

Summary
Escherichia coli DksA, GreA, and GreB have similar structures and bind to the same location on RNA polymerase (RNAP), the secondary channel. We show that GreB can fulfill some roles of DksA in vitro, including shifting the promoter-open complex equilibrium in the dissociation direction, thus allowing rRNA promoters to respond to changes in the concentrations of ppGpp and NTPs. However, unlike deletion of the dksA gene, deletion of greB had no effect on rRNA promoters in vivo. We show that the apparent affinities of DksA and GreB for RNAP are similar, but the cellular concentration of GreB is much lower than that of DksA. When overexpressed and in the absence of competing GreA, GreB almost completely complemented the loss of dksA in control of rRNA expression, indicating its inability to regulate rRNA transcription in vivo results primarily from its low concentration. In contrast to GreB, the apparent affinity of GreA for RNAP was weaker than that of DksA, GreA only modestly affected rRNA promoters in vitro, and even when overexpressed GreA did not affect rRNA transcription in vivo. Thus, binding in the secondary channel is necessary but insufficient to explain the effect of DksA on rRNA transcription. Neither Gre factor was capable of fulfilling two other functions of DksA in transcription initiation: co-activation of amino acid biosynthetic gene promoters with ppGpp and compensation for the loss of the ω subunit of RNAP in the response of rRNA promoters to ppGpp. Our results provide important clues to the mechanisms of both negative and positive control of transcription initiation by DksA.
Keywords: Transcription initiation, DksA, GreA, GreB, Secondary Channel of RNA polymerase
Introduction
Transcription initiation in bacteria begins with the reversible binding of RNA polymerase (RNAP) to promoter DNA.1 The RNAP-promoter complex then undergoes several isomerizations, eventually resulting in a transcriptionally-competent open complex. At most Escherichia coli promoters, the open complex (or an intermediate on the pathway to its formation) is long-lived even in the presence of competitor for free RNAP. However, the competitor-resistant complex formed at rRNA and some other promoters is short-lived, shifting equilibrium occupancy from the open complex back to earlier, competitor-sensitive intermediates.2,3 A non-optimal interaction between region 1.2 of the σ subunit of RNAP and a non-template strand base two positions downstream of the -10 promoter element is one of the major determinants of the short-lived nature of the rRNA promoter complex and is required for rRNA transcription regulation.4
Two small molecules regulate many promoters that form short-lived complexes with RNAP: the initiating NTP (iNTP) and guanosine 5′-diphosphate 3′-diphosphate (ppGpp). The concentrations of NTPs and ppGpp change at distinct times in the bacterial growth cycle, regulating rRNA promoters,5 many tRNA promoters,3 and certain mRNA promoters6,7 to meet the translational needs of the cell. By binding to RNAP and pairing with the template DNA, the iNTP transiently stabilizes the rRNA promoter complex, thereby driving transcription initiation forward by mass action.8–10 ppGpp decreases the half-life of the complex with RNAP at all promoters, inhibiting transcription from intrinsically short-lived complexes.11 ppGpp not only directly inhibits rRNA promoters, but in combination with the transcription factor DksA, a 17.5 kDa protein with pleiotropic effects on gene expression in a number of bacterial species, it also directly co-activates some other promoters.12
Although effects of changing iNTP and ppGpp concentrations are observed on rRNA promoters in vitro with purified components, the magnitudes of these effects are much smaller than those observed in vivo or in extracts.11,13 DksA accounts for the differences in regulation observed in vitro versus in vivo.12,14 DksA shifts the rRNA promoter-open complex equilibrium in the dissociation direction, increases the dependence of the promoter on the iNTP concentration, and increases the sensitivity of the promoter to ppGpp.12,14 Furthermore, DksA and ppGpp together positively regulate promoters for certain amino acid biosynthesis genes and virulence factors.12,15,16 Thus, depending on the intrinsic kinetics of the promoter, DksA can decrease transcription of some promoters and increase transcription of others.
DksA also rescues a defect in rRNA transcription regulation by ppGpp exhibited by RNAP lacking the ω subunit in vitro.17 Since mutants lacking rpoZ (the gene coding for ω) respond to ppGpp in vivo,18 we proposed that DksA masks a defect in regulation caused by the absence of ω.17
The crystal structure of DksA revealed that the protein has two domains, a coiled-coil with two acidic residues at one end and a globular domain at the end opposite the acidic tip.19 DksA exhibits an overall similarity in structure to the transcription elongation factors GreA and GreB, despite a lack of sequence homology. The Gre factors bind in the secondary channel of RNAP, a pore though which NTP substrates gain access to the active site of the enzyme.20,21,22 The acidic tips of GreA and GreB are adjacent to the RNAP active site and stimulate an intrinsic RNA cleavage activity of RNAP, releasing RNA that has backtracked into the secondary channel. However, GreA activates cleavage of 2–3 nt, whereas GreB can activate cleavage of up to ~18 nt from the 3′ end of backtracked transcripts. Both Gre factors inhibit pausing and prevent the formation of arrested elongation complexes, but only GreB rescues already-arrested complexes.
Although there is no co-crystal structure of DksA bound to RNAP, there is considerable biochemical support that DksA binds in the secondary channel, as predicted from its resemblance to the Gre factors19 (I. Toulokhonov, J. Mukhopadhyay, R.H. Ebright, and R.L.G., unpublished data). Like the Gre factors, DksA prevents the formation of arrested complexes, although it cannot activate the intrinsic cleavage activity of RNAP or rescue arrested elongation complexes.19
To provide further insight into the mechanism of DksA function, we compared the effects of DksA and the Gre factors on transcription initiation in vitro and in vivo. If one or both Gre factors could perform the same functions as DksA, this would suggest that a shared property of the factors might explain the effects of DksA. If the Gre factors could not perform functions of DksA, this would focus future efforts on understanding properties unique to DksA.
We show here that, like DksA, GreB affects rRNA promoters in vitro, but its low concentration prevents it from controlling rRNA transcription in vivo. In contrast, GreA has little or no effect on rRNA promoters either in vitro or in vivo. Furthermore, unlike DksA, neither Gre factor co-activates amino acid biosynthesis gene promoters with ppGpp nor rescues the ability of RNAP lacking the ω subunit to respond to ppGpp in vitro and in vivo. We conclude that properties shared between DksA and GreB, including their ability to increase the rate of RNAP-promoter complex dissociation, are sufficient to explain most of the effect of DksA on rRNA transcription. However, these properties are insufficient to account for the role of DksA in positive control of transcription initiation or in compensation for the loss of the ω subunit.
Results
rrnB P1 transcription is regulated similarly by GreB and DksA in vitro
DksA is essential for regulation of rRNA transcription initiation.14 GreA and GreB are structurally similar to DksA, and all three factors bind in the RNAP secondary channel19,20,21,22 (I. Toulokhonov, J. Mukhopadhyay, R.H. Ebright, and R.L.G., unpublished data). To determine whether GreA or GreB can regulate rRNA transcription like DksA, we first compared the effects of the three factors on transcription from rrnB P1 in vitro (Figure 1(a)). Both GreB and DksA strongly and specifically inhibited rrnB P1 transcription (~35 and 50-fold inhibition with 2 μM GreB and DksA, respectively). At the same concentration, GreA inhibited rrnB P1 transcription less than 2-fold. Consistent with our previous work on DksA, none of the three proteins inhibited transcription from the lacUV5 promoter or from the RNA-I promoter (encoded by the plasmid) (Figure 1(b)). Inhibition of transcription was specific to rrnB P1 even though the lacUV5 promoter construct produced exactly the same RNA transcript (both plasmids were constructed with the same DNA sequences downstream of +1; see Materials and Methods). This suggested that the mechanism of inhibition most likely derived from an effect on transcription initiation, not elongation (see also below).
Figure 1.

GreB and DksA specifically inhibit transcription from rrnB P1 in vitro, enhance inhibition of transcription by ppGpp, and increase the iNTP concentration required for maximal transcription. (a) Effect of DksA, GreA or GreB (2μM) on transcription from rrnB P1. Single-round in vitro transcription reactions were carried out in duplicate in transcription buffer containing 30 mM NaCl with plasmid templates containing rrnB P1 (endpoints −61/+1; pRLG5944) (see Materials and Methods). RNA-I is a plasmid-encoded transcript. The amount of transcript from rrnB P1 in the presence of each factor relative to that with no factor is shown beneath the gel lanes. (b) Same as (a) except the plasmid contained the lacUV5 promoter (endpoints −46/+1; pRLG3422) (c) Effect of low concentrations of DksA, GreA or GreB on transcription from rrnB P1. Single-round in vitro transcription reactions were carried out in duplicate in transcription buffer containing 50 mM NaCl with plasmid template pRLG6555. Reactions contained 0.1 μM DksA, GreB, or GreA. (d) Effects of ppGpp and DksA, GreA, or GreB on transcription from rrnB P1. When indicated, reactions contained 100 μM ppGpp and 100 μM ppGpp with DksA (0.1 μM), GreB (0.1 μM) or GreA (6 μM). (e) Effect of DksA, GreA, or GreB on ATP concentration-dependence of transcription from rrnB P1. Multiple-round in vitro transcription reactions were carried out at increasing concentrations of the iNTP, ATP. The plasmid template was pRLG6555, and the buffer contained 170 mM KCl (see Materials and Methods). Transcription was normalized to the maximum (plateau) level for each condition. The DksA, GreA, and GreB concentrations were 3.5 μM, 3.5 μM, and 0.5 μM, respectively (see text).
DksA greatly amplifies the inhibitory effect of ppGpp in vitro and in vivo.14 To address whether the Gre factors, like DksA, could synergize with ppGpp to inhibit transcription, we compared the effects of the three factors at concentrations (0.1 μM) where alone they had little or no effect on rrnB P1 transcription (Figure 1(c)) and under conditions where ppGpp by itself had only a small effect on transcription (~30% inhibition; Figure 1(d)). Under these conditions, ppGpp and GreB together inhibited transcription ~5-fold, similar to the effect of ppGpp and DksA together (Figure 1(d)). However, ppGpp and GreA together inhibited transcription little more than ppGpp alone, even with 60-fold more GreA than GreB or DksA (6 μM, Figure 1(d)), sufficient to saturate GreA binding to RNAP (see below).
rRNA promoters need high concentrations of the first NTP to initiate transcription, 5,8 and DksA further increases this NTP requirement.14 Like DksA, GreB increased the NTP concentration required for transcription initiation from rrnB P1 (Figure 1(e)). Surprisingly, GreB had a larger effect than DksA (see Discussion). GreA had no effect on the iNTP requirement.
GreB and DksA similarly decrease the half-life of the RNAP-promoter complex
We next addressed the mechanism responsible for the effect of GreB on rrnB P1 transcription. Since the effects of GreB and DksA on rrnB P1 were so similar, we reasoned they might have the same kinetic basis. We showed previously that the half-life of the RNAP-rRNA promoter complex correlated with its regulation in vitro and in vivo.4,5,9,11 DksA decreased the half-life of the RNAP complex with all promoters tested.14 We proposed that DksA inhibits transcription from rRNA promoters, but not from most other promoters, because rRNA promoters make unusually short-lived complexes with RNAP. By further decreasing the half-life of the rRNA promoter complex, DksA reduces transcription and further sensitizes the promoter to changing concentrations of the iNTP and ppGpp. Therefore, rrnB P1 mutations that increased the lifetime of the complex with RNAP eliminated the effect of DksA on transcription initiation in vitro and in vivo.3,4,14
We compared the effects of the Gre factors on promoter complex half-life with that of DksA. RNAP, with or without GreA, GreB, or DksA, was added to a supercoiled plasmid containing rrnB P1 (Figure 2(a)) or to a DNA fragment containing lacUV5, a promoter with which RNAP forms a long-lived complex that is not rate-determining for transcription (Figure 2(b)). Transcription (Figure 2(a)) or filter retention (Figure 2(b)) were then used to quantify the fraction of complexes remaining at different times after competitor addition. We note that the filter binding assay does not depend on transcript formation as a read-out of the fraction of competitor-resistant complexes remaining. Therefore, in this assay complexes would not be subject to potential effects of Gre factors on transcript cleavage or elongation rates that might confound interpretation of effects on initiation (see below and Materials and Methods). Consistent with the results of a previous report on the λPR promoter,23 GreB decreased the half-lives of the complexes formed on both rrnB P1 (Figure 2(a)) and lacUV5 (Figure 2(b)). Furthermore, its effects were almost identical to those of DksA. In contrast, GreA had a much smaller effect on the complexes formed by both promoters.
Figure 2.
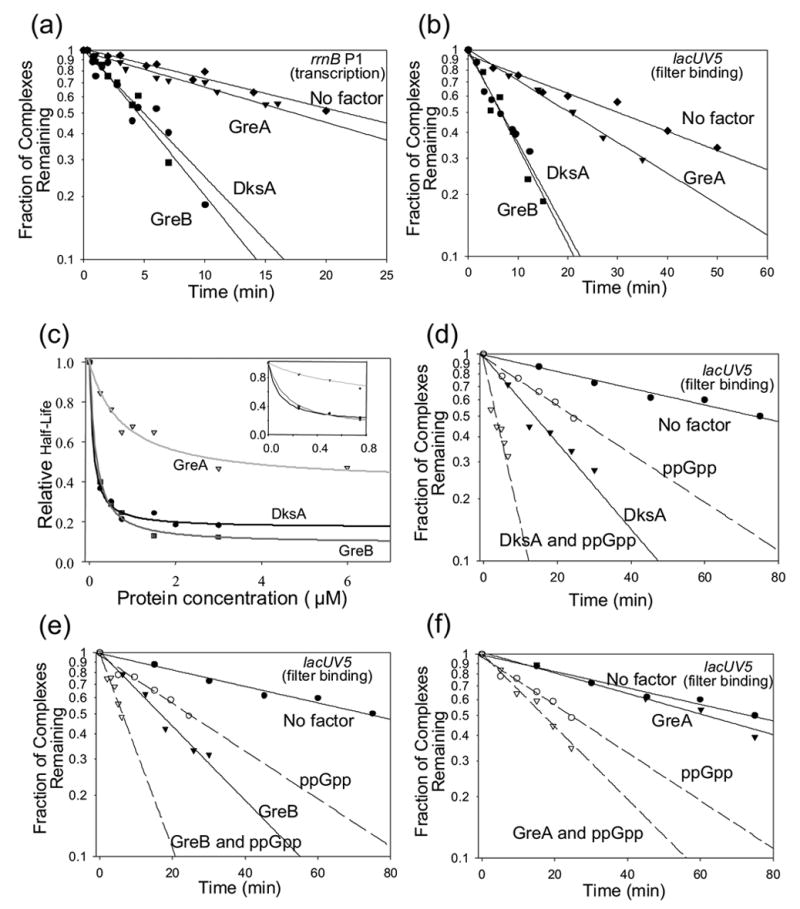
DksA, GreB, and GreA reduce the RNAP-promoter complex half-life. (a) Half-life of the competitor-resistant rrnB P1 promoter complex in the presence of 0.1 μM DksA, 0.8 μM GreA, 0.1 μM GreB, or buffer only (no factor) was determined using a transcription based assay at 30ºC containing 30 mM NaCl and plasmid pRLG5944 (see Materials and Methods). Transcription at different times after addition of competitor (consensus promoter DNA) was normalized to the total amount of transcription at time zero. (b) Half-life of the competitor-resistant lacUV5 complex in the presence of DksA, GreA, or GreB, or buffer only (no factor) was determined using a filter binding assay at 25ºC in 100 mM NaCl (see Materials and Methods). The representative decay curves (0.75 μM of each factor) show the fraction of complexes remaining versus time after heparin addition. (c) Apparent affinities of each factor for RNAP. Half-lives of the competitor-resistant lacUV5 complex were determined at a range of DksA, GreA, and GreB concentrations or with no factor by the filter binding assay. The results are plotted as a function of the factor concentration. Best fit exponential decay curves were drawn using SigmaPlot. The inset shows an expanded view of the low concentration range. The apparent binding constants for each factor (i.e. the concentration of each factor needed to decrease the half-life of the complex by 50%) were ~0.1 μM DksA, ~0.1 μM GreB, and ~0.8 μM GreA. (d) – (f) Effects of DksA, GreB, and GreA on half-life in the presence of ppGpp. Half-lives of the competitor-resistant lacUV5 complex in the absence of factor, with 80 μM ppGpp alone, with 0.15 μM DksA (d), GreA (e), or GreB (f) alone, or with 80 μM ppGpp together with 0.15 μM DksA (d), GreA (e), or GreB (f). Half-lives were determined as in (b) (see also Materials and Methods) except the experiments were performed at 30ºC.
By measuring the half-lives of lacUV5 promoter complexes at a range of DksA, GreA, and GreB concentrations, we determined the maximal decrease in complex half-life at saturating factor concentration and the factor concentration required to reduce the half-life by 50% (the apparent Kd). Saturating concentrations of DksA and GreB reduced complex half-life to the same extent, whereas GreA reduced complex half-life to a much smaller extent (Figure 2(c)). The apparent Kds of GreB and DksA for RNAP were virtually identical (Kd ~ 0.1 μM), whereas the apparent affinity of GreA for RNAP (Kd ~0.8 μM) was 8-fold weaker than that of GreB or DksA (see also24).
DksA and GreB also similarly enhanced the effect of ppGpp on decay (Figure 2(d) and (e); see also14,17). ppGpp and GreA together reduced complex lifetime much less than ppGpp with DksA or GreB (Figure 2(f)), as expected from the fact that GreA had a smaller effect on rrnB P1 transcription and on complex lifetime. Thus, the apparent binding affinities of the three factors for RNAP, their relative effects on promoter complex half-life, and their effects on half-life in conjunction with ppGpp are consistent with their effects on rrnB P1 transcription in vitro.
We think that two other previously-characterized activities of GreB are less likely to account for its inhibitory effect on rrnB P1 transcription. First, GreB can stimulate the intrinsic RNA cleavage activity of RNAP,20 but no transcript cleavage was detected at the GreB concentrations where promoter-specific inhibition of rrnB P1 transcription was observed (Figure 1). (Shorter migrating transcripts were detected at very high GreB concentrations in vitro, however, where GreB inhibited transcription promoter-nonspecifically; data not shown). Furthermore, the identity of the sequence upstream, rather than downstream, of the transcription start site was responsible for the inhibitory effect of GreB, suggesting that it affected transcription initiation (Figure 1(a) and (b)). Finally, DksA and GreB similarly inhibited transcription of rrnB P1, yet DksA does not stimulate the intrinsic RNA cleavage activity of RNAP,19 indicating that cleavage is not a requirement for transcription inhibition.
Second, Gre factors have been reported to reduce abortive initiation and enhance promoter clearance.25 However, these effects increase transcription,20 opposite of the effect of GreB observed on rrnB P1. Taken together, the data suggest that GreB, like DksA, exerts its promoter-specific effects on rrnB P1 during transcription initiation rather than elongation.
Deletion of greA, greB, or both has little or no effect on rRNA promoter activity in vivo
We next determined whether effects of Gre factors on rRNA promoters could be detected in vivo by comparing the activities of promoter-lacZ fusions in wild-type, dksA, greA, or greB deletion mutant strains. As observed previously, rrnB P1 promoter activity increased in strains lacking dksA14 (~3 to 6.5-fold in rich medium in log and stationary phase, respectively; ~4 to 10-fold in defined medium; Figure 3(a)–(d)), reflecting a loss of regulation by ppGpp and iNTP concentration. In contrast, a control promoter, lacUV5, increased much less in the dksA mutant (1.2 to 1.4-fold in rich medium; Figure 3(e) and (f)).
Figure 3.
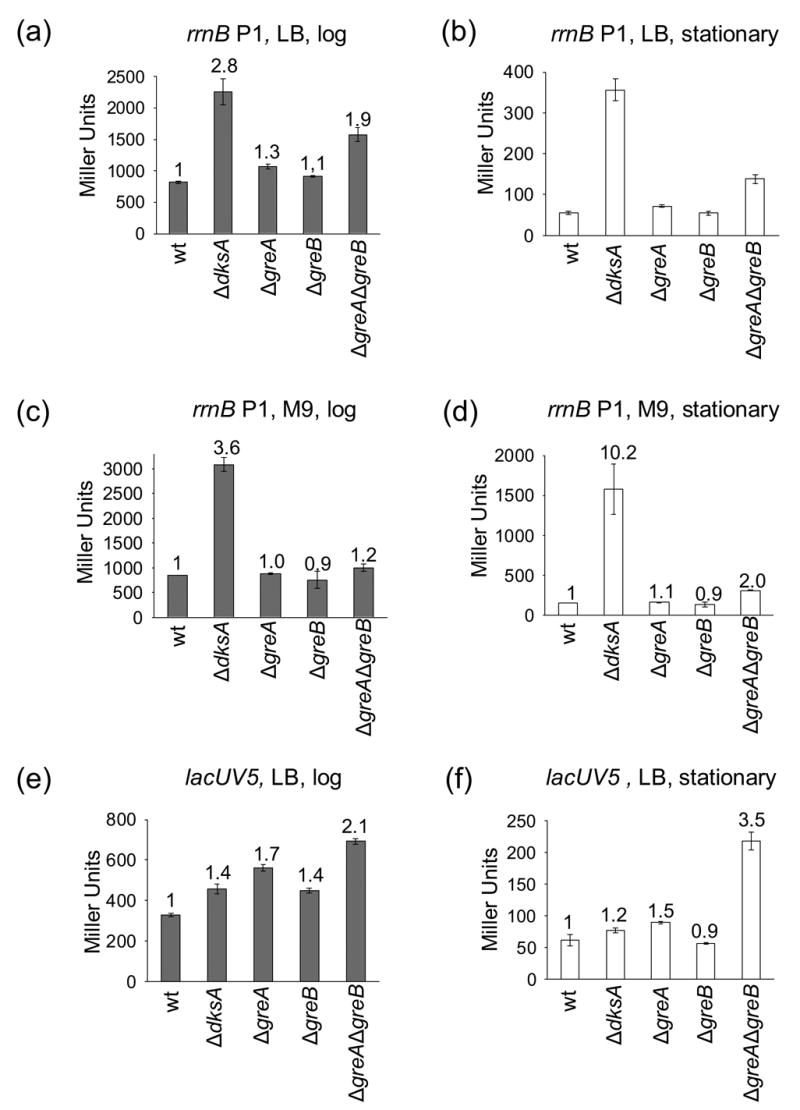
dksA, but not greA or greB, is required for regulation of rRNA synthesis in vivo. β-galactosidase activities (expressed in Miller Units) were determined in strains containing promoter-lacZ fusions. (a) – (d) rrnB P1 promoter (sequence endpoints −61 to +1; RLG5950). (e) and (f) lacUV5 promoter (−46 to +1; RLG5022). In (a), (b), (e), and (f), cells were grown in LB at 30ºC. In (c) and (d), cells were grown in M9 defined medium (see Materials and Methods) at 30ºC. (a), (c), (e) log phase (OD600~0.4). (b), (d), (f) stationary phase. Numbers above each bar refer to the fold-change in promoter activity relative to the same promoter in the same growth condition in the wild-type strain. β-galactosidase activities from ≥ 2 cultures were averaged, and ranges or standard errors are shown.
Deletion of either greA or greB alone increased rrnB P1 promoter activity only slightly in any of the growth conditions tested (Figure 3(a)–(d)), and these effects were similar to those on the control promoter (Figure 3(e) and (f)). Effects of deletion of both greA and greB together (ΔgreAΔgreB) on rrnB P1 were slightly larger than with the ΔgreA or ΔgreB single mutants in stationary phase and were exaggerated even slightly more on the lacUV5-lacZ fusion (Figure 3(a)–(f)). We have not investigated the mechanism(s) responsible for these effects, since they were promoter-nonspecific. In theory, they could derive from direct or indirect effects on transcription elongation or on translation of the reporter rather than on transcription initiation (see also20,26).
Strains were also constructed containing a deletion of the dksA gene in combination with deletions of greA, greB or both (ΔdksAΔgreA,ΔdksAΔgreB, or ΔdksAΔgreAΔgreB). In general, the effects on rrnB P1 or lacUV5 in log phase and stationary phase were similar to that of the ΔdksA mutation alone (data not shown). The triple mutant strain displayed growth defects, saturated at a lower optical density, and accumulated suppressor mutations. We reproducibly observed small increases in rrnB P1 and lacUV5 promoter activity in stationary phase in the ΔdksAΔgreAΔgreB mutant relative to the ΔdksA mutant. However, these ratios represent comparisons of accumulated β-galactosidase activity in stationary phase cultures that had undergone different numbers of cell divisions and therefore should be interpreted with caution.
In summary, our results suggest that GreA and/or GreB have little specific influence on rRNA promoter activity in vivo. These results contrast sharply with the large effect of DksA.
The DksA concentration is higher than that of GreA or GreB at all times in growth
The cellular concentrations of DksA, GreA, and GreB, as well as their RNAP binding affinities, would influence their effects on transcription in vivo. Therefore, we determined the amounts of the three factors in cell lysates at different times during growth using quantitative Western blots (Figure 4). The DksA concentration was ~137 fmoles/μg total cell protein in cells growing exponentially (Figure 4(a) and legend) and remained at almost this level for several hours in stationary phase, after which it declined by ~50% (Figure 4(b); V. Shingler, Umea University, personal communication). Since DksA is a negative regulator of rRNA transcription, and rRNA promoter activity decreases in stationary phase,5 this change in DksA concentration could not be responsible for rRNA promoter inhibition. These results are consistent with our previous conclusion that changes in ppGpp and iNTP concentrations, rather than in DksA concentration, account for regulation of rRNA promoter activity.14
Figure 4.
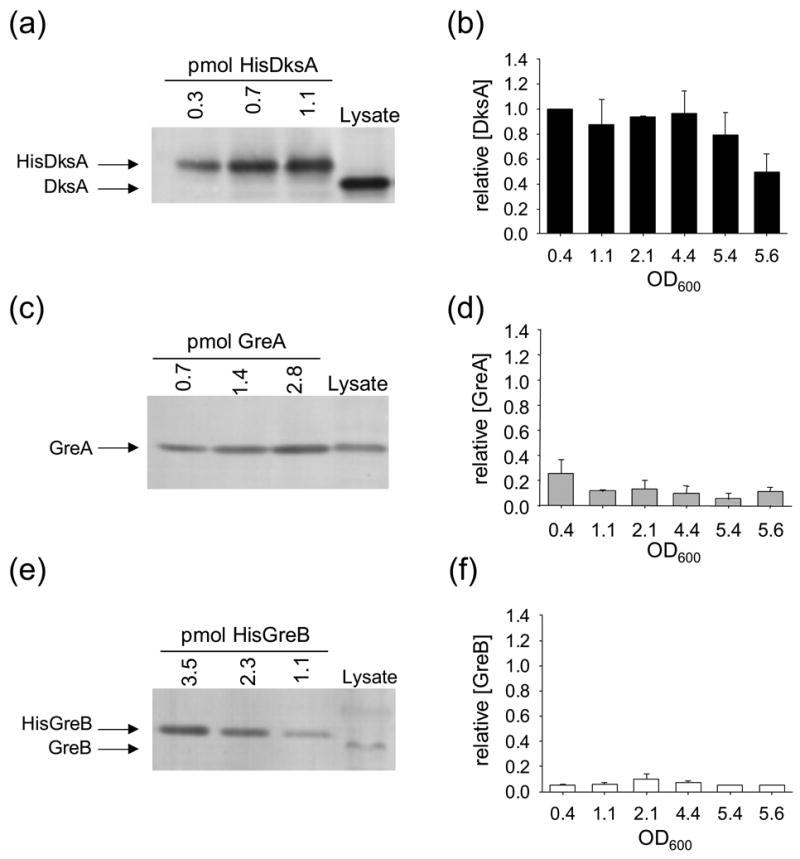
DksA concentrations are higher than GreA and GreB concentrations at all points in the growth curve. Panels (a), (c), and (e) show representative Western blots from cell lysates of a wild-type strain (RLG5950) grown in MOPS defined medium (see Materials and Methods) in log phase (OD600 = 0.4). Panels (b), (d), and (f) plot relative amounts of the factors versus OD600 at different times in a growth curve. (Concentrations are expressed relative to DksA at OD600 ~ 0.4, defined as 1.0). Standard curves made from blots of each purified protein were used to quantify the amounts of the factors in cell lysates. The DksA and GreB standards migrate slightly slower than the native proteins from cell lysates, because they contain hexahistidine tags. Concentrations (fmol factor/μg total protein) were determined from at least 3 separate experiments at each of six cell densities, as indicated. The absolute concentrations at OD600 ~ 0.4 were: DksA, 137 ± 34 fmol/μg. GreA, 53 ± 17 fmol/μg. GreB, 13 ± 5 fmol/μg. Similar results were obtained for cells grown in LB medium (data not shown).
The GreA concentration (~53 fmoles/μg total protein in log phase; Figure 4(c)) was ~2 to 3-fold lower than the DksA concentration at all times (Figure 4(d)). The combination of its low cellular concentration and its weak effect on RNAP-promoter complexes, even at saturating concentrations, would be sufficient to account for the lack of an effect of the ΔgreA mutation on rRNA promoter activity.
The GreB concentration (~13 fmoles/μg total protein in log phase; Figure 4(e)) was ~10-fold lower than the DksA concentration throughout the growth curve (Figure 4(f)). Thus, even though GreB and DksA have similar effects on rrnB P1 in vitro (Figures 1–2), the low cellular GreB concentration would be sufficient to explain the lack of an effect of the ΔgreB mutation on rRNA promoter activity in vivo.
We also measured the amounts of RNAP to determine its approximate stoichiometry with DksA, GreA, and GreB at different times. The RNAP concentration was ~12 fmoles/μg total protein in log phase (data not shown), consistent with previous estimates (e.g.27), and decreased 25% or less in stationary phase. Thus, DksA and GreA are in excess of RNAP in vivo, and GreB is approximately equimolar with RNAP.
Overexpressed GreB, but not GreA, complements a ΔdksA strain for regulation of rRNA transcription
The above results showed that GreB (and to a much lesser extent GreA) affected rRNA promoters like DksA in vitro, but that the concentrations of GreB were much lower than that of DksA in vivo, accounting for the lack of specific effects of deleting greB on rrnB P1 activity. To determine whether there were differences in mechanistic properties, in addition to differences in amounts, of the different factors that affected their abilities to inhibit rrnB P1 activity in vivo, we determined whether overexpression of the Gre factors complemented the ΔdksA mutation (i.e. reduced rrnB P1 promoter activity to its wild-type level).
In the ΔdksA mutant, rrnB P1 promoter activity was higher than in the wild type strain (3.6-fold higher in log phase, Figure 5(b), and 17-fold higher in stationary phase, Figure 5(c)). The medium was different than that used in Figure 3, likely accounting for the slightly different fold-increase compared to that observed previously in stationary phase (see also14). A plasmid that supplied DksA (~50% as much DksA as in wild-type cells; Figure 5(a)) was sufficient to almost fully complement the ΔdksA mutation, reducing rRNA promoter activity to near wild-type levels (Figures 5(b) and (c), ΔdksA + pdksA; see also14).
Figure 5.
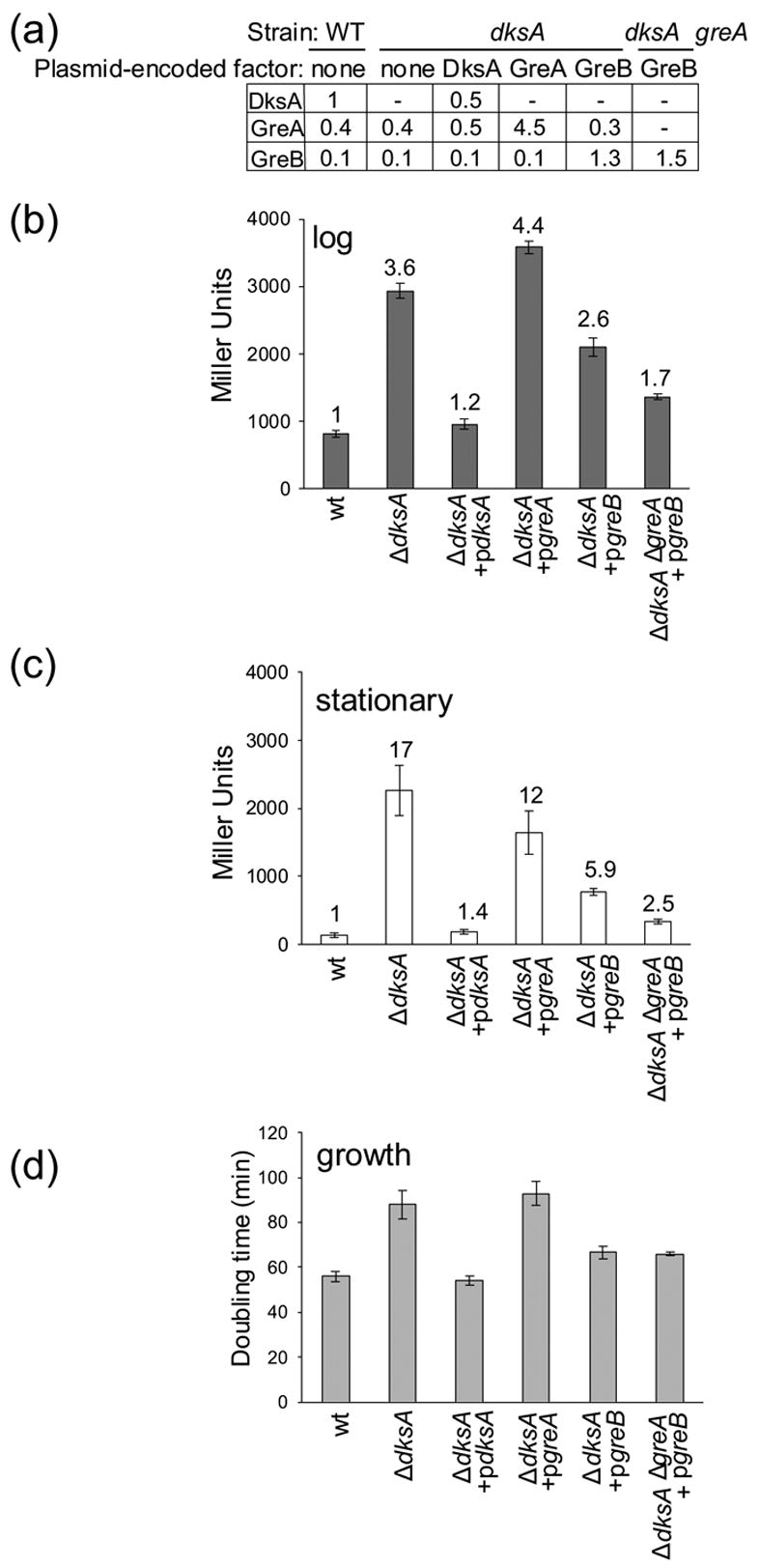
Overexpression of GreB, but not GreA, can partially compensate for the absence of DksA. In the indicated strain backgrounds, DksA, GreA, or GreB were expressed without IPTG induction from an lpp-lac promoter on plasmids derived from pINIIIAI (pRLG6333, pRLG8229, pRLG8242; see Materials and Methods; Table 1). Cells were grown in MOPS medium supplemented with 0.4% glycerol, 20 amino acids (see Materials and Methods), and 100 μg/ml ampicillin. (a) Protein concentrations, relative to the concentration of DksA in the wild-type (WT) strain, were determined by quantitative Western blots at OD600 = 0.4 (log phase). Promoter activities were determined (b) in log (OD600 ~0.4) and (c) in stationary phase (24 hr of growth). Strains were derivatives of RLG5950, carrying an rrnB P1-lacZ reporter on a λ prophage (see Materials and Methods). Averages and standard deviations were calculated from at least 3 experiments, except for the ΔdksA greA + pgreB strain, which was measured in duplicate. The number above each bar indicates the fold-increase relative to the rrnB P1 promoter activity of the wild-type strain in the same condition. (d) Doubling times in log phase in the MOPS medium used above were determined by monitoring OD600 (wild-type, 56 min. ΔdksA, 88 min. ΔdksA + pdksA, 54 min. ΔdksA + pgreA, 93 min. ΔdksA + pgreB, 67 min. ΔdksA ΔgreA + pgreB, 66 min.)
When GreA was expressed 11-fold higher than its wild-type level, enough to compensate for its 8-fold lower apparent affinity for RNAP than DksA, there was little effect on rrnB P1 promoter function in the ΔdksA mutant (Figures 5(b) and (c), ΔdksA + pgreA). In contrast, when GreB was expressed to a level 13-fold higher than its wild-type concentration, 1.3-fold higher than the concentration of DksA in a wild-type strain (Figure 5(a)), there was partial complementation of the ΔdksA mutant: rrnB P1 promoter activity was reduced relative to that in the ΔdksA strain, although it was still higher than in the wild-type strain or in the ΔdksA strain complemented with pdksA (Figures 5(b) and (c), ΔdksA + pgreB). We conclude that, when expressed at high enough levels in vivo, GreB partially restores regulation of rrnB P1 in the absence of DksA. Parallel experiments with the lacUV5 control promoter-lacZ fusion showed that expression of DksA fully complemented and overexpression of GreB partially complemented the small (~2-fold) effect of the ΔdksA mutation on this promoter, but overexpression of GreA had no effect (data not shown).
Based on their sequence and structural similarities, GreA likely interacts with the same site on RNAP as GreB. Although the details of their interactions with RNAP must differ enough to account for the differences in their effects on rrnB P1 transcription, we reasoned that binding of GreA to RNAP might compete with binding of GreB in the absence of DksA, and thereby lessen the ability of GreB to complement the ΔdksA mutant. To test this possibility, GreB was overexpressed in a ΔdksAΔgreA double mutant strain. Under these conditions, complementation by GreB was more effective than in the ΔdksA strain (Figure 5(b) and (c)): rRNA promoter activity was reduced almost to wild-type levels in both log and stationary phase (i.e. only ~1.7-fold higher than in the wild-type strain in log phase, and only ~2.5-fold higher in stationary phase, lower than the derepression of rrnB P1 observed with GreB overexpression in the ΔdksA mutant strain wild-type for greA, and much lower than that observed without GreB overexpression).
Overexpression of GreB also complemented most of the effect of the ΔdksA mutation on growth rate (Figure 5(d)). The generation time of the ΔdksA mutant in this medium (MOPS/glycerol/amino acids; see Materials and Methods) was considerably longer than that of the wild-type strain (Figure 5(d); see legend for doubling times). Plasmid-encoded DksA (pdksA) fully complemented the ΔdksA mutant with respect to effects on growth rate. Overproduction of GreA did not complement the ΔdksA mutant at all, whereas overproduction of GreB partially complemented the ΔdksA mutant, qualitatively consistent with the effects observed on rRNA promoter activity.
We emphasize that the low concentration of GreB in wild-type cells likely limits the physiological significance of its effect on rRNA promoter activity. However, taken together with the effects of GreB described in vitro (Figures 1–2), our results strongly suggest that GreB and DksA share mechanistic properties that account for their effects on rrnB P1 transcription. Furthermore, given the similar effects of GreB and DksA on promoter complex half-life, our results are consistent with the model that shifting the rRNA promoter-RNAP equilibrium in the dissociation direction is a major component of the role of DksA in regulating rRNA transcription initiation in vivo.
The Gre factors do not facilitate positive control by ppGpp in vitro
In addition to their role in regulation of rRNA promoter activity, DksA and ppGpp together can co-activate transcription, greatly increasing the effect of ppGpp on a subset of promoters for amino acid biosynthesis including Phis and PthrABC.12 ppGpp or DksA alone had little effect on these promoters in vitro, but ppGpp together with DksA increased Phis activity ~3-fold (Figure 6(a)) and PthrABC activity ~5-fold (Figure 6(b)). In contrast, ppGpp together with GreA or GreB were unable to affect these promoters. Thus, positive control of transcription by ppGpp requires distinct properties of DksA that are not supplied by the Gre factors.
Figure 6.

ppGpp and DksA, but not the Gre factors, co-activate transcription at amino acid biosynthesis gene promoters. Multiple round in vitro transcription from Phis and PthrABC was carried out in duplicate in transcription buffer containing 165 mM NaCl and 2 μM DksA, 2 μM GreB, or 7 μM GreA with or without 100 μM ppGpp. The templates were (a) Phis (promoter endpoints −60 to +1; pRLG4413) or (b) PthrABC (promoter endpoints −72 to +16; pRLG5073). Transcription from duplicate lanes was averaged and is quantified as the ratio ± ppGpp. Each of the three factors increased transcription slightly in the absence of ppGpp, as described previously for DksA.12
The Gre factors, unlike DksA, cannot synergize with ppGpp to regulate rRNA promoters in the absence of the ω subunit of RNAP
We reported previously that ppGpp failed to inhibit transcription by RNAP lacking the ω subunit in vitro, but ppGpp did inhibit the enzyme without ω when DksA was included in the reaction.17 Consistent with these results, DksA at 0.5 μM together with ppGpp at 400 μM, conditions where neither alone decreased transcription of rrnB P1 by RNAP lacking ω, together inhibited transcription by almost 60% (Figure 7(a)). In contrast, neither Gre factor worked with ppGpp to inhibit transcription by RNAP lacking ω. Thus, ppGpp and DksA could synergistically inhibit transcription even in the absence of ω, but GreA or GreB were deficient in functioning without ω. This deficiency did not result from an ω requirement for Gre factor binding (data not shown).
Figure 7.

DksA, but not Gre factors, can compensate for the absence of the ω subunit of RNAP. (a) Single round in vitro transcription reactions were carried out in duplicate in transcription buffer with 30 mM NaCl using RNAP purified from a ΔrpoZ strain (see Materials and Methods). Reactions contained 0.5 μM DksA, GreA, GreB, or no factor and either 400 μM ppGpp or water. (b) and (c) Plasmid-encoded GreB cannot complement a strain lacking both dksA and rpoZ for regulation of rrnB P1. ΔdksA rpoZ (RLG8327) strains were transformed with an empty vector (pINIIIA1), pINIIIA1 encoding DksA (pdksA; pRLG6333), or pINIIIA1 encoding GreB (pgreB; pRLG8242). Cells were diluted into fresh LB with 100 μg/ml ampicillin at 30°C to an OD600~0.025, and β-galactosidase activity was measured after 4 generations (OD600~0.4) (b) or after 24 hr (c). The OD600 after 24 hr was ~2.5 in the ΔdksA ΔrpoZ strain and ~5 in the other strains (see Results). Reported values are averages from at least 2 experiments.
To address whether ω was required for the observed partial complementation of the ΔdksA mutation in vivo by plasmid-encoded GreB (Figure 5), we measured the activity of an rrnB P1 promoter-lacZ fusion in rich medium in a ΔdksA strain that was also deleted for rpoZ, the gene encoding ω. Consistent with previous reports, the ΔrpoZ mutation by itself had little or no effect on rrnB P1 promoter activity in vivo,18 and the ΔdksA mutation derepressed rrnB P1 transcription14 (almost 3-fold in log and almost 6-fold in stationary phase; Figure 7(b) and (c)). Plasmid-encoded GreB partially complemented the effect of the ΔdksA mutation on rrnB P1 transcription, as also shown in Figure 5 in defined medium.
Plasmid-encoded GreB decreased rrnB P1 transcription less in the ΔdksAΔrpoZ mutant than in the ΔdksA mutant: GreB reduced rrnB P1 activity only 10% in log and 18% in stationary phase in the double mutant compared to 29% and 46%, respectively, in the single mutant (Figure 7(b) and (c)). Thus, consistent with the results in vitro (Figure 7(a)), GreB required ω to affect rrnB P1 transcription in vivo.
We note that the ΔdksAΔrpoZ double mutant displayed a pronounced growth defect; although its doubling time was only about 15% longer than the ΔdksA or ΔrpoZ single mutants in rich medium, it reached an optical density only about one-half that of the single mutant strains. Since β-galactosidase is stable, enzyme activity accumulates per total protein when cells stop growing. This complicates interpretation of differences in β-galactosidase activity when strains saturate at different optical densities (i.e. in the comparison of the double mutant with the single mutant strains in stationary phase; Figure 7(c)). To our knowledge, the decrease in growth of the double mutant strain represents the first in vivo phenotype reported for an E. coli ΔrpoZ strain.
Discussion
Implications for the mechanism of DksA action
Previous work demonstrated that the competitor-resistant complex formed by RNAP on rRNA promoters is short-lived.2 Promoter or RNAP mutants that alter the equilibrium between this and earlier complexes on the pathway to open complex formation alter regulation of rRNA promoters by ppGpp and the iNTP in vitro and in vivo (e.g.4,9,28). Since DksA further decreases the intrinsically short half-life of the rRNA promoter complex and amplifies the effects of ppGpp and the iNTP on rRNA transcription, we suggested that its effect on half-life accounts, at least in part, for its role in control of rRNA transcription.14 We show here that DksA and GreB have similar apparent binding affinities and similar effects on complex half-life, although only DksA can affect rrnB P1 at the concentrations of these factors present in wild-type cells. Expression of GreB to a concentration similar to that of DksA in wild-type cells partially complemented the defect in rRNA regulation in vivo caused by deletion of dksA, and almost fully complemented the absence of dksA in a strain also lacking greA. However, GreB was unable to mimic the effect of DksA in positive control of transcription or to function with ppGpp on RNAP lacking ω, implying that these effects require another property of DksA.
How could GreB mimic some activities of DksA and not others? Differences in the interactions of DksA versus GreB with RNAP could result in effects on different microscopic rate constants in the overall mechanism of transcription initiation, even though they appear to have identical effects on the composite rate constant that determines the half-life of the promoter complex. In theory, DksA and GreB could differentially alter a step in the association direction during open complex formation, thereby differently affecting positive regulation of amino acid biosynthesis promoters.12 We also note that GreB had a greater effect than DksA on the NTP-dependence of rrnB P1 (Figure 1(e) and legend), although differential effects on blocking NTP access, rather than effects on different kinetic intermediates, could be an alternative explanation for this observation.
The low concentration of GreB (relative to DksA) apparently is sufficient to account for the lack of an effect of the ΔgreB mutation on rRNA promoters in otherwise wild-type cells. Whereas the low concentration of GreA, in conjunction with its weak affinity for RNAP, is able to account for its inability to substitute for DksA in vivo, we also show that GreA lacks mechanistic properties of DksA that prevent complementation even when overproduced to compensate for its low affinity. Therefore, binding in the secondary channel is necessary but insufficient to account for the effects of DksA on transcription.
A recent study29 proposed that GreA stimulates rRNA transcription and that by balancing antagonistic effects of DksA and GreA, the cell regulates rRNA promoters in vivo. The apparent stimulatory effects of GreA on transcription in vivo29 were observed only when it was dramatically overproduced from an induced tac promoter designed for protein purification purposes. In contrast, our results suggest that when the GreA concentration is in the physiologically relevant range (at least under the conditions examined here), it does not compete with DksA for binding to the promoter complex and that GreA does not positively control rRNA promoters in vitro or in vivo.
We emphasize, however, that we have not ruled out the possibility that Gre factors might be expressed at higher levels under some other growth conditions and thereby either relieve the effect of DksA on rRNA transcription (in the case of GreA) or substitute for it (in the case of GreB). We also point out that, in theory, either GreB or DksA could suffice to increase the NTP-sensitivity of rRNA promoters under some conditions, since less GreB than DksA was needed to alter the NTP-dependence of rrnB P1 in vitro (Figure 1(e)).
Structural perspective on differences in DksA and Gre factor function
DksA shows little or no primary sequence homology to the Gre factors. Although DksA, GreA, and GreB have similar sizes and comparable folds in their coiled-coil domains,19,22,30 there are substantive differences in amino acid sequence in this domain, and there are even greater differences in sequence and structure of their globular domains.
The globular domains of both DksA and the Gre factors likely dock on a coiled-coil in β′ near the entrance to the RNAP secondary channel19–22 (I. Toulokhonov, J. Mukhopadhyay, R.H. Ebright, and R.L.G., unpublished data). However, the globular domains of DksA and the Gre factors are so different that it is unlikely they bind to RNAP in an analogous fashion. The globular domain of DksA contains a zinc finger and a C-terminal α-helix, neither of which is present in the Gre factors.19 Potential roles for these features of DksA in RNAP binding and/or function remain to be determined.
Furthermore, it is also unclear that the coiled-coil domains of the Gre factors and DksA bind in the RNAP secondary channel in an analogous fashion. If the coiled-coils of the Gre factors and DksA were to superimpose,19 then their acidic tips would be oriented in opposite directions with respect to the RNAP active site region. Conversely, if their acidic tips were to superimpose, then their coiled-coils would be oriented differently with respect to the secondary channel wall. Differences in charge distribution in the coiled-coils of GreA and GreB appear to play a role in the differences in the RNA cleavage activities stimulated by these two proteins.31,32 However, it is not yet clear whether charge distribution in the DksA coiled-coil is important for its effects on transcription initiation. In depth structure-function analyses will be required to determine which parts of GreB and DksA are responsible for their mechanistic similarities and differences.
Two different orientations of ppGpp were identified crystallographically at a single binding location at the bottom of the secondary channel of Thermus thermophilus RNAP, and it was proposed that these differing orientations might be related to positive versus negative regulation by ppGpp.33 It was also proposed that the acidic tip of DksA might chelate a ppGpp-bound Mg++ and this was crucial for DksA function. It is not yet clear that either of the ppGpp binding sites identified in the co-crystal are the ones responsible for the biologically significant effects of ppGpp on transcription initiation. Nevertheless, since DksA and GreB both synergize with ppGpp to inhibit transcription, but only DksA synergizes with ppGpp to stimulate transcription, determination of the residues in DksA and GreB required for positive versus negative control by ppGpp may improve our understanding of these control mechanisms.
Role of ω in regulation of rRNA promoters
Previously, we showed that the rrnB P1 promoter was not inhibited by ppGpp in vitro when transcribed by RNAP lacking ω, although DksA restored the response of the rRNA promoter complex to ppGpp.17 Since ΔrpoZ mutants grow like wild-type cells and display normal stringent responses,18 indicating they respond to ppGpp, we suggested that DksA might mask the effect of the absence of ω on rRNA expression in vivo.17 The properties of the ΔdksAΔrpoZ double mutant (Figure 7) provide the first in vivo phenotype associated with the absence of ω. These results suggest the intriguing possibility that undersaturation of RNAP with ω, under certain conditions, might influence cellular regulatory responses.
Perspective
Since overexpressed GreB can provide almost the same degree of rRNA transcription regulation as DksA, our results suggest that, ultimately, interactions with RNAP that are shared between the two factors will account for much of their effects on rRNA transcription initiation. Conversely, interactions with RNAP that are not shared between DksA and GreB may account for their differing capabilities in positive control. There are ample differences in the structures of DksA and the Gre factors that could account for their functional specificities. Understanding which DksA interactions with RNAP are shared and which are unique will be invaluable in elucidating the molecular mechanisms responsible for its effects on transcription initiation.
Materials and Methods
Strains, plasmids, and proteins
Strains and plasmids are listed in Table 1. Promoter-lacZ fusions are on λ prophages integrated at the λ attachment site in VH1000.34 Promoter sequence endpoints are indicated in the figure legends. The ΔdksA mutation (dksA::tet) has been described previously.14 The ΔgreA and ΔgreB mutations (greA::cm and greB::kan) originated from CLT252 and CLT253, respectively and were obtained from R. Landick, University of Wisconsin. greA::cm and greB::kan are insertion-deletions that replace the entire coding regions with chloramphenicol or kanamycin-resistance cassettes, respectively (109 bp upstream of the AUG and 177 bp downstream of the stop codon for the greA mutation and 7 bp upstream of the AUG and 119 downstream of the stop codon for the greB mutation). The mutations were transduced to reporter strains by standard procedures using phage P1vir.
Table 1.
Strains and Plasmids.
| Strain or Plasmid | Genotype or Description | Source or Reference |
|---|---|---|
| Strain | ||
| VH1000 | MG1655 pyrE+ lacI lacZ (RLG3499) | 8 |
| RLG830 | JV554 rpoZ::kan | 18 |
| RLG5022 | VH1000 λlacUV5 (−46/+1)-lacZ | C. Hirvonen and R.L.G. |
| RLG5950 | VH1000 λrrnB P1 (−61/+1)-lacZ | 39 |
| RLG7238 | RLG5950 dksA::tet | this work |
| RLG7239 | RLG5950 greA::cm | this work |
| RLG7240 | RLG5950 greB::kan | this work |
| RLG7241 | RLG5950 dksA::tet greA::cm | this work |
| RLG7242 | RLG5950 greA::cm greB::kan | this work |
| RLG7500 | RLG5022 dksA::tet | this work |
| RLG8050 | RLG5022 greA::cm | this work |
| RLG8051 | RLG5022 greB::kan | this work |
| RLG8054 | RLG5022 greA::cm greB::kan | this work |
| RLG8325 | RLG5950 rpoZ::cm | this work |
| RLG8327 | RLG5950 rpoZ::cm dksA::tet | this work |
| Plasmid | ||
| pDNL278 | GreA overproduction plasmid | 26 |
| pMO1.4His | His6GreB overproduction plasmid | 24 |
| pRLG770 | transcription vector | 35 |
| pRLG3422 | PRLG770 containing lacUV5 (−46/+1) | 8 |
| pRLG4264 | pSL6 containing lacUV5 (−60 to +38) | 40 |
| pRLG4413 | PRLG770 containing Phis (−60/+1) | B. Paul and R.L.G. |
| pRLG5073 | PRLG770 containing PthrABC (−72/+16) | 41 |
| pRLG5944 | PRLG770 containing rrnB P1 (−61/+1) | C. Hirvonen and R.L.G. |
| pRLG6332 | pINIIIA1 | 42 |
| pRLG6333 | PRLG6332 encoding DksA | 14 |
| pRLG6555 | PRLG770 containing rrnB P1 (−66/+9) | 9 |
| pRLG7067 | pET28a encoding HisDksA | 14 |
| pRLG8229 | PRLG6332 encoding GreA | this work |
| pRLG8242 | PRLG6332 encoding GreB | this work |
Plasmids expressing GreA or GreB from the lpp-lac promoter were constructed by amplifying the gene of interest by PCR and ligating the products between the XbaI and HindIII sites of pINIIIA1, as described previously for DksA.14 The constructs contained sequences from 16, 21, or 16 bp upstream of the ATG to 28, 22, or 28 bp downstream of the termination codon for greA, greB, and dksA, respectively.
Plasmid templates used for in vitro transcription were derivatives of pRLG770.35 Promoter sequence endpoints are indicated in Table 1. The plasmids contain rrnB T1T2 transcription terminators ~160 bp downstream of the site of insertion of the promoter fragment and also encode the RNA-I promoter.
Hexahistidine-tagged DksA was overexpressed from a phage T7 promoter in BL21(DE3) and purified as described.14 GreA was overexpressed from pDNL278 in JM109 and purified as described.26 His-tagged GreB was overexpressed from the trc promoter in pMO1.4His in BL21 after induction with IPTG, and purified as described.24 Protein concentrations were determined with the Bradford Assay Reagent (BioRad), using BSA as a standard, and diluted into storage buffer (40 mM Tris HCl pH 7.5, 0.8 M NaCl, 1 mM EDTA, 1 mM DTT) before use.
Native RNAP was purified by standard procedures.36 Core RNAP from an rpoZ mutant strain (Table 1) was purified using the polyol-responsive antibody 8RB13 (NeoClone, Inc. Madison, WI) as described37 and was then reconstituted with 3-fold molar excess σ70. σ70 was purified by standard procedures.38
In vitro transcription
Multiple or single round in vitro transcription was performed as described14 with 1 nM supercoiled plasmid and 10 nM RNAP at 30ºC in transcription buffer (40 mM Tris HCl, pH 7.9, 10 mM MgCl2, 1 mM DTT, 0.1 mg/ml BSA) and the salt concentrations indicated in the figure legends. For single round transcription (Figure 1(a,b)), complexes were formed by incubation of RNAP and template DNA at 30ºC for 10 min. NTPs (500 μM ATP, 200 μM GTP, 200 μM CTP, 10 μM UTP, and 1.0 μCi [α-32P] UTP, final concentrations) were added simultaneously with 200 nM double-stranded full consensus promoter DNA as competitor for free RNAP.38 After 10 min for transcription elongation, the reactions were stopped with an equal volume of formamide gel loading solution. Multiple round NTP-dependence assays (Figure 1(e)) were carried out at 25°C using 200 μM GTP, 10 μM CTP, 10 μM UTP and [α-32P] UTP, and varying ATP concentrations from 25–5000 μM, as described.9 For the multiple round reactions shown in Figure 6, the buffer and NTP concentrations were as described above for the single round experiments, and reactions were initiated by addition of RNAP. Transcripts were separated on 7M urea, 6% polyacrylamide gels and quantified by phosphorimaging using ImageQuant. We emphasize that only reactions carried out under the same conditions of salt and temperature can be compared directly. Concentrations of factors, ppGpp, or both in each reaction are indicated in the figure legends. Control reactions contained protein storage buffer.
β-galactosidase assays
Cells were grown without IPTG in the media indicated in the figure legends for ~4 generations to an OD600 of ~0.4 (for log phase) or for ~24 hr to an OD600 of ~5.0 (stationary phase). The ΔdksAΔrpoZ double mutant strain reached an OD600 of only ~2.5 in stationary phase; see Results. The MOPS-based medium contained 0.4% glycerol, 40 μg/ml each of tryptophan and tyrosine, and 80 μg/ml each of the other 18 amino acids. The M9-based medium contained 0.4% glycerol, 40 μg/ml tryptophan, and 0.8% casamino acids. Cultures containing plasmids were supplemented with 100 μg/ml ampicillin. Cells were harvested, chilled on ice for ~20 min, lysed by sonication, and β-galactosidase activity was determined by standard methods.11
RNAP-promoter complex decay assays
Half-lives of RNAP-promoter complexes (Figure 2) were determined using either a transcription based assay or a filter-binding assay.11 For the transcription based assay, 10 nM of RNAP was incubated with 1 nM supercolied DNA template containing the rrnB P1 promoter (endpoint −61 to +1, pRLG5944) in transcription buffer (40 mM Tris HCl pH 7.9, 10 mM MgCl2, 1 mM DTT, 0.1 mg/ml BSA) containing 30 mM NaCl and DksA, GreA, GreB, or storage buffer (as indicated in the figure legends) at 30ºC for 10 min. Reactions were initiated with NTPs (500 μM ATP, 200 μM GTP, 200 μM CTP, 10 μM UTP, and 1.0 μCi [α-32P] UTP, final concentrations) at time points after the addition of 200 nM double-stranded full consensus promoter DNA (as competitor for free RNAP).36 Reactions were stopped, the RNA products were separated on gels, and quantified as described above. The amount of transcription relative to that at the time of competitor addition was plotted as a function of time.
For the filter binding assay (Figure 2(b)–(f)), ~0.2 nM of a 242 bp XhoI restriction fragment containing the lacUV5 promoter (endpoints −60 to +40) from pRLG4264 was 32P end-labeled and incubated with 10 nM RNAP in transcription buffer (40 mM Tris HCl pH 7.9, 10 mM MgCl2, 1 mM DTT, 0.1 mg/ml BSA) containing 100 mM NaCl and the indicated concentration of ppGpp, DksA, GreA, GreB, or storage buffer at 25ºC (or 30ºC for the assays done in the presence of ppGpp) for 30 min. Heparin (to 10 μg/ml) was added, aliquots were removed at the indicated times, and RNAP-promoter complexes were captured on nitrocellulose filters. The radioactivity retained was determined by phosphorimaging, and half-lives were calculated as described.11 DNase I footprinting of lacUV5 complexes formed in the presence and absence of the three factors under these conditions confirmed that the complexes retained by the filters were specific and competitor-resistant. In addition, control experiments confirmed that the factors alone (in the absence of RNAP) were not able to capture the promoter DNA fragment on the filters (data not shown).
To calculate the apparent Kd (Figure 2(c)), half-lives were determined at a range of DksA, GreA, and GreB concentrations relative to the half-lives in the absence of factor and plotted versus the factor concentration. Each point represents the average half-life determined from at least two time courses. The curves were fit to an exponential decay equation using SigmaPlot. The apparent Kd represents the factor concentration required for half the maximal decrease in complex lifetime. In theory, these Kds could underestimate the actual affinities of the proteins for RNAP, since the half-life assay includes step(s) in addition to binding.
Quantitative Western blots
Protein concentrations were determined from 50–100 ml cultures grown under the same conditions used for the β-galactosidase assays (see above). Briefly, after growth to the indicated optical densities and 20 min on ice, cells were harvested by centrifugation, suspended in ≤ 1 ml 10 mM Tris HCl pH 7.9, 1 mM EDTA, 5% glycerol, 0.1 mM PMSF, lysed by sonication, and centrifuged to remove insoluble material. The total soluble protein concentration was determined with Bradford Assay Reagent using BSA as a standard. A range of purified GreA, GreB, and DksA concentrations was used to construct standard curves (see Figure 4). Lysates and standards were separated on 4–12% SDS-polyacrylamide gels (Invitrogen), transferred electrophoretically to PVDF membranes (Bio-Rad), and Western blots were performed as described.14 ~5 μg, ~40 μg, and ~100 μg protein lysate were used for detection of DksA, GreA, and GreB, respectively. Rabbit polyclonal anti-DksA antiserum was a generous gift from D. Downs (UW-Madison) and was precleared with total lysate from a strain lacking dksA. Mouse monoclonal anti-GreA and anti-GreB antisera were produced by NeoClone, Inc (Madison, WI) and showed no cross-reactivity to each other. Mouse monoclonal anti-β′ RNAP antiserum was a gift from R. Burgess (UW-Madison). An HRP-conjugated secondary antibody (specific to rabbit IgG for polyclonal or to mouse IgG for monoclonal antibodies; Santa Cruz Biotechnology) was detected using ECL+ reagent (GE Healthcare). Western blots were scanned with a Typhoon phosphorimager [GE Healthcare, 520 BP 40 Cy2, ECL+ Blue family (emission), ECL+ excitation (laser)], quantified with ImageQuant (Molecular Dynamics), and protein amounts in the cell lysates were interpolated from the standard curve.
Supplementary Material
Acknowledgments
We thank Dick Burgess for antibody to β′, Diana Downs for antibody to DksA, Mike Zwick and NeoClone, Inc. for making monoclonal antibodies to GreA and GreB, Erin Jones for purifying DksA, Richard Ebright and Sergei Borukhov for the HisGreB overexpression plasmid, and Bob Landick for the GreA overexpression plasmid. We also thank Vicky Shingler for sharing information prior to publication. This work was supported by a grant (RO1 GM37048) from the National Institutes of Health to R.L.G., by a fellowship from the N.I.H. Molecular Biosciences Training Grant and a predoctoral fellowship from the University of Wisconsin-Madison to S.T.R., by a Hilldale undergraduate fellowship to J.J.L., and by a predoctoral fellowship from the Howard Hughes Medical Institute to C.E.V.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Record MT, Jr, Reznikoff WS, Craig ML, McQuade KL, Schlaz PJ. Escherichia coli RNA polymerase (Eσ70), promoters, and the kinetics of the steps of transcription initiation. In: Neidhardt FC, editor. Escherichia coli and Salmonella. ASM Press; Washington, D.C: 1996. pp. 792–820. [Google Scholar]
- 2.Gourse RL. Visualization and quantitative analysis of complex formation between E. coli RNA polymerase and an rRNA promoter in vitro. Nucleic Acids Res. 1988;16:9789–9809. doi: 10.1093/nar/16.20.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paul BJ, Ross W, Gaal T, Gourse RL. rRNA transcription in Escherichia coli. Annu Rev Genet. 2004;28:749–770. doi: 10.1146/annurev.genet.38.072902.091347. [DOI] [PubMed] [Google Scholar]
- 4.Haugen SP, Berkman MB, Ross W, Gaal T, Ward C, Gourse RL. rRNA promoter regulation by nonoptimal binding of σ region 1.2: an additional recognition element for RNA polymerase. Cell. 2006;125:1069–1082. doi: 10.1016/j.cell.2006.04.034. [DOI] [PubMed] [Google Scholar]
- 5.Murray HD, Schneider DA, Gourse RL. Control of rRNA expression by small molecules is dynamic and nonredundant. Mol Cell. 2003;12:125–134. doi: 10.1016/s1097-2765(03)00266-1. [DOI] [PubMed] [Google Scholar]
- 6.Walker KA, Mallik P, Pratt TS, Osuna R. The Escherichia coli fis promoter is regulated by changes in the levels of its transcription initiation nucleotide CTP. J Biol Chem. 2004;279:50818–50828. doi: 10.1074/jbc.M406285200. [DOI] [PubMed] [Google Scholar]
- 7.Mallik P, Paul BJ, Rutherford ST, Gourse RL, Osuna R. DksA is required for growth phase-dependent regulation, growth rate-dependent control, and stringent control of fis expression in Escherichia coli. J Bacteriol. 2006;188:5775–5782. doi: 10.1128/JB.00276-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaal T, Bartlett MS, Ross W, Turnbough CL, Jr, Gourse RL. Transcription regulation by initiating NTP concentration: rRNA synthesis in bacteria. Science. 1997;278:2092–2097. doi: 10.1126/science.278.5346.2092. [DOI] [PubMed] [Google Scholar]
- 9.Barker MM, Gourse RL. Regulation of rRNA transcription correlates with nucleoside triphosphate sensing. J Bacteriol. 2001;183:6315–6323. doi: 10.1128/JB.183.21.6315-6323.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider DA, Gaal T, Gourse RL. NTP-sensing by rRNA promoters in Escherichia coli is direct. Proc Natl Acad Sci USA. 2002;99:8602–8607. doi: 10.1073/pnas.132285199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barker MM, Gaal T, Josaitis CA, Gourse RL. Mechanism of regulation of transcription initiation by ppGpp. I Effects of ppGpp on transcription initiation in vivo and in vitro. J Mol Biol. 2001;305:673–688. doi: 10.1006/jmbi.2000.4327. [DOI] [PubMed] [Google Scholar]
- 12.Paul BJ, Berkmen MB, Gourse RL. DksA potentiates direct activation of amino acid promoters by ppGpp. Proc Natl Acad Sci USA. 2005;102:7823–7828. doi: 10.1073/pnas.0501170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choy HE. The study of guanosine 5′-diphsosphate 3′-diphosphate-mediated transcription regulation in vitro using a coupled transcription-translation system. J Biol Chem. 2000;275:6783–6789. doi: 10.1074/jbc.275.10.6783. [DOI] [PubMed] [Google Scholar]
- 14.Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell. 2004;118:311–322. doi: 10.1016/j.cell.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Nakanishi N, Abe H, Ogura Y, Hayashi T, Tashiro K, Kuhara S, Sugimoto N, Tobe T. ppGpp with DksA controls gene expression in the locus of enterocyte effacement (LEE) pathogenicity island of enterohaemorrhagic Escherichia coli through activation of two virulence regulatory genes. Mol Microbiol. 2006;61:194–205. doi: 10.1111/j.1365-2958.2006.05217.x. [DOI] [PubMed] [Google Scholar]
- 16.Sharma AK, Payne SM. Induction of expression of hfq by DksA is essential for Shigella flexneri virulence. Mol Microbiol. 2006;62:469–479. doi: 10.1111/j.1365-2958.2006.05376.x. [DOI] [PubMed] [Google Scholar]
- 17.Vrentas CE, Gaal T, Ross W, Ebright RH, Gourse RL. Response of RNA polymerase to ppGpp: requirement for the omega subunit and relief of this requirement by DksA. Genes Dev. 2005;19:2378–2387. doi: 10.1101/gad.1340305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gentry D, Xiao H, Burgess R, Cashel M. The omega subunit of Escherichia coli K-12 RNA polymerase is not required for stringent RNA control in vivo. J Bacteriol. 1991;173:3901–3903. doi: 10.1128/jb.173.12.3901-3903.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG. Regulation through the secondary channel - structural framework for ppGpp-DksA synergism during transcription. Cell. 2004;118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 20.Borukhov S, Lee J, Laptenko O. Bacterial transcription elongation factors: new insights into molecular mechanism of action. Mol Microbiol. 2005;55:1315–1324. doi: 10.1111/j.1365-2958.2004.04481.x. [DOI] [PubMed] [Google Scholar]
- 21.Laptenko O, Lee J, Lomakin I, Borukhov S. Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. EMBO J. 2003;22:6322–6334. doi: 10.1093/emboj/cdg610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Opalka N, Chlenov M, Chacon P, Rice WG, Wriggers W, Darst SA. Structure and function of the transcription elongation factor GreB bound to bacterial RNA polymerase. Cell. 2003;114:335–345. doi: 10.1016/s0092-8674(03)00600-7. [DOI] [PubMed] [Google Scholar]
- 23.Sen R, Nagai H, Shimamoto N. Conformational switching of Escherichia coli RNA polymerase-promoter binary complex is facilitated by elongation factor GreA and GreB. Genes Cells. 2001;6:389–401. doi: 10.1046/j.1365-2443.2001.00436.x. [DOI] [PubMed] [Google Scholar]
- 24.Koulich D, Orlova M, Malhotra A, Sali A, Darst SA, Borukhov S. Domain organization of Escherichia coli transcript cleavage factors GreA and GreB. J Biol Chem. 1997;272:7201–7210. doi: 10.1074/jbc.272.11.7201. [DOI] [PubMed] [Google Scholar]
- 25.Hsu LM, Vo NV, Chamberlin MJ. Escherichia coli transcript cleavage factors GreA and GreB stimulate promoter escape and gene expression in vivo and in vitro. Proc Natl Acad Sci USA. 1995;92:11588–11592. doi: 10.1073/pnas.92.25.11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng G, Lee DN, Wang D, Chan CL, Landick R. GreA-induced transcript cleavage in transcription complexes containing Escherichia coli RNA polymerase is controlled by multiple factors, including nascent transcript location and structure. J Biol Chem. 1994;269:22282–22294. [PubMed] [Google Scholar]
- 27.Grigorova IL, Phleger NJ, Mutalik VK, Gross CA. Insights into transcriptional regulation and σ competition for an equilibrium model of RNA polymerase binding to DNA. Proc Natl Acad Sci USA. 2006;103:5332–5337. doi: 10.1073/pnas.0600828103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartlett MS, Gaal T, Ross W, Gourse RL. RNA polymerase mutants that destabilize RNAP polymerase-promoter complexes alter NTP-sensing by rrnB P1 promoters. J Mol Biol. 1998;279:331–345. doi: 10.1006/jmbi.1998.1779. [DOI] [PubMed] [Google Scholar]
- 29.Potrykus K, Vinella D, Murphy H, Szalewska-Palasz A, D’Ari R, Cashel M. Antagonistic regulation of Escherichia coli ribosomal RNA rrnB P1 promoter activity by GreA and DksA. J Biol Chem. 2006;281:15238–15248. doi: 10.1074/jbc.M601531200. [DOI] [PubMed] [Google Scholar]
- 30.Stebbins CE, Borukhov S, Orlova M, Polyakov A, Goldfarb A, Darst SA. Crystal structure of the GreA transcript cleavage factor from Escherichia coli. Nature. 1995;373:636–640. doi: 10.1038/373636a0. [DOI] [PubMed] [Google Scholar]
- 31.Koulich D, Nikiforov V, Borukhov S. Distinct functions of N and C-terminal domains of GreA, and Escherichia coli transcript cleavage factor. J Mol Biol. 1998;20:379–389. doi: 10.1006/jmbi.1997.1545. [DOI] [PubMed] [Google Scholar]
- 32.Kulish D, Lee J, Lomakin I, Nowicka B, Das A, Darst S, Normet K, Borukhov S. The functional role of basic patch, a structural element of Escherichia coli transcript cleavage factors GreA and GreB. J Biol Chem. 2000;275:12789–12798. doi: 10.1074/jbc.275.17.12789. [DOI] [PubMed] [Google Scholar]
- 33.Artsimovitch I, Patlan V, Sekine S, Vassylyeva MN, Hosaka T, Ochi K, Yokoyama S, Vassylyev DG. Structrual basis for transcription regulation by alarmone ppGpp. Cell. 2004;117:299–310. doi: 10.1016/s0092-8674(04)00401-5. [DOI] [PubMed] [Google Scholar]
- 34.Rao L, Ross W, Appleman JA, Gaal T, Leirmo S, Schlax PJ, Record MT, Jr, Gourse RL. Factor independent activation of rrnB P1. An “extended” promoter with an upstream element that dramatically increases promoter strength. J Mol Biol. 1994;235:1421–1435. doi: 10.1006/jmbi.1994.1098. [DOI] [PubMed] [Google Scholar]
- 35.Ross W, Thompson JF, Newlands JT, Gourse RL. E. coli Fis protein activates ribosomal RNA transcription in vitro and in vivo. EMBO J. 1990;9:3733–3742. doi: 10.1002/j.1460-2075.1990.tb07586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burgess RR, Jendrisak JJ. A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving polymin P precipitation and DNA-cellulose chromatography. Biochemistry. 1975;14:4634–4638. doi: 10.1021/bi00692a011. [DOI] [PubMed] [Google Scholar]
- 37.Thompson NE, Hager DA, Burgess RR. Isolation and characterization of a polyol-responsive monoclonal antibody useful for gentle purification of Escherichia coli RNA polymerase. Biochemistry. 1992;31:7003–7008. doi: 10.1021/bi00145a019. [DOI] [PubMed] [Google Scholar]
- 38.Gaal T, Ross W, Estrem ST, Nguyen LH, Burgess RR, Gourse RL. Promoter recognition and discrimination by EσS RNA polymerase. Mol Microbiol. 2001;42:939–954. doi: 10.1046/j.1365-2958.2001.02703.x. [DOI] [PubMed] [Google Scholar]
- 39.Hirvonen CA, Ross W, Wozniak CE, Marasco E, Anthony JR, Aiyar SE, Newburn VH, Gourse RL. Contributions of UP elements and the transcription factor FIS to expression from the seven rrn P1 promoters in Escherichia coli. J Bacteriol. 2001;183:6305–6314. doi: 10.1128/JB.183.21.6305-6314.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross W, Gourse RL. Sequence-independent upstream DNA-αCTD interactions strongly stimulate Escherichia coli RNA polymerase-lacUV5 promoter association. Proc Natl Acad Sci USA. 2005;102:291–296. doi: 10.1073/pnas.0405814102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barker MM, Gaal T, Gourse RL. Mechanisms of regulation of transcription initiation by ppGpp. II Models for positive control based on properties of RNAP mutants and competition for RNAP. J Mol Biol. 2001;305:689–702. doi: 10.1006/jmbi.2000.4328. [DOI] [PubMed] [Google Scholar]
- 42.Masui Y, Mizuno T, Inouye M. Novel high-level expression cloning vehicles: 104-fold amplification of Escherichia coli minor protein. Biotechnology. 1984;2:81–85. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.