

Abstract
Kainic acid (KA) treatment is a well-established model of hippocampal neuron death mediated in large part by KA receptor-induced excitotoxicity. KA-induced, delayed neuron death has been shown previously to follow the induction of seizures and exhibit characteristics of both apoptosis and necrosis. Growing evidence supports a role of autophagic stress-induced death of neurons in several in vitro and in vivo models of neuron death and neurodegeneration. However, whether autophagic stress also plays a role in KA-induced excitotoxicity has not been previously investigated. To examine whether KA alters the levels of proteins associated with or known to regulate the formation of autophagic vacuoles, we isolated hippocampal extracts from control mice and in mice following 2–16h KA injection. KA induced a significant increase in the amount of LC3-II, a specific marker of autophagic vacuoles, at 4–6h following KA, which indicates a transient induction of autophagic stress. Levels of autophagy-associated proteins including ATG5 (conjugated to ATG12), ATG6 and ATG7 did not change significantly after treatment with KA. However, ratios of phospho-mTOR/mTOR were elevated from 6–16h, and ratios of phospho-Akt/Akt were elevated at 16h following KA treatment, suggesting a potential negative feedback loop to inhibit further stimulation of autophagic stress. Together these data indicate the transient induction of autophagic stress by KA which may serve to regulate excitotoxic death in mouse hippocampus.
Keywords: kainic acid, hippocampus, autophagy, ATG5, ATG7, Akt, mTOR, LC3
The excitatory amino acid neurotransmitter glutamate is known to play an important role in a vast array of neuronal activities as well as in the induction of excitotoxic neurodegeneration through massive activation of its receptors [1;2]. Kainic acid (KA) is a potent glutamate receptor agonist with selectivity towards non-N-methyl-D-aspartate (NMDA)-type glutamate receptors [3;4]. KA is well known for its ability to induce seizures within minutes of its administration and is followed by a delayed excitotoxic neuron death in the hippocampus several hours later, in part through an increase in intracellular calcium and activation of calcium-dependent neuron death pathways [5–7]. Both apoptotic and necrotic death of neurons are associated with KA-induced excitotoxicity in vivo [8;9], suggesting the existence of multiple death pathways induced by glutamate receptor neurotoxicity.
Autophagic stress results from alterations in autophagy, a lysosomal degradation pathway that is responsible for the homeostatically regulated turnover of macronutrients and organelles [10]. Macroautophagy, the most prominent form of autophagy in cells, occurs via the formation of double-membraned autophagic vacuoles (AVs) that sequester and shuttle damaged organelles and macronutritents to lysosomes for degradation by acidic lysosomal hydrolases[11]. Our laboratory and others have shown that autophagic stress leads to an accumulation of AVs that occurs either from an induction of their nascent formation, or from an inhibition of their recycling due to a dysfunction of acidic organelles [12–15]. If left unchecked, autophagic stress can lead to autophagic cell death[12–15], which has been shown morphologically to possess elements of both apoptosis and necrosis[16]. Recent reports have implicated a role for the induction of autophagic stress in glutamate receptor-mediated excitotoxicity of motor neurons [17;18]. Although multiple types of cell death have been delineated previously in excitotoxic neuron death, the contribution of autophagic stress in models of glutamate receptor-induced excitotoxicity has not been previously investigated.
The stimulation, assembly and recycling of AVs in macroautophagy is exquisitely regulated by a large group of proteins (ATGs) isolated originally in yeast [11]. The importance of ATG proteins in macroautophagy is emphasized by studies of their genetic manipulation, such that over-expression of ATG5, ATG6 (Beclin) and ATG7 induce macroautophagy [19–23] and their targeted deletion (or haploinsufficiency in the case of Beclin) inhibits macroautophagy [24–26]. Deficiencies in ATG5 and ATG7 in particular have been shown to induce neurodegeneration in mice [25;26], which highlights the importance of autophagy-associated proteins in maintaining neuron survival. In addition, the induction of macroautophagy has been shown to be regulated by different classes of phosphatidylinositol 3-kinase (PI3-K). Class I PI3-K activates pro-survival, Akt-mediated signaling and has been shown to inhibit macroautophagy, whereas the activation of class III PI3-K has been shown to stimulate macroautophagy [27].
All mice were cared for in accordance with the guidelines of the NIH Guide for the Care and Use of Laboratory Animals. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham. Mice with 129SvJ x C57BL/6J background were used in all experiments. The experiments were repeated with multiple litters (n=3 for each time point). Mice were injected with KA (20 mg/kg i.p.) as previously reported [28] and were euthanized from 2–16h following KA administration with subsequent removal of their hippocampi and freezing on dry ice. Sham (0h) mice not receiving KA served as controls for each litter tested. Hippocampi were homogenized in lysis buffer containing 25 mM HEPES, 5 mM EDTA, 5 mM MgCl2, 1% SDS, 1% Triton X-100, 1 mM PMSF, 1% protease inhibitor cocktail and 1% phosphatase inhibitor cocktail (Sigma). 25 μg of protein per lane were resolved via SDS-PAGE and transferred to PVDF. Blots were blocked for 1h, RT (5% milk), followed by overnight incubation at 4°C with primary antibody (goat anti-ATG-5 (Santa Cruz); goat anti-ATG-7 (Santa Cruz); rabbit anti-beclin (Santa Cruz); rabbit anti-phospho-mTOR (Ser 2448) and rabbit anti-mTOR (Cell Signaling); rabbit anti-phospho-Akt (Ser 473) and rabbit anti-total Akt (Cell Signaling); and rabbit anti-LC3 (Uchiyama laboratory). All blots were also probed for rabbit anti-β-tubulin (Santa Cruz), which served as a loading control. Blots were washed with 1X TBS containing 0.1% Tween 20, then incubated with secondary antibody (goat anti-rabbit IgG, 1h, RT) and washed. Signal was detected using Supersignal chemiluminescence (Pierce). Blots were scanned for densitometric analysis using UN-Scan-IT software (Orem, Utah). Blots for phospho-specific proteins (Akt and mTOR) were stripped subsequently using ReStore® Western Blot stripping buffer (Pierce) then re-probed for total Akt and mTOR. Protein levels from hippocampi of KA-treated mice (2–16h) were normalized to control levels (0h) in each western blot experiment, with three different western blot experiments representing three separate time courses. Levels of protein were analyzed for significance over time via 1 factor ANOVA and Tukey’s multiple comparison post hoc test. In all cases a level of p<0.05 was considered significant.
We began our study by assessing the effects of KA on LC3-II, the mammalian homologue of ATG8 and a selective biochemical marker for AVs [29]. LC3-II is the cleaved and lipidated form of the cytosolic LC3-I, and these post-translational modifications allow it to insert into the outer membrane of AVs [30]. Significant increases in the ratio of LC3-II/LC3-I were specific for 4h (2.5 fold increase) and 6h (2.2 fold increase) following KA administration (Fig. 1). These data indicate the transient induction of autophagic stress resulting from acute KA administration.
Fig. 1. LC3II/I ratio in the hippocampus is significantly increased by greater than 2-fold at 4–6h post KA treatment, indicating an increased accumulation of AVs.
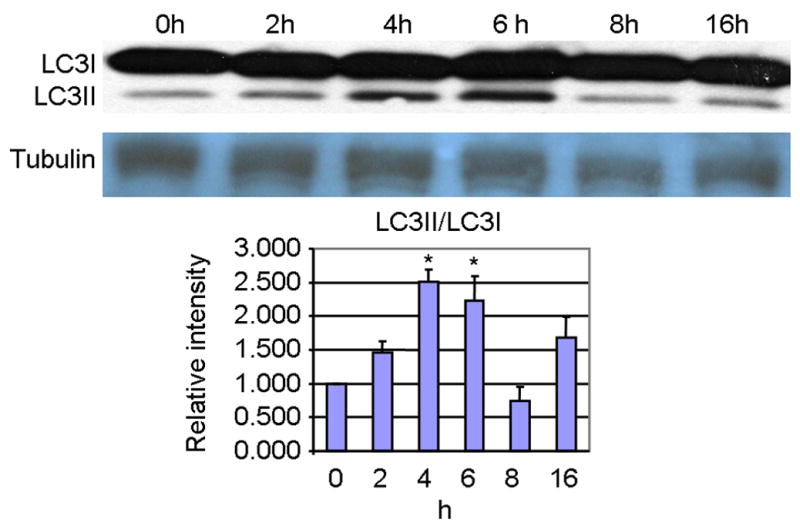
Equal amounts of total hippocampal protein was loaded and detected by western blot (A) followed by scanning and densitometric analysis of the bands. Graphical representation (B) of the ratio of LC3-II relative to LC3-I (mean ± SEM). *p<0.05 compared to 0h sham control by 1 factor ANOVA with Tukey's multiple comparison post-hoc test.
Levels of ATG5, ATG7 and Beclin were also measured following acute KA administration, as these proteins are known to be essential for the induction of autophagy [24–26]. While slight increases in ATG5, ATG6 and ATG7 were observed following KA administration, they were not found to be statistically significant (Fig. 2). These data suggest that a change in steady-state protein levels of ATG proteins is not necessarily required for the transient induction of autophagic stress. In addition, the Beclin-independent induction of autophagic stress has been documented recently in SH-SY5Y cells treated with the dopaminergic neurotoxin MPP+ [31], which suggests that Beclin may not be always required for the induction of autophagic stress through the formation of new vacuoles. Conversely, the transient induction of autophagic stress by KA may result from impairment of lysosomal degradation pathways, which by definition does not involve the ATG5, ATG6 or ATG7-dependent formation of new vacuoles.
Fig. 2. Levels of ATG5 (conjugated to ATG12), ATG7, and Beclin were not significantly altered post KA treatment.
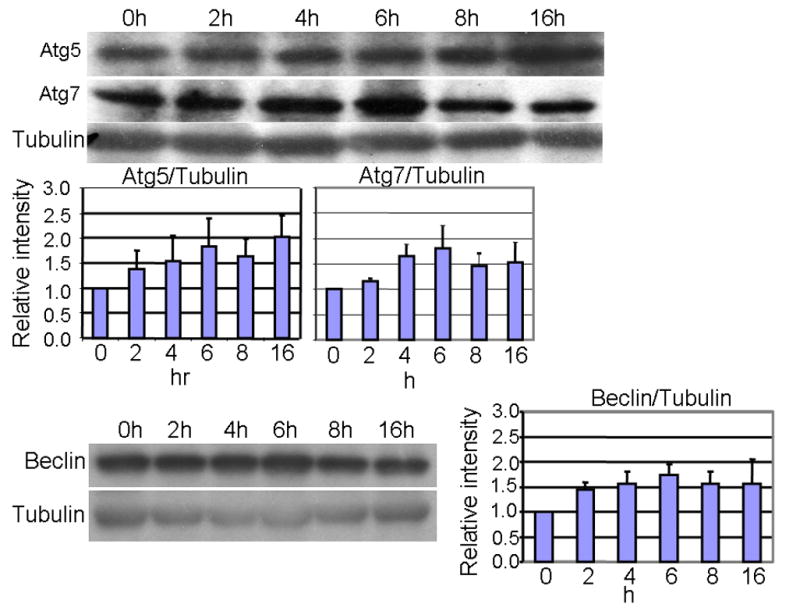
Equal amounts of total hippocampal protein was loaded and detected by western blot (A) followed by scanning and densitometric analysis of the bands. Graphical representation (B) of each band, expressed relative to levels of β-tubulin (mean ± SEM).
Activation of class I PI3-K is known to negatively regulate the induction of autophagy [27]. To determine whether the transient induction of autophagic stress by KA was associated with changes in PI3-K-related signaling molecules, the phosphorylation of mTOR and Akt was measured (Fig. 3). The ratio of phospho-mTOR/mTOR increased significantly by 2–3 fold from 6–16h following KA treatment, and a significant, 2.8-fold increase in the ratio of phospho-Akt/Akt was observed at 16h following KA treatment. The KA-induced activation of Akt and mTOR may result from a stress response in the population of neurons surviving KA treatment. In addition, the KA-induced increase in activation of Akt and mTOR may serve to negatively regulate the induction of autophagy, as evidenced by the decrease in the ratio of LC3-II/I by 8–16h after KA treatment. Previous studies in our laboratory have found a KA-induced, c-fos-regulated increase in BDNF [32], which may induce the activation of Akt [33]. A previous study of intrastriatal KA administration in the rat showed a transient decrease in Akt phosphorylation measured 24–48h following KA [34], but differences in species, route of administration and time course may explain this apparent discrepancy with the results of our study.
Fig. 3. Significant increases in the ratios of phospho-mTOR (2–3 fold, 6–16h) and phospho Akt/Akt (2.8 fold, 16h) were observed post KA treatment.
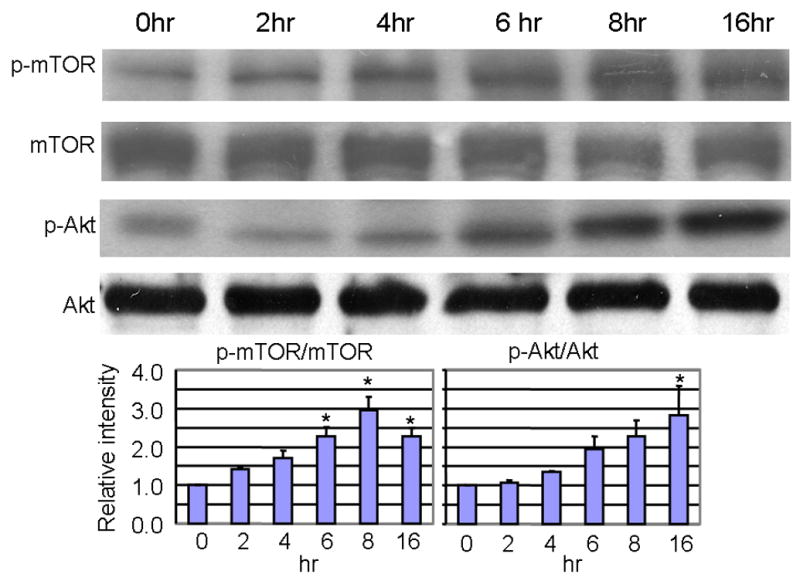
Equal amounts of total hippocampal protein was loaded and detected by western blot (A) followed by scanning and densitometric analysis of the bands. Graphical representation (B) of the ratio of phospho mTOR/mTOR or phospho Akt/Akt (mean ± SEM). *p<0.05 compared to 0h sham control by 1 factor ANOVA with Tukey's multiple comparison post-hoc test.
The present study indicates the early and transient induction of autophagic stress in the mouse hippocampus following KA administration. The induction of autophagic stress by KA may be a stress response of neurons to increase the turnover of proteins and damaged mitochondria under conditions of low trophic support. Furthermore, the transient nature of autophagic stress observed in the present study may be due to the compensatory increase in activation of Akt and mTOR, molecules known to negatively regulate macroautophagy. The compensatory increase in pro-survival Akt signaling may thus serve an important role in regulating the induction of autophagic stress, which if left unchecked has been shown to cause cell death [12]. The transient increase in autophagic stress observed in the present study may result from a stress response of increased seizure-induced activity in the mouse and closely follows the temporal induction in seizures by KA but occurs well before the advent of noticeable neuron death as reported previously by our laboratory [28]. Future studies are warranted to determine the mechanisms and consequences of autophagic stress induction in models of excitotoxic neurodegeneration, and whether its time course or extent can be altered with pharmacological manipulation or additional trophic support.
Acknowledgments
We wish to thank Dr. Liyan Qiao for scientific discussion, an award from the Batten Disease Support and Research Association (JJS), UAB Neuroscience Core Facilities (NS4746 and NS57098), NIH grants NS35107 and NS41962 (KAR), and a UAB faculty development award and an Epilepsy Foundation award (JZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 2.Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719–721. doi: 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- 3.Olney JW, Rhee V, Ho OL. Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 1974;77:507–512. doi: 10.1016/0006-8993(74)90640-4. [DOI] [PubMed] [Google Scholar]
- 4.Greengard P, Jen J, Nairn AC, Stevens CF. Enhancement of the glutamate response by cAMP-dependent protein kinase in hippocampal neurons. Science. 1991;253:1135–1138. doi: 10.1126/science.1716001. [DOI] [PubMed] [Google Scholar]
- 5.Ben Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- 6.Lee HK, Seo YJ, Choi SS, Kwon MS, Shim EJ, Lee JY, Suh HW. Role of gamma-aminobutyricacidB(GABA(B)) receptors in the regulation of kainic acid-induced cell death in mouse hippocampus. Exp Mol Med. 2005;37:533–545. doi: 10.1038/emm.2005.66. [DOI] [PubMed] [Google Scholar]
- 7.Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol. 2005;31:3–16. doi: 10.1385/MN:31:1-3:003. [DOI] [PubMed] [Google Scholar]
- 8.van Lookeren CM, Lucassen PJ, Vermeulen JP, Balazs R. NMDA and kainate induce internucleosomal DNA cleavage associated with both apoptotic and necrotic cell death in the neonatal rat brain. Eur J Neurosci. 1995;7:1627–1640. doi: 10.1111/j.1460-9568.1995.tb01158.x. [DOI] [PubMed] [Google Scholar]
- 9.Humphrey WM, Dong H, Csernansky CA, Csernansky JG. Immediate and delayed hippocampal neuronal loss induced by kainic acid during early postnatal development in the rat. Brain Res Dev Brain Res. 2002;137:1–12. doi: 10.1016/s0165-3806(02)00344-9. [DOI] [PubMed] [Google Scholar]
- 10.Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65:423–432. doi: 10.1097/01.jnen.0000229233.75253.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ, 12 Suppl. 2005;2:1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 13.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Shacka JJ, Klocke BJ, Shibata M, Uchiyama Y, Datta G, Schmidt RE, Roth KA. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol. 2006;69:1125–1136. doi: 10.1124/mol.105.018408. [DOI] [PubMed] [Google Scholar]
- 16.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 17.Matyja E, Taraszewska A, Naganska E, Rafalowska J. Autophagic degeneration of motor neurons in a model of slow glutamate excitotoxicity in vitro. Ultrastruct Pathol. 2005;29:331–339. doi: 10.1080/01913120500214333. [DOI] [PubMed] [Google Scholar]
- 18.Tarabal O, Caldero J, Casas C, Oppenheim RW, Esquerda JE. Protein retention in the endoplasmic reticulum, blockade of programmed cell death and autophagy selectively occur in spinal cord motoneurons after glutamate receptor-mediated injury. Mol Cell Neurosci. 2005;29:283–298. doi: 10.1016/j.mcn.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Kim KW, Mutter RW, Cao C, Albert JM, Freeman M, Hallahan DE, Lu B. Autophagy for cancer therapy through inhibition of proapoptotic proteins and mTOR signaling. J Biol Chem. 2006 doi: 10.1074/jbc.M607094200. [DOI] [PubMed] [Google Scholar]
- 20.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 21.Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281:14474–14485. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- 22.Tanida I, Tanida-Miyake E, Ueno T, Kominami E. The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J Biol Chem. 2001;276:1701–1706. doi: 10.1074/jbc.C000752200. [DOI] [PubMed] [Google Scholar]
- 23.Tanida I, Tanida-Miyake E, Komatsu M, Ueno T, Kominami E. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J Biol Chem. 2002;277:13739–13744. doi: 10.1074/jbc.M200385200. [DOI] [PubMed] [Google Scholar]
- 24.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 26.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 27.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Zhang D, McQuade JS, Behbehani M, Tsien JZ, Xu M. c-fos regulates neuronal excitability and survival. Nat Genet. 2002;30:416–420. doi: 10.1038/ng859. [DOI] [PubMed] [Google Scholar]
- 29.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu CT, Horbinski C, Guo F, Watkins S, Uchiyama Y, Zhu J. Beclin 1-independent regulation of MPP+-induced autophagy, 2006. Neuroscience Meeting Planner. 2006:471.13. [Google Scholar]
- 32.Dong M, Wu Y, Fan Y, Xu M, Zhang J. c-fos modulates brain-derived neurotrophic factor mRNA expression in mouse hippocampal CA3 and dentate gyrus neurons. Neurosci Lett. 2006;400:177–180. doi: 10.1016/j.neulet.2006.02.063. [DOI] [PubMed] [Google Scholar]
- 33.Bhave SV, Ghoda L, Hoffman PL. Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascades and site of ethanol action. J Neurosci. 1999;19:3277–3286. doi: 10.1523/JNEUROSCI.19-09-03277.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez-Navarro E, Gavalda N, Gratacos E, Alberch J. Brain-derived neurotrophic factor prevents changes in Bcl-2 family members and caspase-3 activation induced by excitotoxicity in the striatum. J Neurochem. 2005;92:678–691. doi: 10.1111/j.1471-4159.2004.02904.x. [DOI] [PubMed] [Google Scholar]