after transfection, cells were harvested and Flag-tagged proteins immuno-precipitated and eluted as described. Flag-Dbf4 was incubated with Flag-Chk1WT or Flag-Chk1KD, for 30 min in the presence of [32P]ATP. Control reactions included only one of these proteins. The incubation was terminated upon addition of Laemmli sample buffer and proteins separated by SDS-PAGE. The gel was dried and exposed to a phosphoscreen. B.) HEK-293T cells were transfected with the indicated cDNA’s. Forty eight hours after transfection, cells were harvested and extracts were prepared. Flag-tagged proteins were immuno-precipitated with anti-Flag agarose and eluted with 200 μg/ml Flag peptide. Proteins were separated by SDS-PAGE and immuno-blotted with antibodies against the indicated proteins.
To determine if Dbf4 associates with Chk1 in vivo, Flag-Chk1 and Myc-Dbf4 were expressed in HEK-293T either independently or in combination. Flag-ATR was expressed with Myc-Dbf4 as a negative control. In the experiment depicted in Figure 5B, Myc-Dbf4 and the mediator protein Claspin were immunoprecipitated with Flag-Chk1. Myc-Dbf4 did not interact with Flag-ATR, suggesting that the interaction between Chk1 and Dbf4 in human cells was specific (Fig. 5B). The physical interaction between Myc-Dbf4 and Flag-Chk1 or endogenous Chk1 was observed in two of four experiments suggesting that the interaction between the two proteins was weak or quite transient. Together, these data suggest that Dbf4 may be a target of the Chk1 signaling.
Over-expression of Dbf4 reverses the UVC-induced S checkpoint
In yeast and Xenopus, Dbf4 appears to be rate limiting as over-expression of scDbf4, or addition of ectopic XDbf4 abrogated the S checkpoint response (39,41). To determine if over-expression of human Dbf4 could override the UVC-induced S checkpoint, HeLa cells were transiently transfected with expression vectors for epitope-tagged Dbf4 and S checkpoint function measured 48 h later. Over-expression of human Dbf4, as a Flag- or Myc-fusion protein, severely attenuated UVC-induced inhibition of replicon initiation (Fig. 6A). As HeLa cells express undetectable levels of Dbf4 (Fig. 6A) it was not possible to determine the degree of over-expression of the tagged protein. To determine whether the override of S checkpoint function was specific for the response to UVC and not simply a mass effect of expressing supraphysiologic levels of Dbf4, HeLa cells expressing Flag-Dbf4 were treated with low-dose UVC or IR. The data illustrated in Fig. 6B show that over-expression of Flag-Dbf4 reversed the UVC-induced inhibition of DNA synthesis, but did not affect the response to IR; the same cells that were refractory to inhibition of DNA synthesis by UVC responded to IR with a 40% inhibition of DNA synthesis (Fig. 6B).
Fig. 6. Over-expression of Dbf4 reverses the UVC-induced S checkpoint.
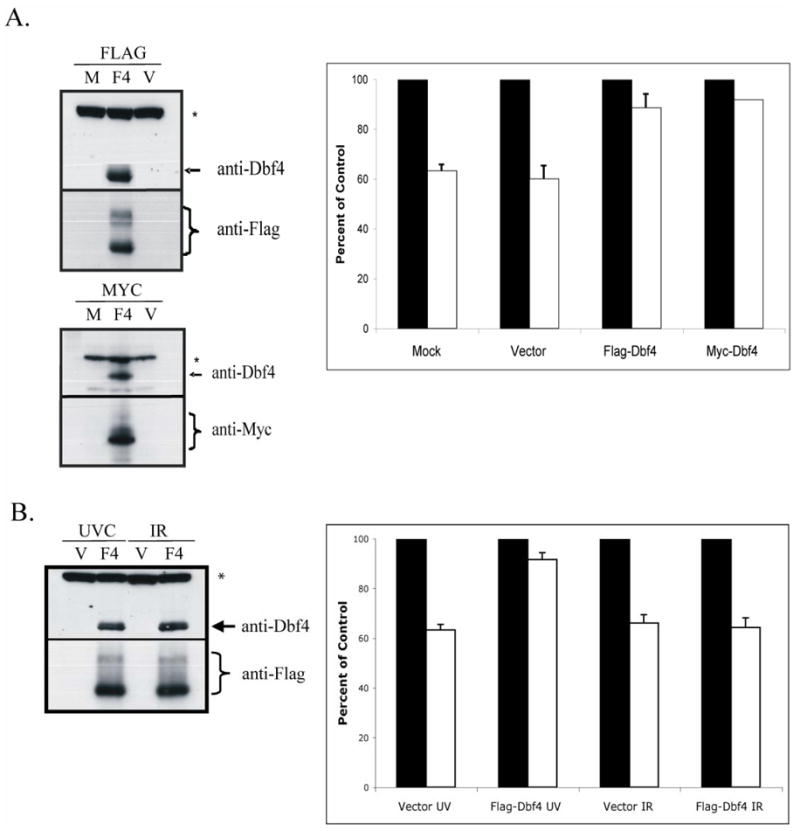
A.) HeLa cells were mock-transfected (M), transfected with an empty vector (V), or transfected with Flag-, or Myc-tagged Dbf4 expression vectors (F4). Cells were harvested 48 h later and extracts prepared for western immunoblot analysis (* = non-specific band). In separate experiments, transfected cultures were incubated with [14C]thymidine to label DNA uniformly; then, cells were treated with either 0 or 1 J/m2 UVC, incubated for 30 min at 37°C and pulsed-labeled with [3H]thymidine for 15 min. DNA synthesis was measured as described in the legend to Fig. 2C and graphed as percentages of paired, sham-treated controls [n=5 (Mock), n=7 (Vector), n=5 (Flag), n=2 (Myc); black bar, sham-treated controls; white bar, UVC-irradiated cells. Error bars correspond to one standard deviation of the mean]. B.) HeLa cells were transiently transfected with either empty vector (V) or a Flag-Dbf4 expressing vector (F4). Cells were harvested 48 h later and extracts prepared for western immunoblot analysis (* = non-specific band). In separate experiments, transfected cultures were incubated with [14C]thymidine to label DNA uniformly; then, cells were sham-treated or irradiated with either 1 J/m2 UVC or 5 Gy IR, incubated for 30 min at 37°C and pulse-labeled with [3H]thymidine for 15 min. DNA synthesis was measured as described in the legend to Fig. 2C and graphed as a percentages of paired, sham-treated controls (n=3; black bar, sham-treated controls; white bar, UVC- or IR-treated cells. Error bars correspond to one standard deviation of the mean).
Since over-expression of Dbf4 reversed specifically the UVC-induced S checkpoint response, studies were undertaken to determine the potential mechanism of this effect. In Xenopus egg extracts, the ATR-dependent S checkpoint inhibits DDK activity by regulating the interaction between Cdc7 and Dbf4, and the magnitude of this response was modulated by increasing the concentrations of DDK (20). Thus, it was hypothesized that over-expressed Dbf4 forms complexes with endogenous Cdc7 and an excess of active DDK prevents checkpoint-dependent inhibition of replicon initiation. To test this hypothesis, Flag-Dbf4-expressing HeLa cells were treated with UVC, etoposide, or HU and Cdc7/Dbf4 complex formation was monitored by co-immunoprecipitation. As shown in Figure 7, endogenous Cdc7 co-precipitated with Flag-Dbf4 from cell extracts after all the DNA damaging treatments. There was no evidence for inhibition of complex formation between Cdc7 and Flag-Dbf4 in UV-treated cells. ATR-dependent checkpoint signaling was evident in damaged cells by the increase in Chk1 phosphorylation (Fig. 7).
Fig. 7. Flag-Dbf4 forms a complex with endogenous Cdc7 following DNA damage.
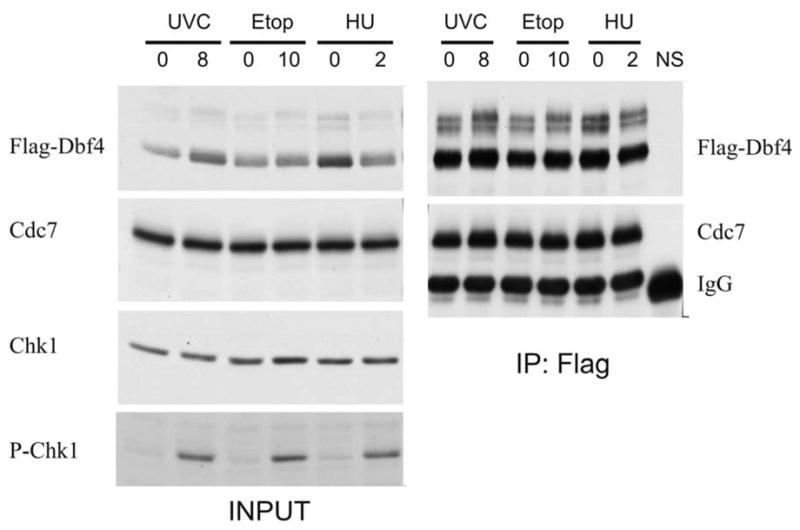
HeLa cells were transiently transfected with a Flag-Dbf4 expression vector. Forty-eight hours later, cells were treated with 8 J/m2 UVC, 10 μM etoposide, or 2 mM HU. Cells were harvested 1 h later and Flag-Dbf4 immuno-precipitated with anti-Flag agarose (NS corresponds to immuno-precipitation with normal mouse IgG). The effect of DNA damage on Cdc7/Dbf4 complex formation was measured by western immunoblot analysis.
DISCUSSION
Cell cycle checkpoints are biochemical signaling pathways that inhibit cell cycle progression when DNA is damaged or when antecedent events are incomplete. Checkpoint signaling networks inhibit cell cycle progression by regulating the activity or location of cyclin-dependent kinase complexes that drive cell cycle transitions (60). For example, the ATM- and p53-dependent DNA damage G1 checkpoint inhibits progression into S phase via transactivation of the Cdk2 inhibitor p21Waf1/Cip1. The IR-induced S checkpoint appears to inhibit replicon initiation by stimulating the ubiquitin-mediated proteolysis of Cdc25A and thereby inhibiting Cdk2. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following treatment with low fluences of UVC. However, the downstream targets of ATR and Chk1 have not been identified, nor has the mechanism that prevents origin firing when S phase cells are damaged. The data presented in this report exclude degradation of Cdc25A and inhibition of Cdk2 as mechanisms by which UV inhibits replicon initiation. Instead, ATR- and Chk1-dependent signaling from sites of UV-induced DNA damage appears to achieve this endpoint by converging on the DDK complex.
The goal of the current study was to investigate the signaling pathway transduced by ATR kinase in human cells exposed to a low fluence of UVC. The data available to date indicate that the UVC-induced S checkpoint inhibits replicon initiation independently of either the ATM-Chk2-Cdc25A-Cdk2 or the ATM-MRN-Smc1 pathways (Figs. 1–3). BPDE, the reactive metabolite of benzo[a]pyrene, produces the same stereotypic inhibition of replicon initiation that is observed after treatment with low-dose UVC (61). The S checkpoint response to low-dose BPDE was also found to be dependent on ATR and Chk1 signaling (62). Consistent with the data reported herein, Vaziri and colleagues have shown that low-dose BPDE does not induce Cdc25A degradation or the inhibition of Cdk2/cyclin E (63). Taken together these studies suggest that low-dose UVC and BPDE inhibit replicon initiation by a mechanism distinct from that of IR.
UV-induced Cdc25A degradation has been observed in cells treated with doses of UVC ranging from 15–100 J/m2 (33–35). These high doses of UV result in saturation of nucleotide excision repair (64), blockage of DNA chain elongation in active replicons, and inhibition of entry of cells into mitosis (65,66). These stress responses induced in S phase cells by UV are reminiscent of those elicited by treatment with HU. Hence, the UV-induced Cdc25A degradation observed in normal human fibroblasts after exposure to high UVC fluences (Fig. 4) could be associated with activation of the replication checkpoint. Cdc25A degradation was correlated also with a dose-dependent increase in UV-induced phosphorylation of Chk1 (Fig. 4B). Work from the Harper and Draetta laboratories suggests that Cdc25A degradation, which is dependent on the F-box protein β-TrCP, requires the concerted action of both Chk1 and Chk2 (57,58). DNA damage-induced Cdc25A degradation has been observed in cells treated with IR, supra-lethal doses of UVC, and HU, all of which result in the activation of both Chk1 and Chk2 (25,30,35). This would explain the observation that a low fluence of UVC did not affect Cdc25A stability, as Chk2 phosphorylation was not observed under the same conditions (Fig. 1B).
Cdc7/Dbf4 is a conserved kinase that has been shown to be essential for the initiation of DNA replication and a target of cell cycle checkpoints in yeast. In S. cerevisiae Dbf4 interacts with, and is phosphorylated by Rad53 in response to treatment with HU (39). Rad53-dependent phosphorylation results in the removal of Dbf4 from chromatin and the inhibition of Cdc7/Dbf4 kinase activity. In S. pombe, the Dbf4 homologue Him1/Dfp1 undergoes Cds1-dependent phosphorylation after treatment with HU (37,38). In Xenopus egg extracts, Cdc7/Dbf4 complexes are targeted by ATR and Chk1 in response to DNA damage induced by either etoposide or exonuclease III treatment (41). Upon activation, ATR, by as yet unknown mechanisms, regulates the interaction between Cdc7 and Dbf4 and thus the activity of the kinase complex. It appears that a common substrate required for activation of ATR-ATRIP includes extended regions of RPA-coated single-stranded DNA (67). Physical blockage of the replication fork at a UV-induced photoproduct results in the uncoupling of leading- and lagging-strand synthesis and extended regions of single-stranded DNA are formed downstream from the lesion (68,69). Treatment of human cells with UVC produces DNA replication intermediates that contain regions of single-stranded DNA (70), which are similar to substrates shown to activate the ATR-Cdc7/Dbf4 checkpoint in Xenopus egg extracts. We determined that human Chk1 could phosphorylate human Dbf4; enhancement of the in vitro phosphorylation signal was absent when Dbf4 was incubated with kinase-dead Chk1. Duncker et al. have shown that scDbf4 interacts with and is phosphorylated by Rad53 in response to treatment with HU (39). This model of checkpoint-dependent regulation of Dbf4 appeared to be conserved as human Chk1 phosphorylated Dbf4 in vitro and these proteins interacted in vivo (Fig. 5). This interaction appeared to be specific since Dbf4 did not co-precipitate with ATR. It would seem plausible that Chk1 activated in response to blocked replication forks could interact with, and phosphorylate Dbf4 transiently, thereby regulating Cdc7/Dbf4 kinase activity. However, the evidence reported herein indicated that over-expression of Flag-Dbf4 did not abrogate the Chk1-dependent, IR-induced S checkpoint. This finding suggests that the Chk1-Dbf4 interaction is not sufficiently strong to compete with the interaction of this kinase with other substrates, such as Cdc25A.
Over-expression of epitope-tagged Dbf4 attenuated the UVC-induced S checkpoint (Fig. 6). These data are in agreement with observations made in yeast and Xenopus systems. In yeast, over-expression of a 248-aminoacid fragment of scDbf4 was shown to override Rad53-dependent HU-induced inhibition of activation of late origins (39). Further, the ATR-dependent S checkpoint in Xenopus egg extracts was reversed by increasing the concentration of recombinant Cdc7/Dbf4 (41). In the same system, the addition of recombinant Cdc25A to egg extracts reversed the ATM-dependent S checkpoint response induced by linear DNA molecules (resembling a DNA DSB) (42). Transient over-expression of Cdc25A in HeLa cells also was shown to reverse the IR-induced S checkpoint (25). In the present study, pretreatment of diploid human fibroblasts with LLnL increased the basal level of Cdc25A, which reversed the IR-induced S checkpoint, but not the response to UVC (Fig. 2). By analogy to the evidence that Cdc25A over-expression attenuated the S checkpoint response to IR, the attenuation of the S checkpoint response to UVC by over-expression of Dbf4 strongly implicates DDK as an effector in the ATR/ATRIP/Claspin/Timeless/Chk1 signaling pathway.
Considering the ability of exogenous Dbf4 to override the S checkpoint, it was expected that endogenous Cdc7 would form active complexes with Flag-Dbf4. Cdc7 was present in Flag-Dbf4 immuno-precipitates, irrespective of the type of induced DNA damage (Fig. 7). These data imply that the ability of Flag-Dbf4 to reverse the S checkpoint was associated with a stable interaction between Cdc7 and Dbf4 in the presence of induced checkpoint signaling. Over-expression of Dbf4 may render ineffective the ATR/Chk1-induced checkpoint signaling that inhibits DDK complexes. An excess of active DDK complexes is then available to activate the Pre-RCs. The observation that an excess of Dbf4 did not attenuate the S checkpoint response to IR suggests that at least one intermediate signaling step might exist between Chk1 and DDK. If the balance between Chk1 and Dbf4 abundance is tilted in the opposite direction by over-expressing Chk1, DNA synthetic activity is almost completely inhibited (27). These observations are consistent with a model in which ATM and ATR actively monitor DNA synthesis and regulate replicon initiation by constitutively regulating the activity of CDK (ATM) and DDK (ATR) (71) (Fig. 8).
Fig. 8.
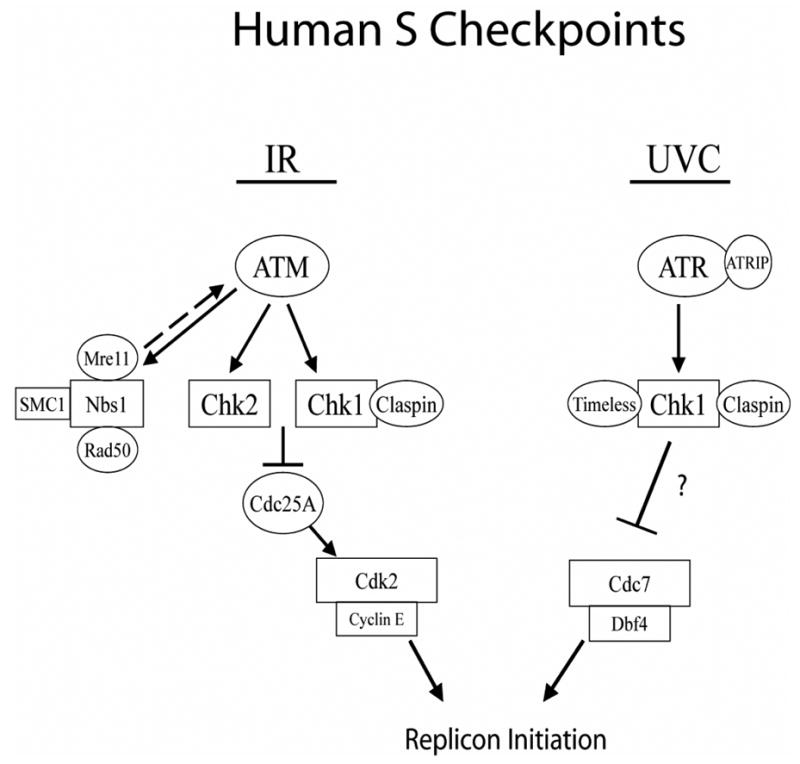
Schematic representation of S checkpoint signaling in human cells.
The data presented here support a model that low fluences of UVC activate an ATR-dependent S checkpoint that inhibits replicon initiation by regulating DDK. It is worth considering, however, that ATR and Chk1 might inhibit replicon initiation by regulating the activity of the pre-replication complex itself. Cortez et al. have shown that the MCM complex is a target of ATM and ATR-dependent phosphorylation in response to IR or UV-induced DNA damage (72). MCM2 is phosphorylated by ATR on serine 108 in response to UV irradiation. However, the biological significance of this phosphorylation has not yet been demonstrated. Ishimi et al. have shown that MCM4 is phosphorylated in an ATR- and Chk1-dependent manner following treatment of cells with UV and HU, and this phosphorylation may inhibit DNA replication by inactivating the MCM helicase (73). Recently, another connection between checkpoint signaling and the pre-replication complex was observed. Tsao et al. have shown that UV-induced activation of ATR and Chk1 requires the interaction between Rad17 and Mcm7 (74). Depletion of either Rad17 or Mcm7 by RNAi-mediated knockdown inhibits ATR-dependent phosphorylation of Chk1 and attenuates the UV-induced S checkpoint. Other potential targets of the S checkpoint include TopBP1 and Mcm10, proteins that are required for the binding of Cdc45 to origins of DNA replication (16,17). However, under conditions in which replicon initiation is inhibited by ~50% there was no significant decrease in either TopBP1 or Mcm10 chromatin binding (data not shown). Additionally, Dunphy and colleagues have reported on a Dbf4-related factor (Drf1) (75). In Xenopus egg extracts, Drf1 binds DNA following DNA damage and replication stress in an ATR-dependent manner. Drf1 binding to DNA is thought to inhibit replicon initiation by preventing the association of Cdc45 with origins of DNA replication. However, little is known about the role of Drf1 in human cells. Taken together, it appears that ATR and Chk1 can regulate proteins involved in replicon initiation, but whether these responses are essential components of the DNA damage S checkpoint remains to be determined.
In summary, the S checkpoint that inhibits replicon initiation in response to sub-lethal irradiation with UVC is biochemically distinct from that induced by IR. The ATR- and Chk1-dependent S checkpoint response to UVC inhibits replicon initiation without degradation of Cdc25A or inhibition of Cdk2/cyclin E. Chk1 phosphorylated Dbf4 in vitro and over-expression of Dbf4 reversed the UVC-induced S checkpoint, suggesting that Chk1 may regulate DDK to control the rates of replicon initiation in human cells. This regulation, however, may include an intermediate between Chk1 kinase and DDK. Taken together, these data suggest that human cells have evolved multiple mechanisms to inhibit replicon initiation in response to DNA damage (Fig. 8).
Acknowledgments
We thank Ying-Chun Zhou and Kathleen Nevis for technical assistance. We also thank Dr. Hisao Masai for providing Cdc7 and Dbf4 expression vectors, and Dr. Cyrus Vaziri for sharing unpublished results and scientific advice.
This work was supported by PHS grants ES011012 (WKK) and CA055065 (MCS), and also by Center grants P30-CA16086 and P30-ES10126. TPH was supported by the Environmental Pathology Training Grant ES07017.
References
- 1.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 2.Dutta A, Bell SP. Annu Rev Cell Dev Biol. 1997;13:293–332. doi: 10.1146/annurev.cellbio.13.1.293. [DOI] [PubMed] [Google Scholar]
- 3.Elsasser S, Chi Y, Yang P, Campbell JL. Mol Biol Cell. 1999;10:3263–3277. doi: 10.1091/mbc.10.10.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jallepalli PV, Tien D, Kelly TJ. Proc Natl Acad Sci U S A. 1998;95:8159–8164. doi: 10.1073/pnas.95.14.8159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang W, Wells NJ, Hunter T. Proc Natl Acad Sci U S A. 1999;96:6193–6198. doi: 10.1073/pnas.96.11.6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perkins G, Drury LS, Diffley JF. Embo J. 2001;20:4836–4845. doi: 10.1093/emboj/20.17.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen BO, Lukas J, Sorensen CS, Bartek J, Helin K. Embo J. 1999;18:396–410. doi: 10.1093/emboj/18.2.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saha P, Chen J, Thome KC, Lawlis SJ, Hou ZH, Hendricks M, Parvin JD, Dutta A. Mol Cell Biol. 1998;18:2758–2767. doi: 10.1128/mcb.18.5.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang W, McDonald D, Hope TJ, Hunter T. Embo J. 1999;18:5703–5713. doi: 10.1093/emboj/18.20.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lei M, Kawasaki Y, Young MR, Kihara M, Sugino A, Tye BK. Genes Dev. 1997;11:3365–3374. doi: 10.1101/gad.11.24.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masai H, Matsui E, You Z, Ishimi Y, Tamai K, Arai K. J Biol Chem. 2000;275:29042–29052. doi: 10.1074/jbc.M002713200. [DOI] [PubMed] [Google Scholar]
- 12.Aparicio OM, Stout AM, Bell SP. Proc Natl Acad Sci U S A. 1999;96:9130–9135. doi: 10.1073/pnas.96.16.9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mimura S, Takisawa H. Embo J. 1998;17:5699–5707. doi: 10.1093/emboj/17.19.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walter J, Newport J. Mol Cell. 2000;5:617–627. doi: 10.1016/s1097-2765(00)80241-5. [DOI] [PubMed] [Google Scholar]
- 15.Zou L, Stillman B. Mol Cell Biol. 2000;20:3086–3096. doi: 10.1128/mcb.20.9.3086-3096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makiniemi M, Hillukkala T, Tuusa J, Reini K, Vaara M, Huang D, Pospiech H, Majuri I, Westerling T, Makela TP, Syvaoja JE. J Biol Chem. 2001;276:30399–30406. doi: 10.1074/jbc.M102245200. [DOI] [PubMed] [Google Scholar]
- 17.Wohlschlegel JA, Dhar SK, Prokhorova TA, Dutta A, Walter JC. Mol Cell. 2002;9:233–240. doi: 10.1016/s1097-2765(02)00456-2. [DOI] [PubMed] [Google Scholar]
- 18.Bartek J, Lukas C, Lukas J. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 19.Bakkenist CJ, Kastan MB. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 20.Goldberg M, Stucki M, Falck J, D’Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- 21.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 22.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. Science. 2002;3:3. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 23.Xu B, Kim S, Kastan MB. Mol Cell Biol. 2001;21:3445–3450. doi: 10.1128/MCB.21.10.3445-3450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. Nat Genet. 2002;30:290–294. doi: 10.1038/ng845. [DOI] [PubMed] [Google Scholar]
- 25.Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 26.Sorensen CS, Syljuasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK, Zhou BB, Bartek J, Lukas J. Cancer Cell. 2003;3:247–258. doi: 10.1016/s1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 27.Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M, Kaufmann WK. Mol Cell Biol. 2002;22:8552–8561. doi: 10.1128/MCB.22.24.8552-8561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chini CC, Chen J. J Biol Chem. 2003;278:30057–30062. doi: 10.1074/jbc.M301136200. [DOI] [PubMed] [Google Scholar]
- 29.Unsal-Kacmaz K, Mullen TE, Kaufmann WK, Sancar A. Mol Cell Biol. 2005;25:3109–3116. doi: 10.1128/MCB.25.8.3109-3116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molinari M, Mercurio C, Dominguez J, Goubin F, Draetta GF. EMBO Rep. 2000;1:71–79. doi: 10.1093/embo-reports/kvd018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao Z, Xue J, Sowin TJ, Rosenberg SH, Zhang H. Oncogene. 2005;24:1403–1411. doi: 10.1038/sj.onc.1208309. [DOI] [PubMed] [Google Scholar]
- 32.Agner J, Falck J, Lukas J, Bartek J. Exp Cell Res. 2005;302:162–169. doi: 10.1016/j.yexcr.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 33.Hassepass I, Voit R, Hoffmann I. J Biol Chem. 2003;278:29824–29829. doi: 10.1074/jbc.M302704200. [DOI] [PubMed] [Google Scholar]
- 34.Goloudina A, Yamaguchi H, Chervyakova DB, Appella E, Fornace AJ, Jr, Bulavin DV. Cell Cycle. 2003;2:473–478. [PubMed] [Google Scholar]
- 35.Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 36.Jares P, Donaldson A, Blow JJ. EMBO Rep. 2000;1:319–322. doi: 10.1093/embo-reports/kvd076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takeda T, Ogino K, Tatebayashi K, Ikeda H, Arai K, Masai H. Mol Biol Cell. 2001;12:1257–1274. doi: 10.1091/mbc.12.5.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown GW, Kelly TJ. Proc Natl Acad Sci U S A. 1999;96:8443–8448. doi: 10.1073/pnas.96.15.8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duncker BP, Shimada K, Tsai-Pflugfelder M, Pasero P, Gasser SM. Proc Natl Acad Sci U S A. 2002;99:16087–16092. doi: 10.1073/pnas.252093999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weinreich M, Stillman B. Embo J. 1999;18:5334–5346. doi: 10.1093/emboj/18.19.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. Mol Cell. 2003;11:203–213. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 42.Costanzo V, Robertson K, Ying CY, Kim E, Avvedimento E, Gottesman M, Grieco D, Gautier J. Mol Cell. 2000;6:649–659. doi: 10.1016/s1097-2765(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 43.Shackelford RE, Innes CL, Sieber SO, Heinloth AN, Leadon SA, Paules RS. J Biol Chem. 2001;4:4. doi: 10.1074/jbc.M011303200. [DOI] [PubMed] [Google Scholar]
- 44.Jordan M, Schallhorn A, Wurm FM. Nucleic Acids Res. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaufmann WK, Cleaver JE. J Mol Biol. 1981;149:171–187. doi: 10.1016/0022-2836(81)90297-7. [DOI] [PubMed] [Google Scholar]
- 46.Cistulli CA, Kaufmann WK. Cancer Research. 1998;58:1993–2002. [PubMed] [Google Scholar]
- 47.Cordeiro-Stone M, Frank A, Bryant M, Oguejiofor I, Hatch SB, McDaniel LD, Kaufmann WK. Carcinogenesis. 2002;23:959–965. doi: 10.1093/carcin/23.6.959. [DOI] [PubMed] [Google Scholar]
- 48.Bao S, Tibbetts RS, Brumbaugh KM, Fang Y, Richardson DA, Ali A, Chen SM, Abraham RT, Wang XF. Nature. 2001;411:969–974. doi: 10.1038/35082110. [DOI] [PubMed] [Google Scholar]
- 49.Chaturvedi P, Eng WK, Zhu Y, Mattern MR, Mishra R, Hurle MR, Zhang X, Annan RS, Lu Q, Faucette LF, Scott GF, Li X, Carr SA, Johnson RK, Winkler JD, Zhou BB. Oncogene. 1999;18:4047–4054. doi: 10.1038/sj.onc.1202925. [DOI] [PubMed] [Google Scholar]
- 50.Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K, Savage K, Lukas J, Zhou BB, Bartek J, Khanna KK. J Biol Chem. 2003;278:14806–14811. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- 51.Gatei M, Young D, Cerosaletti KM, Desai-Mehta A, Spring K, Kozlov S, Lavin MF, Gatti RA, Concannon P, Khanna K. Nat Genet. 2000;25:115–119. doi: 10.1038/75508. [DOI] [PubMed] [Google Scholar]
- 52.Kim ST, Xu B, Kastan MB. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH, Kastan MB. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 54.Matsuoka S, Huang M, Elledge SJ. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 55.Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J. Genes Dev. 2002;16:571–582. doi: 10.1101/gad.970702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 57.Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, Harper JW. Genes Dev. 2003;17:3062–3074. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- 59.Kim JM, Yamada M, Masai H. Mutat Res. 2003;532:29–40. doi: 10.1016/j.mrfmmm.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 60.Abraham RT. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 61.Cordeiro-Stone M, Boyer JC, Smith BA, Kaufmann WK. Carcinogenesis. 1986;7:1775–1781. doi: 10.1093/carcin/7.10.1775. [DOI] [PubMed] [Google Scholar]
- 62.Guo N, Faller DV, Vaziri C. Cell Growth Differ. 2002;13:77–86. [PubMed] [Google Scholar]
- 63.Liu P, Barkley LR, Day T, Bi X, Slater DM, Alexandrow MG, Nasheuer HP, Vaziri C. J Biol Chem. 2006;281:30631–30644. doi: 10.1074/jbc.M602982200. [DOI] [PubMed] [Google Scholar]
- 64.Kaufmann WK, Wilson SJ. Mutat Res. 1990;236:107–117. doi: 10.1016/0921-8777(90)90038-7. [DOI] [PubMed] [Google Scholar]
- 65.Kaufmann WK, Kies PE. Mutation Research. 1998;400:153–167. doi: 10.1016/s0027-5107(98)00041-4. [DOI] [PubMed] [Google Scholar]
- 66.Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E, Fornace AJ., Jr Nature. 2001;411:102–107. doi: 10.1038/35075107. [DOI] [PubMed] [Google Scholar]
- 67.Zou L, Elledge SJ. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 68.Cordeiro-Stone M, Makhov AM, Zaritskaya LS, Griffith JD. J Mol Biol. 1999;289:1207–1218. doi: 10.1006/jmbi.1999.2847. [DOI] [PubMed] [Google Scholar]
- 69.Lopes M, Foiani M, Sogo JM. Mol Cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 70.Meneghini R, Cordeiro-Stone M, Schumacher RI. Biophys J. 1981;33:81–92. doi: 10.1016/S0006-3495(81)84873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shechter D, Costanzo V, Gautier J. Nat Cell Biol. 2004;6:648–655. doi: 10.1038/ncb1145. [DOI] [PubMed] [Google Scholar]
- 72.Cortez D, Glick G, Elledge SJ. Proc Natl Acad Sci U S A. 2004;101:10078–10083. doi: 10.1073/pnas.0403410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ishimi Y, Komamura-Kohno Y, Kwon HJ, Yamada K, Nakanishi M. J Biol Chem. 2003;278:24644–24650. doi: 10.1074/jbc.M213252200. [DOI] [PubMed] [Google Scholar]
- 74.Tsao CC, Geisen C, Abraham RT. Embo J. 2004;23:4660–4669. doi: 10.1038/sj.emboj.7600463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yanow SK, Gold DA, Yoo HY, Dunphy WG. J Biol Chem. 2003;278:41083–41092. doi: 10.1074/jbc.M307144200. [DOI] [PubMed] [Google Scholar]