

Abstract
We have synthesized novel phosphonic acid analogues of β-diketo acids. Interestingly, the phosphonic acid isostere, 2, of our anti-HIV compound, 1, was an inhibitor of only the strand transfer step, in stark contrast to 1. Compound 2 had lower anti-HIV activity than 1, but was more active and less toxic than the phosphonic acid analogue of L-708906. These isosteric compounds represent the first examples of β-diketo phosphonic acids of structural, synthetic and antiviral interest.
Keywords: Synthesis, Phosphonates, HIV integrase inhibitors, Mechanism of inhibition, Anti-HIV activity
The pol gene of the human immunodeficiency virus (HIV) encodes viral enzymes that are required for HIV replication.1–3 Drug design and discovery work directed at inhibitors of two of these enzymes, HIV reverse transcriptase (RT) and HIV protease (PR), have created clinically-useful compounds for the treatment of acquired immunodeficiency syndrome (AIDS).4–8 However, research efforts on drug discovery pertaining to the third enzyme of the pol gene, HIV integrase, have not produced a single approved drug whose mechanism of action is inhibition of HIV integrase.9–11 As HIV integrase has no human counterpart and is important in HIV replication, it remains a key viral target for the design and discovery of new anti-HIV agents.
HIV-1 integrase is encoded at the 3′-end of the HIV pol gene and is a relatively small (32 kDa) viral protein. Its catalysis results in the incorporation of HIV DNA into host chromosomal DNA.12–16 This process begins in the cytoplasm where the viral DNA, produced by reverse transcription, is assembled on HIV integrase. Following this assembly, there is specific endonucleolytic cleavage of two nucleotides from each 3'-end of double-stranded viral DNA. This step, referred to as 3'-processing, produces tailored and recessed viral DNA. The next step, which occurs in the nucleus, is identified as strand transfer. Following staggered nicking of chromosomal DNA and joining of each 3'-end of the recessed viral DNA to the 5'-ends of the host DNA, there is repair, which completes the integration process. The strand transfer step, occurring in the nucleus, is partitioned from the 3'-processing step in the cytoplasm.
While many structurally diverse compounds have been reported to be inhibitors of HIV integrase,8–10,17–28 only a few compounds of one group, the β-diketo acids, and their related compounds, represent the most convincing, biologically-validated inhibitors23,24 of this viral enzyme. Nair and coworkers have designed a conceptually new β-diketo acid with a nucleobase scaffold, compound 1, which is a powerful inhibitor of both 3′-processing and strand transfer steps of HIV-1 integrase and which exhibits remarkably potent in vitro anti-HIV activity.29,30
Phosphonic acids have been viewed commonly as mimics of carboxylic acids, particularly with reference to biological activity. For example, amino phosphonic acids, isosteres of amino acids, reveal diverse biological properties.31 With this concept in mind, we designed the β-diketo phosphonic acids, 2 and 3, the phosphorus-based isosteres of compounds 1 and L-708906. In this report, we describe the methodology for the syntheses of 2 and 3 and discuss their integrase inhibition and antiviral data.
Although the β-diketo acid 1 was synthesized, using as the key step, the condensation of acetyl uracil 4 with dimethyl oxalate under basic conditions,29 the related reaction of 4 with trimethyl phosphonoformate was unsuccessful (Scheme 1). The reason for this may be the instability of the C(O)-P(O) bond of trimethyl phosphonoformate under basic conditions.32 Therefore, an alternative synthetic route was devised (Scheme 2).
Scheme 1.
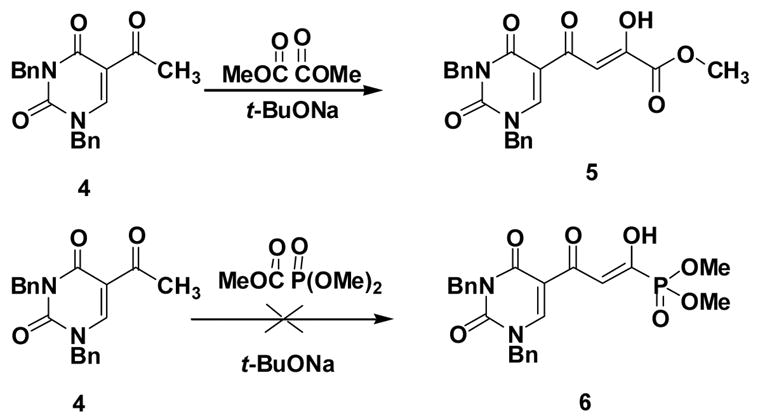
The difference in the reactivity of 4 with dimethyl oxalate compared to trimethyl phosphonoformate.
Scheme 2.
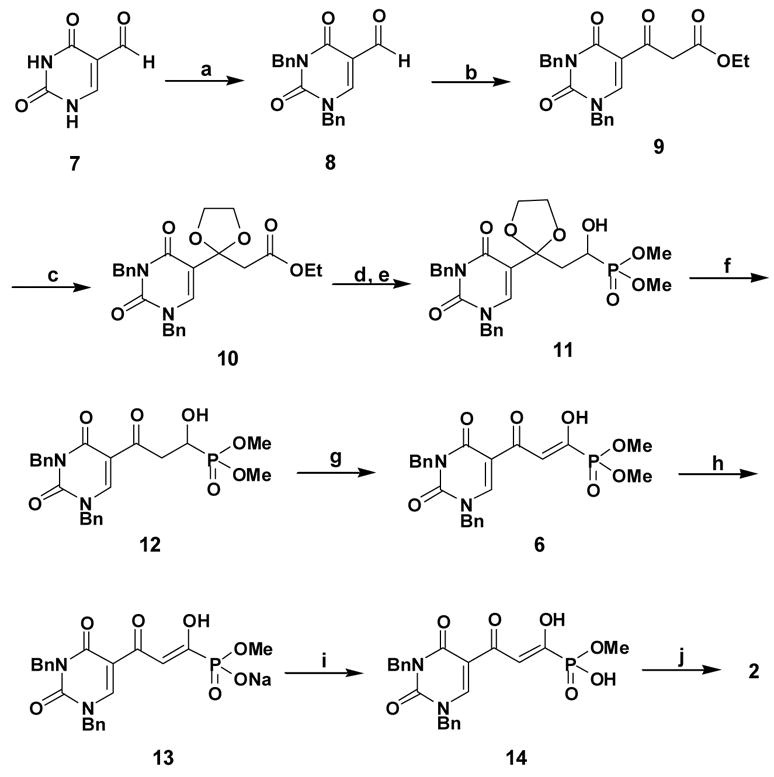
Synthesis of integrase inhibitor 2.
Reagents and conditions: a) BnBr/DMF/K2CO3; b) N2CHCO2Et/SnCl2/CH2Cl2; c) Ethylene glycol/CH(OEt)3/PTS; d) Dibal-H/toluene /−78°C; e) HP(O)(OMe)2/Et3N/MeOH; f) PTS/acetone/H2O; g) Dess-Martin periodinane/CH2Cl2; h) NaI/acetone; i) Dowex50Wx8-100/MeOH; j) NaI/acetone/reflux.
Thus, 5-formyl uracil 7 was benzylated to give 8 (89%). Compound 8 was condensed with ethyl diazoacetate in the presence of tin (II) chloride33 to afford 9, which was transformed into its protected form 10 (28% yield from 8). Reduction of 10 to the corresponding aldehyde with diisobutylaluminium hydride at −78°C, followed by a Pudovik reaction with dimethyl phosphite and triethylamine, proceeded to form the phosphonate 11 (50% for two steps). The ketal group of 11 was deprotected to produce the key intermediate 12, which was purified by HPLC (65% yield) and fully characterized by multinuclear NMR and HRMS data.34 Dess-Martin oxidation of 12 produced diketone 6, which exists largely in the enolic form. Compound 6 was deprotected by stirring with NaI in acetone for 3 days.35 The resulting precipitate was collected and washed with acetone to afford the sodium methyl phosphonate 13 (67% yield). However, compound 13 could not be further deprotected with NaI, even under acetone reflux conditions, possibly because of the presence of the negative charge on the phosphonyl group and the poor solubility of 13 in acetone.35 The problem was circumvented by conversion of 13 to its protonated form by ion-exchange chromatography with Dowex 50×8 in methanol to afford 14. When compound 14 was heated under reflux with 5 equivalents of NaI in acetone for 24 h and the resulting crystalline precipitate was collected and washed with acetone, the target phosphonate 2 (monosodium salt) was produced in a highly purified form (51% yield). Compound 3 was synthesized by a similar procedure to compound 2. The structures of 2 and 3 were confirmed by 1H, 13C and 31P NMR and HRMS data.34
Integrase inhibition studies were conducted with recombinant wild-type HIV-1 integrase and a 21-mer oligonucleotide substrate following a previously described procedure.36,37 Target compound 2 was not an inhibitor of the 3'-processing step of HIV integrase (IC50 >333 μM) but inhibited the strand transfer step (IC50 20.2 ±7.2 μM).38 The integrase inhibition activity of 2 is in sharp contrast to that of compound 1, which is a potent inhibitor of both the 3'-processing and strand transfer steps of integrase (IC50 3.7 ±1.0 μM and 0.2 ±0.1μM, respectively29,39 and 14.4 ± 1.6 μM and 4.5 ± 2.5 ± μM, respectively38).
Compound 2 was tested in a PBMC cell-based, microtiter anti-HIV assay against the clinical isolate, HIV-1TEKI (NSI phenotype) and HIV-1NL4-3 (SI phenotype).29 Antiviral determinations were performed in triplicate with serial ½log10 dilution of the test materials (six to nine concentrations total). The overall performance of both assays was validated by the MOI-sensitive positive control compound, AZT. As summarized in Table 1, in vitro anti-HIV studies against HIV-1 isolates in PBMC showed that compound 2 (highest test concentration = 200 μM) was active with IC50 values in the low μM range and with antiviral efficacy data [therapeutic indices, TI = CC50/IC50 of >50 (HIV-1TEKI) and >62 (HIV-1NL4-3)]. Cell viability data showed only mild cellular cytotoxicity at the highest test concentrations; however, a CC50 (>200 μM) was not reached. In contrast, compound 3 was significantly less active and more toxic [TI = 5.8 (HIV-1TEKI) and 4.0 (HIV-1NL4-3)]. The control compound, AZT (highest test concentration = 1 μM), gave therapeutic indices of >504 (HIV-1TEKI) and >573 (HIV-1NL4-3).
Table 1.
Antiviral data for compounds of Figure 1 in PBMC
| Compds | HIV-1 Isolate Cell Line | IC50 | CC50 | TI |
|---|---|---|---|---|
| 1# | HIV-1TEKI (PBMC) | 50 nM | >200 μM | >4,000 |
| HIV-1NL4-3 (PBMC) | <20 nM | >200 μM | >10,000 | |
| L708906† | HIV-1 (IIIB) (MT-4) | 5.5 μM | 88.3 μM | 16 |
| 2 | HIV-1TEKI (PBMC) | 4.0 μM | >200 μM | >50 |
| HIV-1NL4-3 (PBMC) | 3.2 μM | >200 μM | >62 | |
| 3 | HIV-1TEKI (PBMC) | 8.1 μM | 46.9 μM | 5.8 |
| HIV-1NL4-3 (PBMC) | 11.8 μM | 46.9 μM | 4.0 |
In summary, we have developed a methodology for the synthesis of a new β-diketo phosphonic acid, 2, an isostere of our highly-active, anti-HIV compound, 1. In complete contrast to 1, compound 2 specifically inhibits only the strand transfer step of WT HIV-1 integrase. Anti-HIV data show that the exchange of the carboxylate functionality with a phosphonate results in a reduction in in vitro anti-HIV activity. The anti-HIV data in PBMC may be a reflection of the difference in integrase inhibitory activity of 1 compared to 2 and the result of decreased cellular permeability of phosphonate 2 compared to the less polar carboxylate 1. Interestingly, compound 2 is more anti-HIV active and less toxic than the phosphonic acid analogue of L-708906 (compound 3). Finally, these isosteric compounds represent the first examples of β-diketo phosphonic acids of structural, synthetic and antiviral interest.
Figure 1.
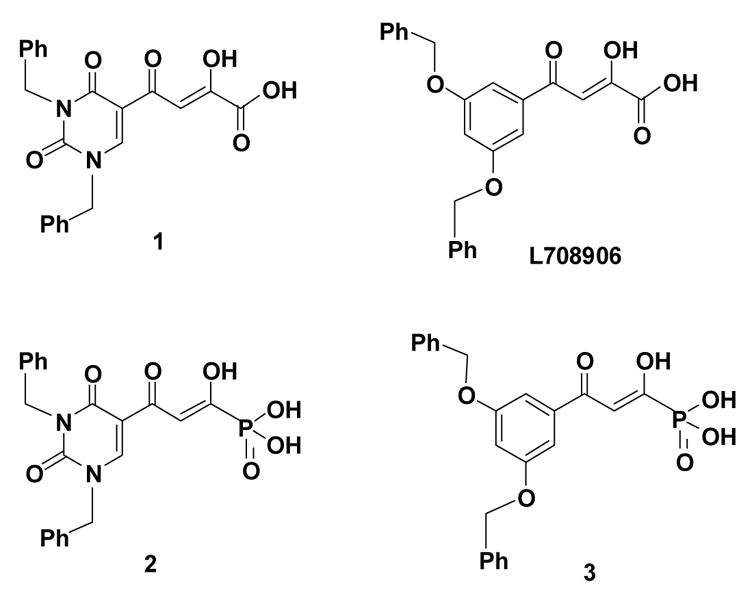
Structures of two biologically-active diketo acids (top) and their phosphonic acid isosteres (bottom).
Acknowledgments
This project was supported by Grant No. RO1 AI 43181 from the National Institutes of Health (NIAID) and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. We thank the SRI for the anti-HIV screening data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Fauci AS. Science. 1988;239:617. doi: 10.1126/science.3277274. [DOI] [PubMed] [Google Scholar]
- 2.Katz RA, Skalka AM. Annu Rev Biochem. 1994;63:133. doi: 10.1146/annurev.bi.63.070194.001025. [DOI] [PubMed] [Google Scholar]
- 3.Frankel AD, Young JAT. Annu Rev Biochem. 1998;67:1. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 4.De Clercq E. Nature Rev Drug Discovery. 2002;1:13. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 5.De Clercq E. Clin Microbiol Rev. 1997;10:674. doi: 10.1128/cmr.10.4.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson SC, Gerber JG. In: Advances in Internal Medicine. Schrier RW, Baxter JD, Dzau VJ, Fauci AS, editors. Vol. 45. Mosby; St. Louis: 2000. pp. 1–40. [PubMed] [Google Scholar]
- 7.Nair V, St Clair MH, Reardon JE, Krasny HC, Hazen RJ, Paff MT, Boone LR, Tisdale M, Najera I, Dornsife RE, Averett DR, Borroto-Esoda K, Yale JL, Zimmerman TP, Rideout JL. Antimicrob Agents Chemother. 1995;39:1993. doi: 10.1128/aac.39.9.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Clercq E. J MedChem. 2005;48:1297. [Google Scholar]
- 9.Pommier Y, Johnson AA, Marchand C. Nature Rev Drug Discovery. 2005;4:236. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 10.Nair V. Frontiers in Med Chem. 2005;2:3. [Google Scholar]
- 11.Dayam R, Neamati N. Curr Pharm Design. 2003;9:1789. doi: 10.2174/1381612033454469. [DOI] [PubMed] [Google Scholar]
- 12.Asante-Appiah E, Skalka AM. Adv Virus Res. 1999;52:351. doi: 10.1016/s0065-3527(08)60306-1. [DOI] [PubMed] [Google Scholar]
- 13.Esposito D, Craigie R. Adv Virus Res. 1999;52:319. doi: 10.1016/s0065-3527(08)60304-8. [DOI] [PubMed] [Google Scholar]
- 14.Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Science. 1994;266:1981. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 15.Wu Y, Marsh JW. Science. 2001;293:1503. doi: 10.1126/science.1061548. [DOI] [PubMed] [Google Scholar]
- 16.Espeseth AS, Felock P, Wolfe A, Witmer M, Grobler J, Anthony N, Egbertson M, Melamed JY, Young S, Hamill T, Cole JL, Hazuda DJ. Proc Natl Acad Sci USA. 2000;97:11244. doi: 10.1073/pnas.200139397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazumder A, Uchida H, Neamati N, Sunder S, Jaworska-Maslanka M, Wickstrom E, Zeng F, Jones RA, Mandes RF, Chenault HK, Pommier Y. Mol Pharmacol. 1997;51:567. doi: 10.1124/mol.51.4.567. [DOI] [PubMed] [Google Scholar]
- 18.Taktakishvili M, Neamati N, Pommier Y, Pal S, Nair V. J Am Chem Soc. 2000;122:5671. [Google Scholar]
- 19.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Science. 2000;287:646. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 20.Wai JS, Egbertson MS, Payne LS, Fisher TE, Embrey MW, Tran LO, Melamed JY, Langford HM, Guare JP, Jr, Zhuang L, Grey VE, Vacca JP, Holloway MK, Naylor-Olsen AM, Hazuda DJ, Felock PJ, Wolfe AL, Stillmock KA, Schleif WA, Gabryelski LJ, Young SD. J Med Chem. 2000;43:4923. doi: 10.1021/jm000176b. [DOI] [PubMed] [Google Scholar]
- 21.Marchand C, Zhang X, Pais GCG, Cowansage K, Neamati N, Burke TR, Jr, Pommier Y. J Biol Chem. 2002;277:12596. doi: 10.1074/jbc.M110758200. [DOI] [PubMed] [Google Scholar]
- 22.Hazuda DJ, Young SD, Guare JP, Anthony NJ, Gomez RP, Wai JS, Vacca JP, Handt L, Motzel SL, Klein HJ, Dornadula G, Danovich RM, Witmer MV, Wilson KAA, Tussey L, Schleif WA, Gabryelski LS, Jin L, Miller MD, Casimiro DR, Emini EA, Shiver JW. Science. 2004;305:528. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- 23.Grobler JA, Stillmock K, Hu B, Witmer M, Felock P, Espeseth AS, Wolfe A, Egbertson M, Bourgeois M, Melamed J, Wai JS, Young S, Vacca J, Hazuda DJ. Proc Natl Acad Sci USA. 2002;99:6661. doi: 10.1073/pnas.092056199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldgur Y, Craigie R, Cohen GH, Fujiwara T, Yoshinaga T, Fujishita T, Sugimoto H, Endo T, Murai H, Davies DR. Proc Natl Acad Sci USA. 1999;96:13040. doi: 10.1073/pnas.96.23.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshinaga T, Sato A, Fujishita T, Fujiwara T. 9th Conference on Retroviruses and Opportunistic Infections; Seattle, WA. February 24–28, 2002; Abstract 8, p 55. [Google Scholar]
- 26.Pannecouque C, Pluymers W, Van Maele B, Tetz V, Cherepanov P, De Clercq E, Witvrouw M, Debyser Z. Curr Biol. 2002;12:1169. doi: 10.1016/s0960-9822(02)00952-1. [DOI] [PubMed] [Google Scholar]
- 27.Pais GCG, Zhang X, Marchand C, Neamati N, Cowansage K, Svarovskaia ES, Pathak VK, Tang Y, Nicklaus M, Pommier Y, Burke TR., Jr J Med Chem. 2002;45:3184. doi: 10.1021/jm020037p. [DOI] [PubMed] [Google Scholar]
- 28.Sato M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, Kawakami H, Matsuzaki Y, Watanabe W, Yamataka K, Ikeda S, Kodama E, Matsuoka M, Shinkai H. J Med Chem. 2006;49:1506. doi: 10.1021/jm0600139. [DOI] [PubMed] [Google Scholar]
- 29.Nair V, Chi G, Ptak R, Neamati N. J Med Chem. 2006;49:445. doi: 10.1021/jm0508890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nair V, Uchil V, Neamati N. Bioorg Med Chem Lett. 2006;16:1920. doi: 10.1016/j.bmcl.2005.12.093. [DOI] [PubMed] [Google Scholar]
- 31.Kukhar VP, Hudson HR, editors. Aminophosphonic and Aminophosphinic Acids, Chemistry and Biological Activity. John Wiley and Sons, Ltd; Chichester: 2000. [Google Scholar]
- 32.Sekine M, Kume A, Nakajima M, Hata T. Chem Lett. 1981:1087. [Google Scholar]
- 33.Holmquist CR, Roskamp EJ. J Org Chem. 1989;54:3258. [Google Scholar]
- 34.Data for dimethyl 3-(1,3-dibenzyl-2,4-dioxo-1,2,3,4-tetra-hydropyrimidin-5-yl)-1-hydroxy-3-oxopropylphosphonate (12). 1HNMR (CDCl3): 8.29 (s, 1H), 7.30–7.50 (m, 10H), 5.18 (s, 2H), 5.05 (d, 1H, J=15), 5.02 (d, 1H, J=15), 4.61 (m, 1H), 3.87 (d, 3H, J=11.0), 3.86 (d, 3H, J=10.5), 3.57–3.64 (m, 1H), 3.42–3.48 (m, 1H). 13CNMR (CDCl3): 194.9 (d, J=17.6), 160.8, 150.9, 149.3, (136.2, 134.3, 129.5, 129.2, 129.1, 128.7, 128.5, 128.1, phenyl ring), 111.7 (d, J=2.31), 64.3 (d, J=169.4), 53.8, (53.72, 53.68, 53.66, 53.61, P-OCH3), 45.1, 43.9 (d, J=3.82). 31PNMR (CDCl3): 26.5. FAB-HRMS: calcd for C23H26N2O7P (M+H)+ 473.1478, found 473.1469. Data for 3-(1,3-dibenzyl-2,4-dioxo-1,2,3,4-tetrahydro-pyrimidin-5-yl)-1-hydroxy-3-oxoprop-1-enylphosphonic acid (2) as its monosodium salt: mp 224°C(dec.). 1HNMR (CD3OD): 8.58 (s, 1H), 7.22–7.39 (m, 11H), 5.12 (s, 2H), 5.10 (s, 2H). 13CNMR (CD3OD): 188.1 ( d, J=14.5), 185.9 (d, J=190.4), 161.5, 152.5, 150.6, (138.3, 137.1, 130.2, 129.6, 129.5, 129.3, 128.7, phenyl ring), 111.2 (d, J=4.90), 104.5 (d, J=24.4), 54.4, 45.7. 31PNMR (CD3OD): 0.82. FAB-HRMS: calcd for C21H19N2NaO7P (M+H)+ 465.0828, found 465.0840. Data for 3-(3,5-bis(benzyloxy)phenyl)-1-hydroxy-3-oxoprop-1-enyl-phosphonic acid (3) as its monosodium salt: mp 201°C(dec.). 1HNMR (CD3OD): 7.29-7.46 (m, 10H), 7.25 (d, 2H, J=2.5), 6.89 (d, 1H, J=7.0), 6.85 (t, 1H, J=2.5), 5.12 (s, 4H). 13CNMR (CD3OD): 193.3 ( d, J=13.4), 185.0 (d, J=191.3), [161.8, 140.0 (d, J=4.27), 138.4, 129.7, 129.2, 128.9, 108.3, 107.8, phenyl ring], 101.6(weak), 71.5. 31PNMR (CD3OD): 0.91. FAB-HRMS: calcd for C23H21NaO7P (M+H)+ 463.0923, found 463.0927.
- 35.Karaman R, Goldblum A, Breuer E, Leader H. J Chem Soc, Perkin Trans 1. 1989:765. [Google Scholar]
- 36.Mazumder A, Neamati N, Sundar S, Owen J, Pommier Y. In: Antiviral Methods and Protocols. Kinchington D, Schinazi R, editors. Vol. 24. Humana; Totowa: 1999. pp. 327–338. [Google Scholar]
- 37.Marchand C, Johnson AA, Karki RG, Pai GC, Zhang X, Cowansage K, Burke TR, Pommier Y. Mol Pharmacol. 2003;64:600. doi: 10.1124/mol.64.3.600. [DOI] [PubMed] [Google Scholar]
- 38.Magnesium chloride-based assay.
- 39.Manganese chloride-based assay.