

Abstract
S-Nitrosothiols have been implicated as intermediary transducers of nitric oxide bioactivity; however, the mechanisms by which these compounds affect cellular functions have not been fully established. In this study, we have examined the effect of S-nitrosothiol transport on intracellular thiol status and upon the activity of a target protein (caspase-3), in bovine aortic endothelial cells. We have previously demonstrated that the specific transport of amino acid-based S-nitrosothiols (S-nitroso-L-cysteine and S-nitrosohomocysteine) occurs via amino acid transport system L to generate high levels of intracellular protein S-nitrosothiols (Zhang, Y., and Hogg, N. (2004) Proc. Natl. Acad. Sci. U. S. A. 101, 7891–7896). In this study, we demonstrate that the transport of S-nitrosothiols is essential for these compounds to affect intracellular thiol levels and to modify intracellular protein activity. Importantly, the ability of these compounds to affect intracellular processes occurs independently of nitric oxide formation. These findings suggest that the major action of these compounds is not to liberate nitric oxide in the extracellular space but to be specifically transported into cells where they are able to modify cellular functions through nitric oxide-independent mechanisms.
S-Nitrosothiols (RSNO),2 thioesters of nitrite, have been implicated to play a role in the complex biological responses of nitric oxide (NO). S-Nitrosation has been observed in whole animals and in the cell systems under basal (1–3) and inflammatory conditions (4, 5). It has been suggested that S-nitrosation of enzyme cysteine residues represents a post-translation modification involved in signal transduction and the alteration of enzyme function in a similar way to phosphorylation (6). Indeed, there are numbers of protein targets that have been reported to be nitrosated on cysteine residues including p21ras (7), L-type Ca2+ channels (8), transcription factor NF-κB (9), inhibitory κB kinase (10), and caspase-3 (11, 12).
Although the evidence is strong, in test systems, that thiol nitrosation can affect many cellular processes and enzyme activities, it is less clear that this represents a physiological or a pathophysiological effect of NO formation. NO per se is a poor and inefficient nitrosating agent (13), and intracellular systems that catalyze nitrosation from NO have not yet been defined. Although the plasma copper-protein ceruloplasmin has been shown to catalyze RSNO formation (14), this has not been demonstrated in whole blood containing high concentrations of hemoglobin, a potent NO scavenger (15). From a pharmacological perspective, however, it has been demonstrated in many studies that the exposure of cells to low molecular weight RSNOs, such as S-nitrosoglutathione (GSNO), S-nitroso-N-acetylpenicillamine (SNAP), and S-nitroso-L-cysteine (L-CysNO), can affect cellular processes (16). Although it is often assumed that these compounds spontaneously release NO in cell culture and that the effects of these compounds can be attributed to NO release, this assumption is almost never tested. In contradiction to this assumption, it has been shown that the effects of some RSNOs have a chiral dependence (17), that the decomposition of RSNOs depends upon the presence of cells (18, 19), and that RSNOs of simple amino acids can be taken up into cells via the amino acid transport system L (L-AT) (13, 20, 21). In addition, we have previously demonstrated that the nitrosation of intracellular proteins by low molecular weight RSNOs does not depend upon the release of NO (13). Taken together, these observations suggest that low molecular weight RSNOs, such as L-CysNO, affect cellular processes not through the release of NO in the extracellular space but by transport into the cell and direct transfer of the nitroso functional group to a target thiol.
In this study, we have tested the idea that low molecular weight RSNOs affect intracellular thiol modification via a transport mechanism and without the intermediacy of NO. In addition, we have examined whether NO can alter intracellular processes in the same manner as low molecular weight RSNOs. We show, by measuring total cellular nitrosation, cellular GSH levels, and the activity of a specific target protein (caspase-3), that the effects of L-CysNO on thiolic targets depend exclusively on the transport of L-CysNO into the cell and are not dependent on the formation of NO. In addition, we show that these functions cannot be recapitulated by exposing cells to NO. We conclude that amino acid-based RSNOs access a different set of signaling opportunities than NO and that the formation of such RSNOs could be thought of as a bifurcation in NO signaling pathways.
EXPERIMENTAL PROCEDURES
Materials
Acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (Ac-DEVD-AMC), acetyl-Asp-Glu-Val-Asp-aldehyde (Ac-DEVD-CHO), caspase-3, L-cysteine, L-cystine, sodium nitrite, diethylenetriamine pentaacetic acid (DTPA), GSH, D/L-homocysteine, N-acetyl-D/L-penicillamine, N-ethylmaleimide (NEM), CHAPS, EDTA, iodine, mercury chloride, and protease inhibitor mixture were purchased from Sigma. Dithiothreitol (DTT), HEPES, hydrochloric acid, potassium iodide, potassium phosphate monobasic, potassium phosphate dibasic, and sodium hydroxide were obtained from Fisher. L-glutamic acid monosodium salt monohydrate and sulfanilamide were supplied by Aldrich. (Z)-1-[N-(3-Ammoniopropyl)-N-[4-(3-aminopropylammonio)butyl]-amino]diazen-1ium-1,2-diolate (Sper/NO) was from Cayman Chemical. 2-(N,N-Diethylamino)-diazenolate-2-oxide (Dea/NO) was obtained from Alexis Biochemicals. Bovine aortic endothelial cells (BAECs) were purchased from Cambrex Corp. Dulbecco’s modified Eagle’s medium, streptomycin/penicillin, Hepes buffer solution, PBS, trypsin EDTA, and Hanks’ balanced salt solution (HBSS) were obtained from Invitrogen, and fetal bovine serum was obtained from HyClone. Oxyhemoglobin (oxyHb) was purified from fresh human blood as described previously (22) and was reacted with NEM to block free thiols followed by purification on G25 Sephadex column. OxyHb concentration was determined by measuring the absorbance at 577 nm (ε577 = 14.4 mM−1 cm−1). GSNO, SNAP, and L-CysNO were synthesized as described previously (3, 13, 23, 24).
Cell Culture and Treatment
BAECs were routinely cultured as described previously (25). For each experiment, cells were seeded into 6-well plates and grown overnight to reach 70–80% confluence. The old medium was removed by double wash with PBS. Specific compounds were added to cells either in full medium or in HBSS (supplemented with 10 mM Hepes). Cells used in this study were between passages 5–12.
S-Nitrosothiol Determinations
For detection of RSNOs, cells were washed twice with PBS, and 250 μl of lysis buffer (50 mM phosphate, 1 mM DTPA, 50 mM NEM, pH 7.4) was added to the cells. Cells were scraped and sonicated (550 Sonic Dismembrator from Fisher Scientific, power level 2 for 15 s) followed by centrifugation (12,000 ×g for 5 min). The supernatant was used for measurement of RSNOs in the triiodide-dependent ozone-based chemiluminescence method as described previously (3, 13).
Thiol Determination
The HPLC-based ammonium 7-fluorobenzo-2-oxa-1,3-diazole-4-sulfonate method was used to measure intracellular thiols (26). Briefly, BAECs were washed twice with PBS and scraped into 200 μl of buffer (50 mM phosphate, 1 mM DTPA, pH 7.4) followed by sonication and centrifugation. 25 μl of sample was mixed with 25 μl of a chilled solution of 10% trichloroacetic acid and spun down to precipitate protein. 25 μl of resulting supernatant was mixed with 75 μl of ammonium 7-fluorobenzo-2-oxa-1,3-diazole-4-sulfonate (0.66 mg/ml in 2.5 M borate, pH 9.5) and incubated for 1 h at 60 °C. The analysis was performed on Kromasil C-18 column (5 μm, 250 × 4.6 mm inner diameter, Alltech) using solvent A (0.1 M sodium acetate, pH 4, methanol; 98:2), solvent B (0.1 M sodium phosphate, pH 6, methanol; 95:5), and methanol. The chromatographic conditions were as follows: solvent A to solvent B, 0–20 min (0–100%); methanol, 20–30 min; solvent A, 30–35 min. The retention times for cysteine, homocysteine, and GSH were 5.6, 6.6, and 10 min, respectively.
Caspase Assay
Caspase-3 activity was induced with doxorubicin (0.5 μM, 8 h in full medium). After double wash with PBS, specific compounds were added to cells in HBSS and incubated for 1 h. For detection of caspase-3 activity, cells were lysed in buffer without or with 5 mM DTT (50 mM HEPES, 5 mM CHAPS, pH 7.4) followed by centrifugation (14,000 × g for 15 min at 4 °C). Caspase-3 activity in resulting supernatants was determined by measuring the cleavage of fluorometric substrate Ac-DEVD-AMC in assay buffer without or with 10 mM DTT (20 mM HEPES, 0.1% CHAPS, 2 mM EDTA, pH 7.4). After incubating at 37 °C for 1 h, fluorescence was monitored (excitation 360 nm, emission 460 nm). The specificity of the assay was confirmed with purified caspase-3 and the caspase-3 inhibitor, Ac-DEVD-CHO.
Statistical Analysis
All data presented in this study represent the mean ± S.E. of at least three experiments. Statistical analyses were performed using the Student’s t test. A two-tailed value of p < 0.05 was considered to indicate statistical significance. All comparatives made in the study were significant to at least this level.
RESULTS
Effects of RSNO and NO on Intracellular S-Nitrosation in BAEC
We have previously demonstrated that exposure of RAW 264.7 cells to the low molecular weight RSNO, L-CysNO, but not the NO donor, Sper/NO, resulted in extensive intracellular RSNO formation. We have also demonstrated that L-CysNO, but not GSNO, is directly transported in RAW 264.7 cells via L-AT transport system (13). We first examined whether BAEC behaved in a qualitatively similar way. As shown in Fig. 1A, incubation of cells with either Sper/NO or GSNO resulted in a modest increase in intracellular RSNO levels as measured by triiodide-based chemiluminescence. However, incubation with L-CysNO resulted in ~100-fold higher levels. The RSNOs in cell lysate were retained by a 3-kDa cut off Centricon filter, indicating that the vast majority of detected RSNOs were protein-associated (data not shown). In all cases, treatment of samples with HgCl2 diminished the signal by greater than 95%, demonstrating that the majority of the chemiluminescence signal was derived from RSNOs and not from other nitroso or nitrosyl species. To examine whether the L-AT system was involved in L-CysNO transport in BAEC, we examined the effects of L-leucine, a competitor for the L-AT system, on intracellular RSNO formation. As shown in Fig. 1A, L-leucine inhibited intracellular RSNO formation. The presence of oxyHb, a potent NO scavenger, did not affect the level of intracellular RSNO formed during incubation of BAEC with L-CysNO. This confirms that L-CysNO directly modifies intracellular thiols without the intermediacy of NO. In this and all following experiments, we have used oxyHb that has been pretreated with NEM to prevent loss of L-CysNO due to transnitrosation to hemoglobin thiols (27). As shown in Fig. 1B, the formation of intracellular RSNO after exposure of cells to L-CysNO is concentration-dependent, approaching a maximum of about 20 nmol of RSNO/mg of protein. These results indicate that BAEC are qualitatively identical to RAW 264.7 cells with respect to RSNO transport.
FIGURE 1. Intracellular RSNO formation after exposure of BAECs to NO and RSNO.
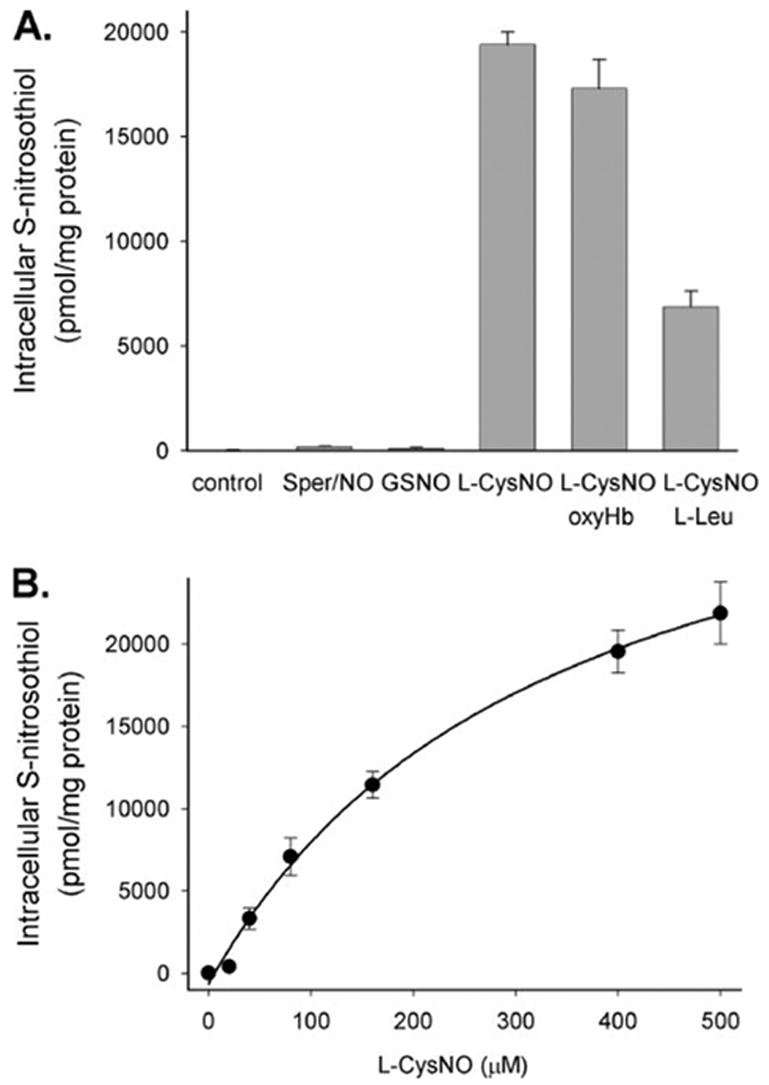
A, BAECs were treated with Sper/NO (400 μM), GSNO (400 μM), or L-CysNO (160 μM) for 60 min in HBSS. In the case of L-CysNO, cells were coin-cubated with or without oxyHb (160 μM) or L-Leu (8 mM), a NO scavenger and L-AT competitor, respectively. B, BAECs were treated with various concentrations of L-CysNO for 60 min in HBSS. Intracellular RSNO levels were measured by chemiluminescence. Data were normalized to protein concentration and represent mean ± S.E. (n = 3).
Effects of L-CysNO Uptake on Intracellular GSH
The concentration dependence of L-CysNO on the level of intracellular GSH is shown in Fig. 2A. At low concentrations, L-CysNO had no significant effect on GSH levels, but above 40 μM L-CysNO, GSH was substantially depleted in a concentration-dependent manner. At 400 μM L-CysNO, less than 5% of the original GSH level remained. In contrast, treatment with 400 μM Sper/NO for 1 h resulted in the drop in GSH concentration to 72.0 ±0.5% of control levels (data not shown). To examine whether L-CysNO-dependent GSH depletion depended upon the transport of L-CysNO, we examined the effect of L- and D-leucine. As shown in Fig. 2B, L-leucine strongly inhibited L-CysNO-mediated GSH depletion, whereas D-leucine was much less effective, indicating that uptake of L-CysNO via the L-AT system was required for GSH depletion. OxyHb did not affect L-CysNO-mediated GSH depletion, excluding the involvement of NO in this process. In combination, these data show that L-CysNO uptake results in the formation of intracellular RSNO and the loss of GSH.
FIGURE 2. Intracellular thiol levels after exposure of BAEC to L-CysNO and the metabolism of GSNO by BAEC lysate.
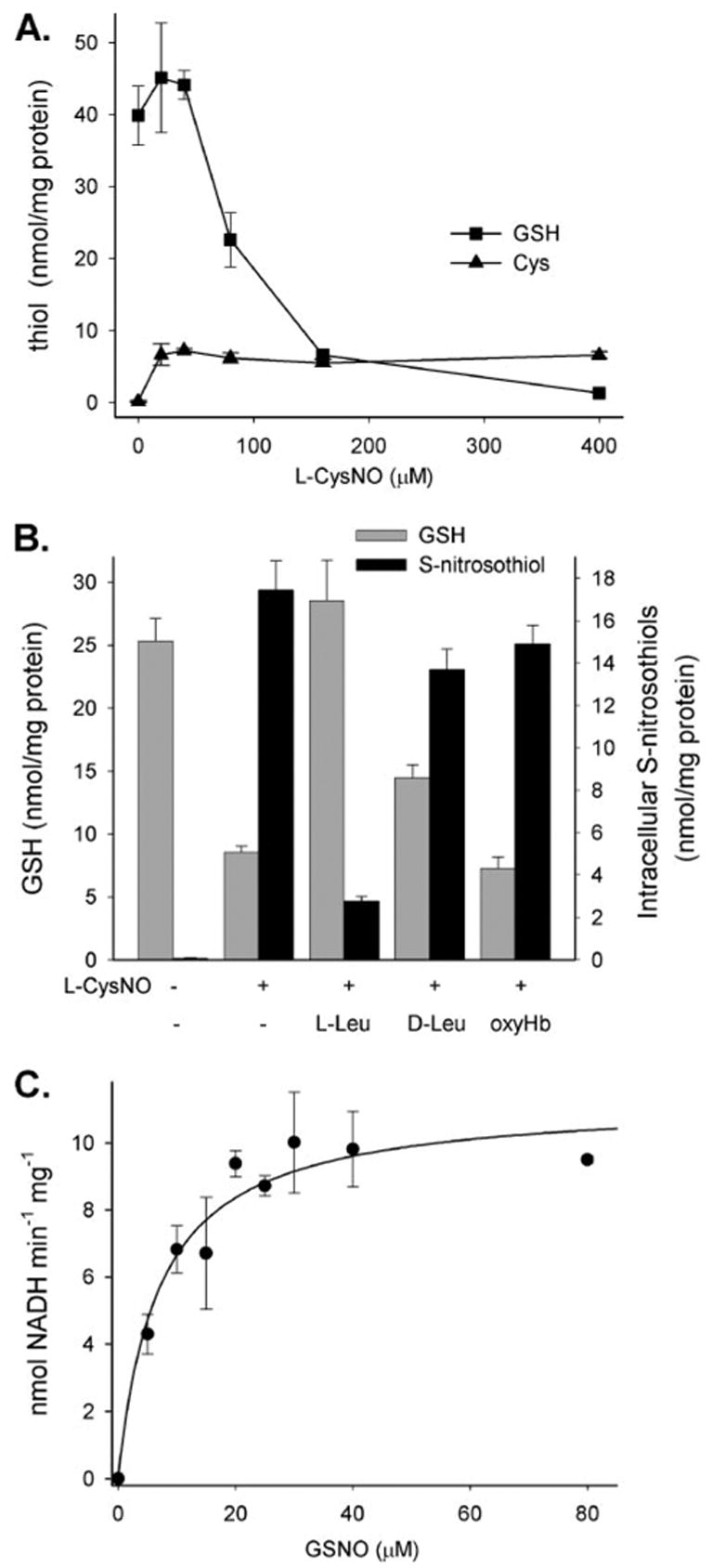
A, BAECs were treated with L-CysNO for 60 min in HBSS. GSH (squares) and cysteine (triangles) levels in cell lysates were determined by HPLC. Data were normalized to protein concentration and represent mean ± S.E. (n = 3). B, BAECs were treated with L-CysNO (160 μM) in the absence or presence of L-AT competitors (L-leucine, D-leucine, 8 mM) and NO scavenger oxyHb (160 μM). GSH level (gray bars) in cell lysates was determined by HPLC, and RSNO levels (black bars) were measured by chemiluminescence. Data were normalized to protein concentration and represent mean ± S.E. (n = 3). C, the rate of NADH (200 μM) consumption at varying concentrations of GSNO was measured as a decrease in absorption at 340 nm. The initial velocity of reaction was calculated as nmol NADH/min/mg of protein. A rectangular hyperbola was fitted to the data points (solid line). The data represent mean ± S.E. (n = 3).
It has previously been demonstrated that GSH-dependent formaldehyde dehydrogenase (GSH-FDH) exhibits an NADH-dependent GSNO reductase activity, which could account for GSH loss (28, 29). By this mechanism, L-CysNO enters the cell and transfers the nitroso group to GSH, forming GSNO, which is a substrate for GSH-FDH. To examine whether BAECs contain a GSNO reductase, we examined GSNO-dependent NADH oxidation by BAEC cell lysate. No increase in NADH oxidation was observed in the absence of cell lysate at all GSNO concentrations tested. In the presence of BAEC cell lysate, we observed a robust GSNO reductase activity that was NADH-but not NADPH-dependent (data not shown). As shown in Fig. 2C, this activity obeyed Michaelis-Menten kinetics, giving a Km value of 7 ± 1.8 μM. This value is somewhat lower than the published value of 28 μM for purified GSH-FDH (28). For these experiments, we monitored the change in absorption of NADH at 340 nm. Although GSNO also absorbs light at this wavelength, its extinction coefficient is about 10% of NADH. When a correction was made for this (assuming a 1:1 stoichiometry for NADH:GSNO), Vmax was reduced by 10%, but there was no effect on the Km.
Effects of L-CysNO and hCysNO on Intracellular Thiol Levels
Incubation of BAEC with L-CysNO resulted in an increase in intracellular cysteine levels (Fig. 2A). This increase exhibited little concentration dependence and was maximal at the lowest concentration tested. To examine whether this increase in cysteine was also attributable to uptake via the L-AT system, we examined the effects of L-leucine on intracellular cysteine levels after exposure to L-CysNO. As shown in Fig. 3, L-leucine significantly inhibited (p = 0.015) the accumulation of intracellular cysteine, although the effect was not dramatic. We have previously demonstrated that homocysteine could facilitate GSNO-dependent intracellular nitrosation, suggesting that hCysNO is also a substrate for the L-AT receptor (13). In Fig. 3, we examined the effect of hCysNO on intracellular thiol levels. In a similar manner to L-CysNO, hCysNO caused a drop in intracellular GSH levels (data not shown) and an increase in intracellular homocysteine levels by a mechanism that can be extensively inhibited by L-leucine. These data suggest that nitrosated thiolic amino acids are taken up intact into cells as opposed to transfer of only the nitroso group across the membrane.
FIGURE 3. Effect of RSNO treatment on intracellular thiol levels.
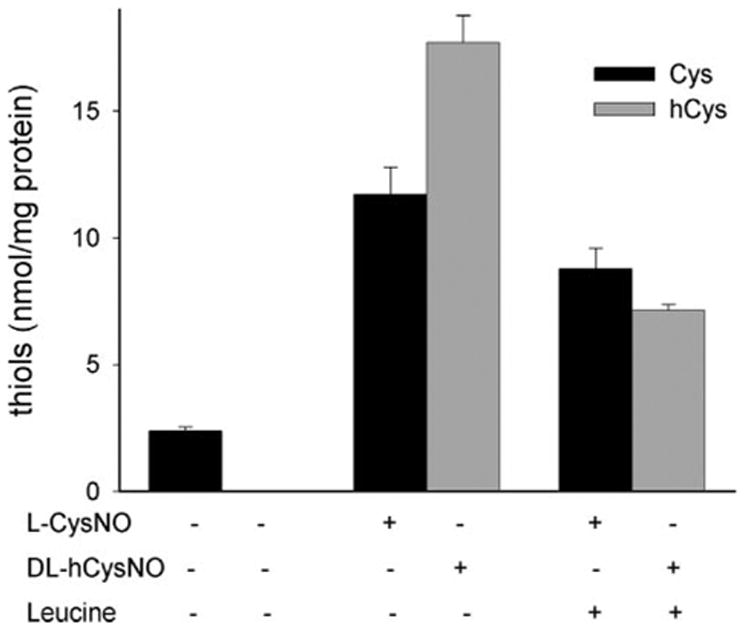
BAECs were treated with L-CysNO (160 μM) or DL-hCysNO (160 μM) in the absence or presence of the L-AT competitor, L-leucine (8 mM). Cysteine (black bars) and homocysteine (gray bars) levels in cell lysates were determined by HPLC. Data were normalized to protein concentration and represent mean ± S.E. (n = 3).
Inhibition of Caspase-3
To examine whether the uptake of L-CysNO can affect the activity of a target protein, we induced the activation of caspase-3 in BAEC by preincubation with doxorubicin (30). Incubation of cells with doxorubicin increased caspase-3 activity ~20-fold over control levels (see legends of Figs. 4 and 5), and the measured activity was reduced to control levels in the presence of the caspase-3 inhibitor, Ac-DEVD-CHO (data not shown). For the following studies, the activity of caspase-3 was assayed both without and with DTT. The ratio of these two values gives an index of the amount of enzyme that was inhibited by reversible modification of the active site thiol. Incubation of doxorubicin-pretreated BAEC with L-CysNO had little effect on caspase-3 activity in the presence of DTT, apart from a 26 ± 5% drop at the highest concentration used (400 μM). As shown in Fig. 4, the ratio of DTT-untreated:DTT-treated caspase-3 activity decreased at 80 μM L-CysNO and above, indicating that higher concentrations of L-CysNO are able to modify caspase-3 at the critical active site thiol and inhibit enzyme activity.
FIGURE 4. Concentration-dependent effects of CysNO on caspase-3-like activity.
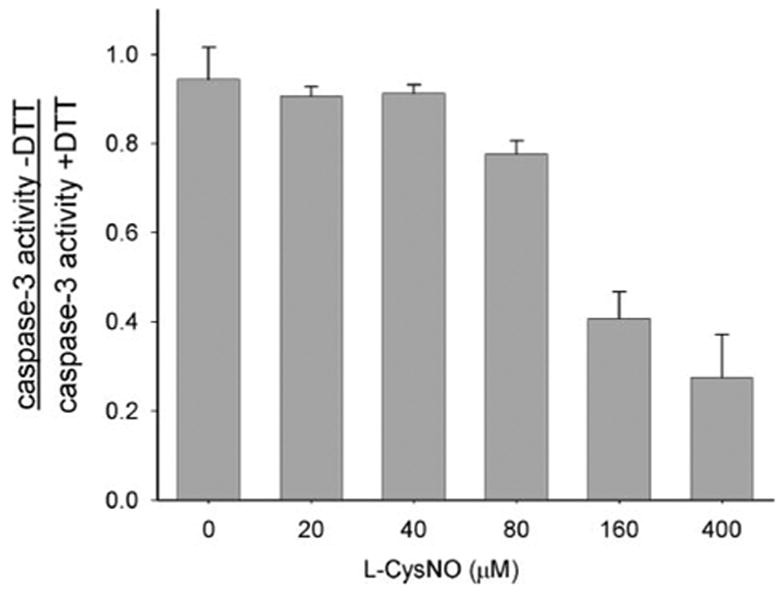
BAECs were incubated with doxorubicin (0.5 μM) for 8 h in medium to induce caspase-3 activity. Cells were washed and incubated for a further 1 h with L-CysNO in HBSS. The caspase-3-like activity was determined in cell lysate in the presence and absence of DTT and expressed as the ratio of caspase-3 activity − DTT:caspase-3 activity + DTT. Caspase-3 activity for untreated cells was 6.8 ± 0.2 pmol of AMC/mg/min (with DTT) and 6.3 ± 0.3 pmol of AMC/mg/min (without DTT); caspase-3 activity for cells treated only with doxorubicin (control) was 122.0 ± 10.0 pmol of AMC/mg/min (with DTT) and 115.2 ± 2.0 pmol of AMC/mg/min (without DTT). The data represent mean ± S.E. (n = 3).
FIGURE 5. Effects of CysNO transport on caspases-3-like activity.
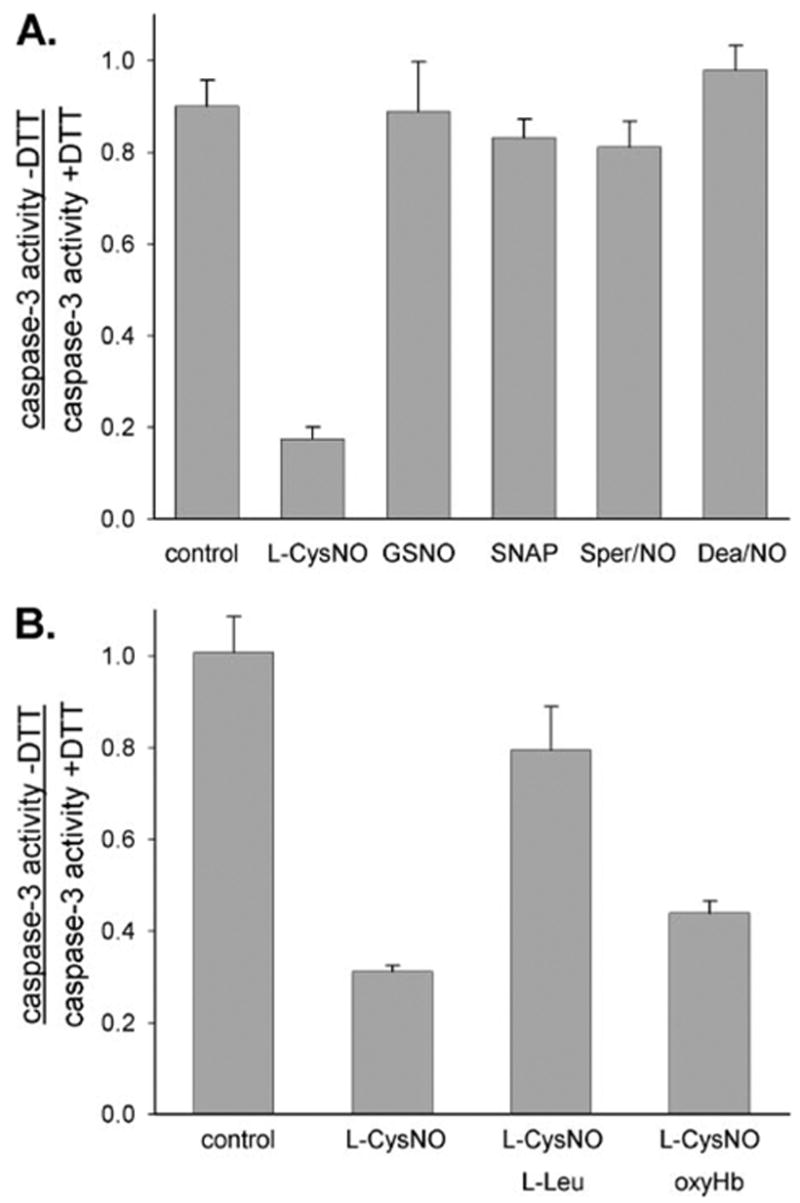
BAECs were incubated with doxorubicin (0.5 μM) for 8 h in medium to induce caspase-3 activity. A, cells were washed and incubated for a further 1 h with various low molecular weight RSNOs (400 μM) and NO donors (400 μM). Caspase-3-like activity was determined in the cell lysates in the presence and absence of DTT and expressed as the ratio caspase-3 activity − DTT:caspase-3 activity + DTT. Caspase-3 activity for untreated cells was 4.3 ± 0.1 pmol of AMC/mg/min (with DTT) and 3.0 ± 0.23 pmol of AMC/mg/min (without DTT); caspase-3 activity for cells treated only with doxorubicin (control) was 115.8 ± 5.8 pmol of AMC/mg/min (with DTT) and 102.0 ± 3.7 pmol of AMC/mg/min (without DTT). The data represent mean ± S.E. (n = 3). B, cells were washed and incubated for a further 1 h with L-CysNO (160 μM) in the absence or presence of L-Leu (8 mM) or oxyHb (160 μM). The caspase-3-like activity was determined in the cell lysates in the presence or absence of DTT and expressed as the ratio caspase-3 activity − DTT:caspase-3 activity + DTT. Caspase-3 activity for untreated cells was 4.6 ± 0.2 pmol of AMC/mg/min (with DTT) and 4.6 ± 0.1 pmol of AMC/mg/min (without DTT); caspase-3 activity for cells treated only with doxorubicin (control) was 99.6 ± 6.2 pmol of AMC/mg/min (with DTT) and 99.4 ± 1.5 pmol of AMC/mg/min (without DTT). The data represent mean ± S.E. (n = 3).
To examine whether RSNOs or NO donors could produce the same level of caspase-3 inhibition, we incubated doxorubicin-pretreated cells with the non-transportable RSNOs, GSNO, and SNAP and with the NO donors, Sper/NO and Dea/NO. As shown in Fig. 5A, these compounds had little or no effect on caspase-3 activity. In addition, oxyHb, had no effect on the L-CysNO-mediated inhibition of caspase-3, indicating that NO release does not contribute to this effect (Fig. 5B). The effect of L-leucine on caspase-3 inhibition is also shown in Fig. 5B. As can be seen, L-leucine substantially reversed caspase-3 inhibition by L-CysNO. In combination, these data indicate that the inhibition of caspase-3 activity by L-CysNO occurs via uptake of L-CysNO by the L-AT system followed by covalent modification of caspase-3 thiols without the necessity of NO release.
The Relationship between Intracellular RSNO Level, GSH Depletion, and the Inhibition of Caspase-3 Activity
Fig. 6 plots the decrease in GSH concentration and the ratio of DTT-untreated:DTT-treated caspase-3 activity as a function of intracellular RSNO level after treatment of BAEC with L-CysNO. Several interesting points can be observed when the data are plotted in this way. Firstly, a significant level of intracellular RSNO can be sustained without either GSH depletion or inhibition of caspase activity. Secondly, the inhibition of caspase activity by L-CysNO parallels the loss of GSH. Thirdly, maximal inhibition of caspase activity occurs when GSH is extensively depleted. These data indicate that caspase-3 is not a sensitive target for inhibition by L-CysNO and that other proteins can be nitrosated at lower levels of L-CysNO and without loss of GSH.
FIGURE 6. GSH depletion and caspase-3 activity as a function of intracellular RSNO level.
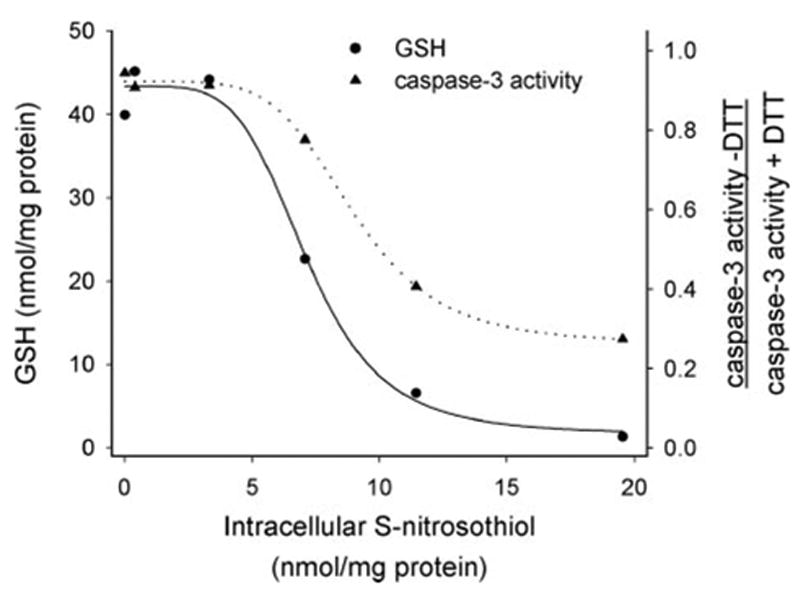
Data from Figs. 1B, 2A, and 4 were replotted to show the depletion of GSH and the increase in the reversible inhibition of caspase-3 activity as a function of the level of intracellular RSNO.
DISCUSSION
It was previously suggested that the L-AT transport system was responsible for mediating the uptake of L-CysNO as L-leucine, a competitor for this system, was able to prevent some L-CysNO-mediated effects (31). We have previously demonstrated that L-CysNO, and not GSNO, is directly transported into RAW 264.7 cells via the L-AT transporter system, resulting in a high level of intracellular nitrosation (13). In addition, we demonstrated that cysteine or cystine was absolutely necessary for the uptake of the nitroso group of GSNO, again via a mechanism involving the L-AT system (13). The importance of the L-AT in L-CysNO transport has since been confirmed by small interfering RNA knockdown techniques (21). L-CysNO and other RSNOs have been used as NO donors in cell culture, and many of the effects of these species have been attributed to the release of NO. However, the above studies indicate that L-CysNO, GSNO, and other RSNOs can affect cells by NO-independent processes. Although this calls into question the interpretation of all studies that have used RSNOs as NO donors in cell culture, it also highlights the presence of a potentially important transmembrane transport mechanism that allows RSNOs to directly influence the intracellular environment without the necessity of NO release.
In this study, we investigated the importance of the L-AT system in mediating the ability of L-CysNO to modify intracellular thiol groups. We demonstrate that L-CysNO is avidly taken up into BAEC and that the mechanism is qualitatively identical to that observed in RAW 264.7 cells. In fact, we have observed this mechanism to be present in many different and varied cell lines and primary cells including pulmonary endothelial cells, red blood cells, smooth muscle cells, and neuroblastoma cells (32), suggesting that the L-AT system represents a universal mechanism of L-CysNO transport. We show that L-CysNO exposure results in a decrease in intracellular GSH levels, an increase in intracellular RSNO levels, and a DTT-reversible decrease in the activity of caspase-3. All these functions of L-CysNO are mediated through transport of this RSNO via the L-AT system and occur independently of NO release. In all cases, the effect could not be recapitulated by a NO donor, nor was it inhibited by the avid NO scavenger oxyHb. This represents a fundamental reassessment of the mechanism by which L-CysNO affects cellular functions.
In this study, we have also assessed the fate of the amino acid carrier of the RSNO rather than just the nitroso functional group. In the case of both L-CysNO and hCysNO, exposure of cells to these RSNOs resulted in an increase in intracellular levels of cysteine and homocysteine, respectively. In the case of hCysNO, this increase was strongly inhibited by L-leucine; however, the inhibition of intracellular cysteine was less robust, although significant. This is likely because L-CysNO partially decays to form cystine, which can be taken up via the xc− transport system. These data strongly suggest that amino acid-based RSNOs are taken up into cells intact and that it is not simply the nitroso functional group that is transported across the cell membrane.
The effect of L-CysNO on GSH depletion was shown to be concentration-dependent with concentrations of L-CysNO of 40 μM or less having little effect on GSH levels. However, above this level, a precipitous loss of GSH occurred, resulting in GSH levels of about 5% of the initial value. Several possibilities exist for the observed loss of GSH. The first, and most likely, involves transnitrosation (the transfer of the nitroso function group) with GSH to form GSNO in the intracellular space followed by enzymatic decomposition of GSNO to form GSSG. An enzymatic reductase that uses NADH but not NADPH as a source of electrons was observed in BAEC cell lysate with a Km of 7 μM. Previous studies have identified glutathione-dependent formaldehyde dehydrogenase (alcohol dehydrogenase class III) as a catalyst of NADH-dependent GSNO reduction with a Km of 28 μM (28). Mice lacking this enzyme had increased RSNO levels in the lungs and were protected from experimental asthma (33). Whatever the nature of the catalyst, it is clear that this activity is robust enough to consume intracellular GSH in the face of high intracellular L-CysNO levels. It is interesting to note that NO per se is a relatively poor GSH depleting agent in BAEC, and others have observed that in cells chronically exposed to inflammatory concentrations of NO, GSH levels have, in some cases, lowered through the inhibition of GSH reductase (34), but in other cases, have increased through the activation of γ-glutamylcysteine ligase (35) or via induction of cystine transport (36). Clearly, the long term effects of NO on GSH levels are complex and involve much more than chemical oxidation and thiol nitrosation.
We chose caspase-3 as a target protein to examine the role of L-CysNO transport in inhibiting intracellular enzyme activities. The idea that caspase-3 can be reversibly modified by S-nitrosation has received significant attention. The studies performed by Mannick et al. (11) first showed that Fas ligand induced caspase-3 activity both by inducing the processing of the enzyme and by stimulating denitrosation of the active site thiol. In a subsequent study, it has been reported that the subpopulation of mitochondrial caspases is mainly controlled by S-nitrosation (37). As a result of these studies, S-nitrosothiols and NO donors have been used in cell culture to examine effects on caspase-3 activity. There are a number of reports suggesting that caspase-3 activity can be inhibited in the presence of NO donors or RSNOs; however, the enzymatic activity was often measured in the presence of DTT, which can reverse S-nitrosation, making the interpretation of the results difficult. It is important to distinguish between direct effects on caspase-3 activity that occur as a result of modification of the active site thiol versus the prolonged effects of NO on the upstream signaling, induction, and processing of the procaspase-3 to the active form. In studies showing the inhibition of caspase-3 activity after long term exposure to NO donors (12), it was concluded that caspase-3 was inhibited by S-nitrosation, although the activity assays were performed in the presence of DTT. Studies on monocyte survival reported that relatively high doses (millimolar range) of GSNO and SNAP lowered caspase-3 activity, whereas NO donors did not. In addition, the authors did not find a correlation between NO liberated during the 24-h incubation period and activities of caspase-3 and -9 (38). Again, cells were exposed to NO and RSNO for long periods of time, and activity of caspases was measured in the presence of DTT. Kim et al. (39) first reported that long term exposure to SNAP and NO donors inhibited caspase-3 activity by two distinct mechanisms: at the level of caspase-3 processing and activation as well as directly via S-nitrosation. Assays were performed in the presence and absence of DTT. When DTT was present, the total caspase-3 activity was reduced by NO exposure. In the absence of DTT, caspase-3 activity dropped further, revealing the fraction of the enzyme that was oxidatively modified. In our studies, we induced cells to produce the active form of caspase-3 and then exposed them briefly (1-h incubation time) to various low molecular weight RSNOs and NO donors. In this situation, the observed inhibition of the enzyme activity resulted solely from the modification of the active site cysteine. L-CysNO, but not GSNO or the NO donors Sper/NO and Dea/NO, inhibited caspase-3 activity through a DTT-reversible modification. The concentration dependence of this inhibition was remarkably similar to GSH depletion in that caspase-3 inhibition was only observed above 40 μM L-CysNO. This indicates that in this system, the inhibition of caspase-3 by thiol modification occurs simultaneously with a decrease in cellular reducing systems and only at high levels of intracellular RSNO, suggesting that L-CysNO-dependent caspase-3 inactivation is not a particularly sensitive or specific process in BAEC. It is likely that GSH can protect caspase-3 inhibition by two related mechanisms. The first is that GSH will be a sink for cellular RSNO as GSNO can be metabolized by an NADH-dependent process, and the second is that GSH may directly repair nitrosated caspase-3 via transnitrosation or reduction of the active site thiol. Caspase-3 inhibition was reversed by competitors of the L-AT system and was not affected by oxyHb, again highlighting that enzyme inhibition occurred through the direct uptake and action of L-CysNO and not through NO formation.
Although casase-3 did not turn out to be a sensitive target for L-CysNO in BAECs, it should be emphasized that our data only examine caspase-3 inhibition via transnitrosation mechanisms from extracellular donors and do not examine the ability of cellular nitric oxide synthase to modulate caspase activity or apoptosis. In addition, Fig. 6 shows that a significant intracellular level of protein RSNO (up to 4 nmol of RSNO/mg of protein) can be sustained without loss of the GSH pool. This suggests that there are more specific and sensitive targets for S-nitrosation that show some stability even in the presence of normal GSH levels. This strongly hints at a hierarchy of thiol sensitivity to this post-translational modification, and a full understanding of this hierarchy will reveal which proteins are susceptible to modification under physiologically meaningful conditions. It should be noted that in this study, we have not determined the nature of the modification that gives rise to the loss of caspase-3 activity. It is possible that both thiol nitrosation and protein S-thiolation could be involved as both of these modification are reversible by DTT. As there is currently no reliable activity-based method to distinguish between these possibilities, specific analysis of the modified protein is required.
In conclusion, we have demonstrated that the ability of L-CysNO to modify intracellular thiolic targets in BAEC depends exclusively upon its uptake into cells via the L-AT system and not upon the generation of NO. Intracellular thiol modification occurs by transnitrosation to form a protein RSNO or by S-thiolation, either directly or indirectly through the increased oxidative stress accompanying L-CysNO exposure. These data demonstrate that the uptake of amino acid RSNOs by cells represents a potential mechanism by which RSNOs, in extracellular environments such as the plasma or lung lining fluid, can influence the intracellular environment in ways distinctly different from NO. The data also suggest that NO has a very poor ability to transmit signals through the modification of thiols in BAEC but requires prior extracellular conversion to an RSNO and uptake before this avenue of intracellular signaling is opened.
Acknowledgments
We thank Matthew Fishman for technical assistance.
The abbreviations used are
- RSNO
S-nitrosothiol
- GSNO
S-nitrosoglutathione
- NO
nitric oxide
- Sper/NO
(Z)-1-[N-(3-ammoniopropyl)-N-[4-(3-aminopropylammonio)butyl]-amino]diazen-1ium-1,2-diolate
- Dea/NO
2-(N,N-diethylamino)-diazenolate-2-oxide
- L-AT
amino acid transport system L
- SNAP
S-nitroso-N-acetylpenicillamine
- L-CysNo
S-nitroso-L-cysteine
- hcysNO
S-nitrosohomocysteine
- AMC
7-amido-4-methylcoumarin
- DTPA
diethylenetriamine pentaacetic acid
- NEM
N-ethylmaleimide
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid
- DTT
dithiothreitol
- BAEC
bovine aortic endothelial cells
- HBSS
Hanks’ balanced salt solution
- OxyHb
oxyhemoglobin
- PBS
phosphate-buffered saline
- HPLC
high pressure liquid chromatography
- FDH
formaldehyde dehydrogenase
Footnotes
This work was supported by Grant GM55792 from the National Institute of General Medicine.
References
- 1.Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, Singel D, Valeri CR, Loscalzo J. Proc Natl Acad Sci U S A. 1992;89:7674–7677. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryan NS, Rassaf T, Maloney RE, Rodriguez CM, Saijo F, Rodriguez JR, Feelisch M. Proc Natl Acad Sci U S A. 2004;101:4308–4313. doi: 10.1073/pnas.0306706101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, Hogg N. Am J Physiol. 2004;287:L467–L474. doi: 10.1152/ajplung.00350.2003. [DOI] [PubMed] [Google Scholar]
- 4.Crawford JH, Chacko BK, Pruitt HM, Piknova B, Hogg N, Patel RP. Blood. 2004;104:1375–1382. doi: 10.1182/blood-2004-03-0880. [DOI] [PubMed] [Google Scholar]
- 5.Jourd’heuil D, Gray L, Grisham MB. Biochem Biophys Res Commun. 2000;273:22–26. doi: 10.1006/bbrc.2000.2892. [DOI] [PubMed] [Google Scholar]
- 6.Stamler JS, Lamas S, Fang FC. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 7.Lander HM, Hajjar DP, Hempstead BL, Mirza UA, Chait BT, Campbell S, Quilliam LA. J Biol Chem. 1997;272:7323–7326. doi: 10.1074/jbc.272.7.4323. [DOI] [PubMed] [Google Scholar]
- 8.Poteser M, Romanin C, Schreibmayer W, Mayer B, Groschner K. J Biol Chem. 2001;276:14797–14803. doi: 10.1074/jbc.M008244200. [DOI] [PubMed] [Google Scholar]
- 9.Marshall HE, Stamler JS. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 10.Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EF, van d V, Janssen-Heininger YM. Proc Natl Acad Sci U S A. 2004;101:8945–8950. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 12.Rossig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher AM, Mulsch A, Dimmeler S. J Biol Chem. 1999;274:6823–6826. doi: 10.1074/jbc.274.11.6823. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Hogg N. Proc Natl Acad Sci U S A. 2004;101:7891–7896. doi: 10.1073/pnas.0401167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoue K, Akaike T, Miyamoto Y, Okamoto T, Sawa T, Otagiri M, Suzuki S, Yoshimura T, Maeda H. J Biol Chem. 1999;274:27069–27075. doi: 10.1074/jbc.274.38.27069. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Tanus-Santos JE, Reiter CD, Dejam A, Shiva S, Smith RD, Hogg N, Gladwin MT. Proc Natl Acad Sci U S A. 2004;101:11477–11482. doi: 10.1073/pnas.0402201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamler JS, Toone EJ, Lipton SA, Sucher NJ. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 17.Davisson RL, Travis MD, Bates JN, Lewis SJ. Circ Res. 1996;79:256–262. doi: 10.1161/01.res.79.2.256. [DOI] [PubMed] [Google Scholar]
- 18.Gordge MP, Addis P, Noronha-Dutra AA, Hothersall JS. Biochem Pharmacol. 1998;55:657–665. doi: 10.1016/s0006-2952(97)00498-x. [DOI] [PubMed] [Google Scholar]
- 19.Zeng H, Spencer NY, Hogg N. Am J Physiol. 2001;281:H432–H439. doi: 10.1152/ajpheart.2001.281.1.H432. [DOI] [PubMed] [Google Scholar]
- 20.Satoh S, Kimura T, Toda M, Maekawa M, Ono S, Narita H, Miyazaki H, Murayama T, Nomura Y. J Neurochem. 1997;69:2197–2205. doi: 10.1046/j.1471-4159.1997.69052197.x. [DOI] [PubMed] [Google Scholar]
- 21.Li S, Whorton AR. J Biol Chem. 2005;280:20102–20110. doi: 10.1074/jbc.M413164200. [DOI] [PubMed] [Google Scholar]
- 22.Antonini E, Brunori M. In: Frontiers of Biology. Neuberger A, Tatum EL, editors. North Holland Publishing; Amsterdam: 1971. pp. 1–12. [Google Scholar]
- 23.Hart T. Tetrahedron Lett. 1985;26:2013–2026. [Google Scholar]
- 24.Field L, Dilts RV, Ravichandran R, Lanhert PG, Carnahan PG. J Chem Soc Chem Commun. 1978:249–250. [Google Scholar]
- 25.Zhang Y, Keszler A, Broniowska KA, Hogg N. Free Radical Biol Med. 2005;38:874–881. doi: 10.1016/j.freeradbiomed.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 26.Toyo’oka T, Imai K. J Chromatogr A. 1983;282:495–500. doi: 10.1016/s0021-9673(00)91626-1. [DOI] [PubMed] [Google Scholar]
- 27.Zaman K, Palmer LA, Doctor A, Hunt JF, Gaston B. Biochem J. 2004;380:67–74. doi: 10.1042/BJ20031687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen DE, Belka GK, Du Bois GC. Biochem J. 1998;331:659–668. doi: 10.1042/bj3310659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, Kalyanaraman B. J Biol Chem. 2004;279:25535–25543. doi: 10.1074/jbc.M400944200. [DOI] [PubMed] [Google Scholar]
- 31.Satoh S, Kimura T, Toda M, Miyazaki H, Ono S, Narita H, Murayama T, Nomura Y. J Cell Physiol. 1996;169:87–96. doi: 10.1002/(SICI)1097-4652(199610)169:1<87::AID-JCP9>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 32.Kettenhofen N, Broniowska KA, Riego J, Chen C, Hogg N. Free Radical Biol Med. 2005;39:S101. doi: 10.1016/j.freeradbiomed.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Que LG, Liu L, Yan Y, Whitehead GS, Gavett SH, Schwartz DA, Stamler JS. Science. 2005;308:1618–1621. doi: 10.1126/science.1108228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butzer U, Weidenbach H, Gansauge S, Gansauge F, Beger HG, Nussler AK. FEBS Lett. 1999;445:274–278. doi: 10.1016/s0014-5793(99)00139-8. [DOI] [PubMed] [Google Scholar]
- 35.Moellering D, Mc AJ, Patel RP, Forman HJ, Mulcahy RT, Jo H, Darley-Usmar VM. FEBS Lett. 1999;448:292–296. doi: 10.1016/s0014-5793(99)00371-3. [DOI] [PubMed] [Google Scholar]
- 36.Li H, Marshall ZM, Whorton AR. Am J Physiol. 1999;276:C803–C811. doi: 10.1152/ajpcell.1999.276.4.C803. [DOI] [PubMed] [Google Scholar]
- 37.Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. J Cell Biol. 2001;154:1111–1116. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeigler MM, Doseff AI, Galloway MF, Opalek JM, Nowicki PT, Zweier JL, Sen CK, Marsh CB. J Biol Chem. 2003;278:12894–12902. doi: 10.1074/jbc.M213125200. [DOI] [PubMed] [Google Scholar]
- 39.Kim YM, Talanian RV, Billiar TR. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]