

Abstract
Fear memories elicit multiple behavioral responses, encompassing avoidance or behavioral inhibition in response to threatening contexts. Context-specific freezing, reflecting fear-induced behavioral inhibition, has been proposed as one of the main risks factors for the development of anxiety disorders. We attempted to define the key hippocampal mediators of extinction in a mouse model of context-dependent freezing. Nine-week-old male C57BL/6J mice were trained and tested for contextual fear conditioning and extinction. Freezing behavior scored by unbiased sampling, was used as an index of fear. Proteomic, immunoblot and immunohistochemical approaches were employed to identify, verify and analyze the alterations of the hippocampal extracellular signal-regulated kinases 1 and 2 (Erk-1/2). Targeted pharmacological inhibition of the Erk-1/2 activating kinase, the mitogen activated and extracellular signal-regulated kinase (Mek), served to establish the role of Mek/Erk signaling in extinction. When compared to acquisition, extinction of contextual freezing triggered a rapid activation of Erk-1/2 showing a distinctive time-course, nuclear localization and subcellular isoform distribution. These differences suggested that the upstream regulation and downstream effects of this pathway might be specific for each process. Dorsohippocampal injections of the Mek inhibitors U0126 (0.5 μg/site) and PD98059 (1.5 μg/site) immediately after the nonreinforced trials prevented Erk-1/2 activation and significantly impaired extinction. This effect was dissociable from potential actions on memory retrieval or reconsolidation. On the basis of these findings, we propose that hippocampal Mek/Erk signaling might serve as one of the key mediators of contextual fear extinction.
Keywords: extinction, Erk, proteome, context, freezing, hippocampus
1. Introduction
Anxiety disorders are thought to result from abnormal emotional responses associated with memories of aversive events. Most forms of anxiety are accompanied by exaggerated and persistent fear in response to threatening environments that can be modeled by contextual fear conditioning (Grillon, 2002). Rodent studies have significantly advanced our understanding of the neurobiology of contextual fear by establishing the essential biochemical pathways within the hippocampal brain area mediating fear conditioning (Kandel, 2001; Sweatt, 2001). However, persistent fear may be also caused by impaired extinction, reflecting a learned reduction of fear responses after subsequent nonreinforced contextual exposures (reviewed by Myers & Davis, 2002).
The molecular basis of extinction and its relevance for anxiety disorders has gained significant attention by discovering the roles of the basolateral amygdala (Lu et al., 2001; Walker et al., 2002) and prefrontal cortex (Morgan et al., 1993; Morgan & LeDoux, 1995; Milad & Quirk, 2002; Sotres-Bayon et al., 2006) in extinction of cue-dependent fear. On the basis of the prominent role of the amygdala in different aspects of the fear response such as acquisition, expression, extinction (Davis et al., 1994; Ledoux, 2000), it could be expected that this brain area would also significantly affect extinction of contextual fear. Such an indiscriminative involvement of the amygdala in fear regulation cannot explain the greater resistance to extinction of contextual rather than cue-dependent fear both in rodent experimental models (Stiedl et al., 1999) and human anxiety disorders (Grillon, 2002). On the other hand, the prefrontal cortex does not seem to be involved in the extinction of contextual fear responses (Morgan & LeDoux, 1999) despite its significant role in extinction of tone-dependent fear (Santini et al., 2004). Thus, the neuroanatomical and molecular substrates of contextual fear extinction remain scarce.
It is increasingly recognized that the hippocampus may contribute to the extinction of fear-motivated behaviors, such as inhibitory avoidance, triggered by environmental contexts (Szapiro et al., 2003). Nevertheless, its role in extinction of contextual freezing, reflecting a generalized behavioral inhibition in response to a context associated with an aversive stimulus, has not been extensively investigated. Notably, among multiple responses elicited by fear states, context-specific freezing has been proposed as one of the main risks factors for the development of anxiety disorders (Buss et al., 2004). Impaired extinction of freezing to contextual stimuli has been observed in mouse strains with reduced hippocampal commissure (Schimanski et al., 2002). Supporting this view, we have recently shown that hippocampal mechanisms encompassing actin rearrangement are required for contextual fear extinction (Fischer et al., 2004) in this paradigm. Because cytoskeletal rearrangment regulates the activation and distribution of a number of intracellular signaling molecules, the underlying mediators remained to be characterized in more detail. In addition to the recently demonstrated role of phosphatidylinositol 3 kinase (PI3; Chen et al., 2005), we identified in the present study the Mek/Erk-1/2 signaling pathway as one of the key mediators of extinction of contextual freezing.
2. Materials and methods
2.1. Animals
Nine-week-old male C57BL/6J mice (Centre d’Elevage Janvier and Jackson Laboratories) were individually housed after 8 weeks of age and maintained in enclosed animal cubicles with their own ventilation system (15 air exchanges/hr), at a 12/12 dark light cycle (7 am-7 pm), 40–50% humidity, and 20 ± 2°C. All studies were approved by the Animal Care and Use Committee of Northwestern University in compliance with National Institutes of Health standards.
2.2. Drugs and antibodies
Immunoblot and immunohistochemical analyses were performed using antibodies specific for Erk-1/2, histone H1, (1:1000, Santa Cruz), Erk 1/2 phosphorylated at Thr-183/Tyr-185 (1:5000, mouse monoclonal IgG1, Sigma), anti pThr-Erk-1/2 (1:8000, mouse monoclonal IgG1, Sigma) and anti-pTyr-Erk-1/2 (1:8000, mouse monoclonal IgG1, Santa Cruz), CREB phosphorylated at Ser-133 (pCREB; rabbit polyclonal IgG, 1:2000, Calbiochem), lactate dehydrohenase (LDH; 1:1000, rabbit polyclonal IgG, Rockland Immunochemicals) and cFos (1:12000, rabbit polyclonal IgG, Oncogene). All antibodies gave clear signals at the predicted molecular sizes of the investigated proteins in total hippocampal lysates.
The Mek inhibitors U0126 (Promega) and PD98059 (2′-amino-3′metoxyflavone, Calbiochem) were employed for treatment.
2.3. Contextual fear conditioning and extinction
Fear conditioning was performed in an automated system (TSE Inc.) and consisted of a single exposure to context (3 min) followed by footshock (2s, 0.7 mA, constant current) as described previously (Radulovic et al., 1998). Context-dependent freezing was measured 24 hr later every 10th s over 180 s by two observers unaware of the experimental conditions and expressed as % of total number of observations. The extinction trials were performed in 24 hr intervals and consisted of nonreinforced 3-min exposures to the context (Fischer et al., 2004).
2.4. Proteomics
Cytoplasmic and nuclear hippocampal lysates were prepared as described below, from naïve mice (N), mice exposed to fear conditioning (FC) or mice exposed to 4 consecutive extinction trials (E4). Hippocampi of the FC and E4 groups were obtained 1 hr after training and last extinction trial, respectively, and pooled from 6 mice/group. Two–dimensional electrophoresis was performed as described in detail previously (Stannard et al., 2003). Briefly, proteins from hippocampal cytoplasmic and nuclear fractions were precipitated with 10% trichloroacetic acid, pellet washed twice in ethanol:ether (1:1; vol/vol) and solubilized in immunoelectrophoresis buffer buffer. Proteins were separated according to their isoelectric point using 18 cm immobilized pH-gradient strips (pH 5.5–6.7, Amersham Biosciences). The proteins were then additionally separated by SDS-PAGE and visualised by silver staining. The resultant protein patterns were analyzed by Melanie software (GenBio SA).
Protein spots were excised from the gel, and processed automatically by Progest (Genomic Solutions). Tryptic digests (0.7 μl) were applied to the target plate and allowed to air-dry, then 0.7 μl of saturated solution of α-cyano-4-hydroxycinnamic acid in 33% acetonitrile and 0.1% TFA (v/v) was overlaid. Mass spectra of the tryptic digests were obtained on an Autoflex matrix-assisted laser desorption ionization-time of flight mass spectrometer (MALDI-TOF MS; Bruker Daltonics, Billerica, MA), calibrated with a mixture of seven standard peptides. Monoisotypic peptide masses were assigned and the mass lists were used for protein identification in the NCBI nonredundant (nr) protein database by Mascot search algorithm (Matrix Science Inc. London, UK).
2.5. Protein extraction and immunoblot
After training or indicated extinction trials (E1-4), the dorsal and ventral hippocampi were dissected according to neuroanatomical coordinates (Franklin and Paxinos, 1997). The rostral 2.5 mm septal pole and caudal 2.5 mm of the temporal pole were employed to prepare dorso- and ventohippocampal lysates, respectively. Hippocampal extracts for dorsal and ventral hippocampi were prepared separately because we expected maximal drug effects in the injected (dorsohippocampal) area. Nuclear extracts were prepared with the modified high salt extraction method (Dignam et al., 1983) as described in detail by Thiels et al. (2002). To control for the purity of the nuclear and cytoplasmic samples, control experiments determined the levels of nuclear (CREB, histone H1) and cytoplasmic (LDH) markers in the extracts. Extracts of the somatosensory cortex overlaying the hippocampus additionally served as a control for the anatomical specificity of the effects of the inhibitor. After determining the protein concentration (Bio-Rad), the lysates (20 μg/well) were subjected to 10% SDS polyacrylamide gel electrophoresis were subsequently blotted to PVDF membranes (Millipore) as described previously (Sananbenesi et al., 2002). The membranes were saturated with I-block (Tropix) and then incubated with the primary and corresponding secondary antibodies, enhancer (Nitro-block II, Tropix) and chemiluminescent substrate (CDP Star, Tropix). Blots were exposed to X-ray films and developed in the range of maximal chemiluminescence emission (10 min). Molecular weight and densitometric calculations of pErk-1/2 were performed with the computer software ImageJ (NIH).
2.6. Immunohistochemistry
After training or indicated extinction trials, mice were anesthetized with an intraperitoneal injection of 240 mg/kg of Avertin, and transcardially perfused with ice-cold 4% paraformaldehyde in phosphate buffer (pH 7.4, 150 ml/mouse). Brains were post-fixed for 48 hours in the same fixative and then immersed for 24 hrs each in 10%, 20% and 30% sucrose in phosphate buffer. After the tissue was frozen by liquid nitrogen, 50 μm thick coronal sections were used for performing free-floating immunocytochemistry with corresponding primary antibodies as described previously (Radulovic et al., 1998). Biotinylated secondary antibody and ABC complex (Vector) was used for signal amplification and DAB (Sigma) as chromogen. The specificity of immunostaining was confirmed by preadsorption of primary antibodies with a tenfold excess of corresponding antigenic peptides. For co-immunolabeling studies the TSA Fluorescence System (NEN Life Science Products) was employed, using fluorescein as substrate and DAPI as a nuclear counterstan. Multicolor immunofluorescence was captured and analyzed with a fluorescent microscope (Leica) provided with a CCD camera connected to the Spot software (Macintosh).
Quantification of immunostaining signals was performed separately for the CA1 hippocampal layers (stratum pyramidale and stratum radiatum) as described previously (Lalonde et al., 2004). Digital images were captured with a cooled color CCD camera (RTKE Diagnostic Instruments) and SPOT software for Macintosh. Adobe Photoshop 5.0 for Macintosh was used for image processing. Cell counts as well as signal intensity measures were performed with ImageJ. Nuclear staining: Cell counts from the pErk-1/2 fluorescent images were made using two sections (1.5 and 1.6 mm posterior to the bregma spaced by 50 μm corresponding to the dorsal hippocampus) per mouse. The separate fluorescein isothiocyanate (FITC) and DAPI captures were digitally combined to produce composite images. Equal cutoff thresholds were applied to all captures to remove background fluorescence digitally. For each capture of the pErk-1/2 signal, nuclear pErk-1/2 cell counts were performed within a 100 μm2 grid three times for the CA1 and two times for the CA3 area. An overlapping signal of FITC and DAPI fluorescence was used as a criterion for nuclear localization of pErk-1/2. The measures for each capture were averaged to give the number of pErk-1/2-positive nuclei per 100 μm2 area. Finally, the percentage of pErk-1/2-immunopositive nuclei per total number of counted nuclei in a single focal plane was established by dividing the number of pErk-1/2-positive nuclei per the total number of nuclei displaying the DAPI signal. The staining for other nuclear proteins was performed identically. Immunoreactive dendrites: Images were digitally contrast stretched for maximum resolution of phospho-Erk-1/2-positive dendrites. The number of dendrites and their density within stratum radiatum were determined for each image (Patterson et al., 2001). If background was unacceptably high or the tissue obviously damaged, images were excluded from analyses.
2.7. Surgery and microinjections
Double cannula was placed into the dorsal hippocampus (AP - 1.5 mm, lateral 1 mm, depth 2 mm) as described (Radulovic et al., 1999). The gauge of the guide and injection cannulae was 26 and 28, respectively. U0126 (0.5 μg/site) and PD9859 (1.5 μg/site) were dissolved in 100% dimethyl sulfoxide (DMSO) and further diluted to 2% and 50% DMSO, respectively, in artificial cerebrospinal fluid (aCSF). The inhibitors were delivered bilaterally (0.25 μl/side) over a 15 s period immediately after individual extinction trials testing unless indicated otherwise. Control mice were implanted with a double cannnula into the somatosensory cortex adjacent to the hippocampus ((AP - 1.5 mm, lateral 1 mm, depth 1 mm). The cannula position was determined for each mouse by histological examination of hippocampi following methylene-blue injection after the end of experiments (Fischer et al., 2002) and only data obtained from mice with correctly inserted cannula were analyzed.
2.8. Statistical analysis
Statistically significant differences were determined by two-way ANOVA (Test and Treatment) followed by Scheffés test for post-hoc comparisons, one-way ANOVA or Student t-test. The results are presented as mean ± S.E.
3. Results
3.1. Up-regulation of pErk-1/2 in the hippocampus after contextual fear conditioning and extinction
To identify proteins that are differentially regulated during extinction of conditioned contextual freezing behavior, mice (n=6/group) were subjected to contextual fear conditioning. Cytoplasmatic and nuclear lysates were either prepared 1h after the training (FC group) or 1h after the fourth extinction trials (E4 group) when freezing was significantly reduced (Fig. 1A; F(5,30) = 6.78, p < 0.01). Analysis of proteins separated by two-dimensional gel electrophoresis revealed alterations of two major components of the Erk signaling pathway: an increased phosporylation of Erk-2 in the cytoplasmic extracts of the FC group and increased level and phosphorylation of Erk-1 in the E4 group, as indicated by electrophoretic shifts of the identified proteins (Figure 1B). These observations were confirmed and extended by immunoblot experiments performed after training and individual extinction trials. In these experiments all extracts were collected 1 hr after behavioral manipulations because this time point previously showed maximal Erk-1/2 activation after conditioning in the hippocampus (Sananbenesi et al., 2003) and amygdala (Schafe et al., 2000). The level of cytoplasmic pErk-2 increased significantly after training, continued to increase after initial extinction trials (F(5,25) = 10.92, p < 0.001) and started to decline after the first trial accompanied by reduction of contextual freezing (E4). The up-regulation of cytoplasmic pErk-1 followed a similar time course (Figure 1C, D), however the overall increase of pErk-1 was less pronounced (F(5,25) = 5.89, p < 0.01). In contrast, nuclear pErk-1 increased significantly with advancing extinction trials (F(5,25) = 8.33, p < 0.001), whereas the alterations of nuclear pErk-2 were less consistent (F(5,25) = 2.89, p = 0.067) (Figure 1C, D). The relative purity of the cytoplasmic and nuclear extracts was demonstrated by enrichment of the cytoplasmic fraction with the predominant cytoplasmic enzyme lactate dehydrogenase and enrichment of the nuclear fraction with the nuclear proteins cyclic-adenosine monophosphate response element binding protein (CREB) and histone H1 (Figure 1E).
Fig. 1.
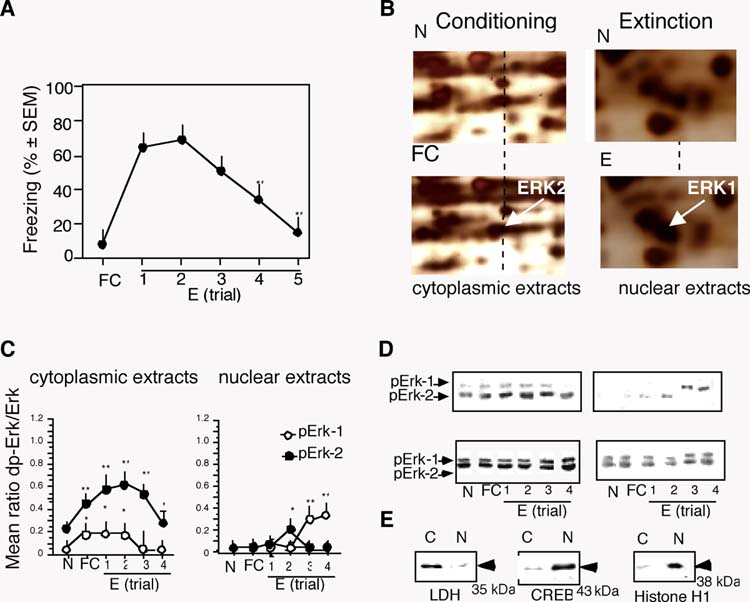
Up-regulation of hippocampal Erk-1 and Erk-2 during fear conditioning and extinction. (A) Time course of fear extinction revealed by decrease of freezing after nonreinforced contextual exposures. (B) Representative proteomic scans of cytoplasmic extracts obtained 1 hr after training (left), demonstrating increased phosphorylation of pErk-2; and nuclear extracts obtained 1 hr after the 4th extinction trial (right) demonstrating nuclear accumulation of Erk-1. (C) The levels of cytoplasmic pErk-1 and pErk-increased significantly after conditioning and extinction (left panel). Nuclear pErk-1 increased significantly after the 3th and 4th extinction trial (right panel). (D) Representative immunoblots. (E) The quality of nuclear extracts was examined by immunoblot experiments with antibodies detecting the nuclear proteins CREB and histone H1 and cytoplasmic protein LDH. Enrichment of the nucleoproteins was observed only in the nuclear extracts. The number of mice per group was 5. Statistically significant differences obtained by one-way ANOVA followed by the Scheffés test for multiple comparisons: *p < 0.01 vs N group; **p < 0.001 vs N group.
3.2. Localization of pErk-1/2 after contextual fear conditioning and extinction
Immunohistochemical examination of the distribution of pErk-1/2 within the hippocampus (Figure 2A–C), demonstrated a significant up-regulation of pErk-1/2 in the apical dendrites of str. radiatum in the CA1 subfield in mice of the FC and E4 groups when compared to naïve mice (F(2,27) = 7.92, p < 0.001) whereas the activation pattern of pErk-1/2 between the FC and E1-4 groups was indistinguishable with these approaches. However, the somatic and nuclear localization of pErk-1/2 showed significant differences between conditioning and extinction (F(2,27) = 9.14, p < 0.001). Mice of the E4 group (subjected to four extinction trials) showed significant somatic and nuclear accumulation of pErk-1/2 (Figure 2A,C), in contrast to the weak immunostaining in these neuronal compartments in the FC and N mice. Erk-1/2 activation subsided to baseline levels when stable reduction of freezing was detectable over 3–4 consecutive trials, as revealed by analyses performed after the 8th extinction trial (E8) (Figure 2B). A time course analysis of the nuclear pErk-1/2 signals after E4 demonstrated a rapid increase at 15 min after the extinction trial, a peak at 60 min and subsequent decrease at 3 hr post-trial (F(5,30) = 12.91, p < 0.001)(Figure 2D).
Fig. 2.
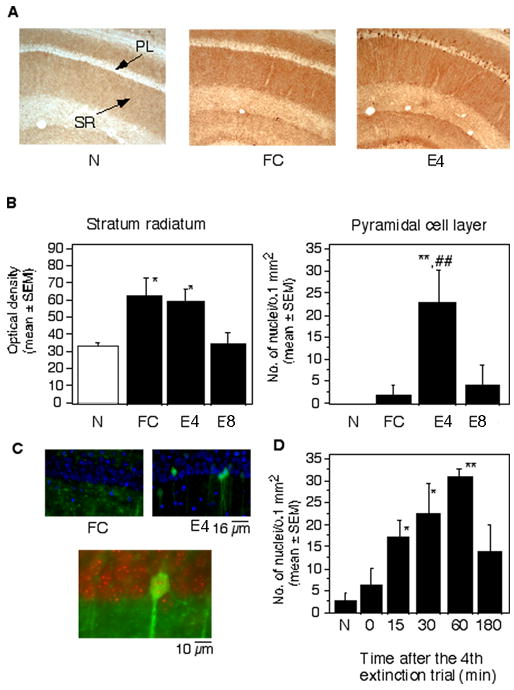
Localization of pErk-1/2 after conditioning and extinction. (A) Representative photomicrographs demonstrating the level of pErk-1/2 in the CA1 hippocampal area of mice of the N, FC and E groups. (B) Mean density of pErk-1/2 immunoreactivity in the stratum radiatum of the experimental groups (left). Mean number of cells per CA1 hippocampal area of the experimental groups (right). Statistically significant differences obtained by one-way ANOVA followed by the Scheffés test for multiple comparisons: *p < 0.01 vs N; **p < 0.001 vs N and T. PL, pyramidal cell layer; SR, Stratum radiatum. (C) Nuclear pErk-1/2 was not observed in mice exposed to training (upper left), whereas extinction triggered a significant increase of nuclear pErk-1/2 in hippocampal pyramidal neurons (upper right), as revealed by merged images with DAPI (blue) and pErk-1/2 (green). Below: DAPI digitized red and pErk-1/2 green show yellow overlapping signals. The mean percentage of Erk-1/2 positive nuclei in the extinction and training groups was 15.4 ± 7.8 and 0.5 ± 1.2, respectively. The number represents a mean ratio of Erk-/2 positive of a total of 200 nuclei counted in a single focal plane in 5 randomly chosen sections of the dorsal hippocampus/mouse. (D) The time course of pErk-1/2 up-regulation after the 4th extinction trial revealed a significant activation already after 15 min whereas 3 hr later the activated kinase was not detectable. Statistically significant differences were revealed by one-way ANOVA followed by Scheffés test : *p < 0.01, **p < 0.001 vs group sacrificed immediately after the extinction trial (0 min). The number of mice per group was 6 for each experiment.
Importantly, the increase of pErk-1/2 was not due to a passage of time, but specifically increased in response to the extinction procedure (Figure 3). There is evidence that upon its translocation to the nucleus, pErk-1/2 is rapidly dephosphorylated and accumulated in a less active monophosphorylated form (Pouyssegur et al., 2002). We subsequently addressed this issue by using antibodies against Erk-1/2 that are monophosphorylated either at Tyr or Thr residues. Brain sections of mice obtained after the 4th extinction trial were stained with anti-pTyr, anti-pThr or anti-pErk-1/2 antibodies and compared. Although pThr-Erk-1/2 and pTyr-Erk-1/2 exhibited distinct immunostaining patterns in hippocampal subfields, only double phosphorylated pErk-1/2 was detected in the pyramidal cell soma within CA1 (data not shown). Because we did not detect any cell bodies positive for monophosphorylated Erk-1/2, these data were not analyzed statistically.
Fig. 3.
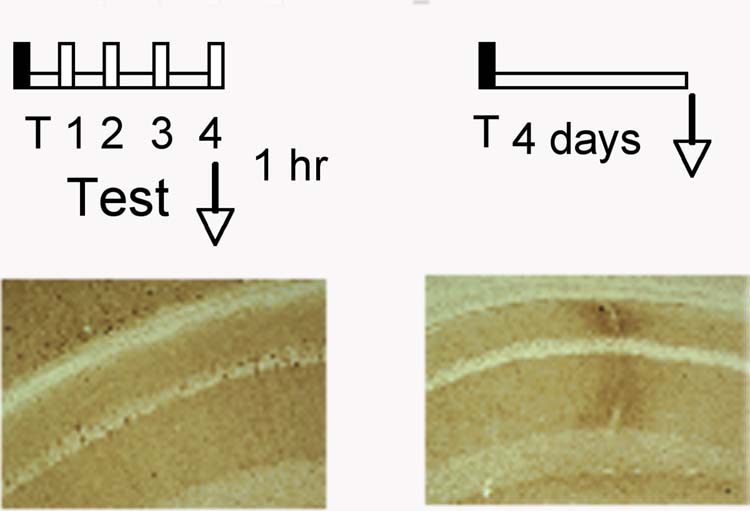
Increased pErk-1/2 after extinction is unrelated to the conditioning experience. Above: experimental design. Below: pErk-1/2 was observed in hippocampi of mice 1 hr after the exposure to the 4th extinction trial but not in trained mice without nonreinforced contextual exposures.
Taken together, although both conditioning and extinction triggered Erk-1/2 activation in the hippocampal CA1 subfield, the localization patterns of the activated Erk isoforms significantly differed between these conditions.
3.3. Effect of the Erk-activating kinase Mek on extinction of contextual freezing
The significance of hippocampal Erk signaling in extinction of contextual fear was investigated by dorsohippocampal inhibition of Erk-1/2 phosphorylation with two inhibitors of the Erk-activating kinase Mek. Both inhibitors profoundly impaired extinction, as revealed by a significant Treatment x Test interaction in mice injected with 1 μg of U0126 (F(5,109) = 18.72; p < 0.001; Figure 4A) or 3 μg of PD9859 (F(5,98) = 14.85; p < 0.001; Figure 4B). Post-hoc Control intracortical (i.c.) injections of these inhibitors (Figure 4C,D) did not affect extinction (U0126: F(1,88) = 0.422, p = 0.614; PD9859: F(1,92) = 0.486, p = 0.72).
Fig. 4.
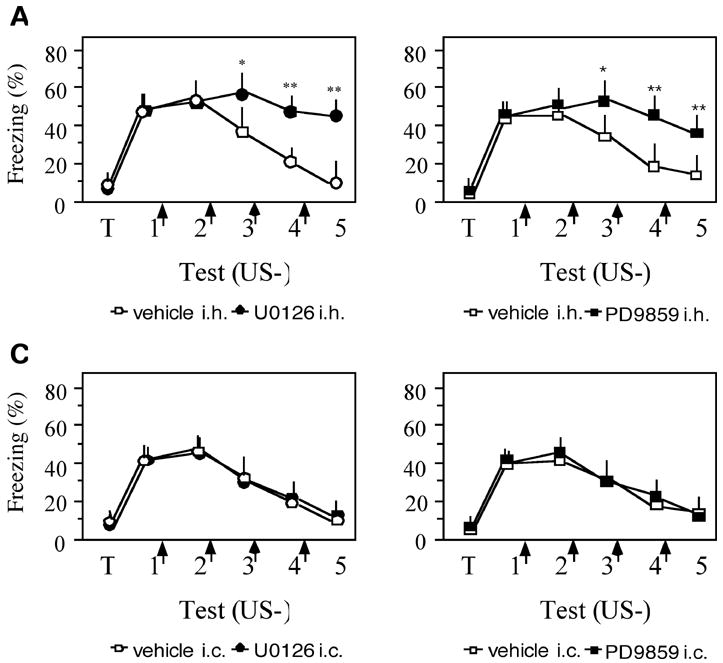
Mek inhibitors impair contextual fear extinction. (A) U0126 injected i.h. immediately after individual extinction trials prevented extinction as indicated by significantly higher freezing of U0126-injected mice when compared to their vehicle controls (p < 0.001). (B) PD9859 injected in the same fashion also impaired extinction (p < 0.001). (C) Injection of U0126 3 into the adjacent somatosensory cortex did not affect extinction when compared to the vehicle-injected control. (D) Similarly, PD9859 was ineffective after i.c. injection. No. of mice/group was 10–15. Statistically significant differences were revealed by one-way ANOVA followed by Scheffés test : *p < 0.01, **p < 0.001 vs vehicle control.
Further experiments, carried out with intrahippocampal (i.h.) injections of U0126 demonstrated that that mice rapidly exhibit extinction after cessation of treatment (day 7: t(1,17) = 0.24; p = 0.535; Fig. 5A). Thus, the elevated freezing behavior was unlikely due to toxic actions of the drug. The specificity of the extinction effect was also supported by the observation that i.h. injections 3 hr post-trial, when pErk-1/2 levels are not significantly elevated (Figure 2D), did not affect freezing behavior (F(5,63) = 0.331, p = 0.714; Figure 5B). In line with the observation that Erk-1/2 is activated after several consecutive nonreinforced trials, a single injection of U0126 after the first trial was not effective in preventing extinction during subsequent trials (F(5,63) = 0.478, p = 0.702; Figure 5C). The efficiency of U0126 to inhibit Erk-1/2 phosphorylation was verified by immunoblot experiments with dorso-, ventrohippocampal and cortical lysates. Mice injected with 1 μg (0.5 μg/site) of U0126 after 4 consecutive extinction tests, and sacrificed 1 hr after the last trial, exhibited a significant decrease of pErk-1/2 in the dorsal hippocampus when compared to vehicle-injected mice (mean O.D. ± SE: U0126, 0.2 ± 0.25; vehicle: 0.9 ± 0.31; t(1, 8) = 5.31, p < 0.01) as well as the ventral hippocampus (t(1, 8) = 7.28, p < 0.01) and somatosensory cortex (t(1, 8) = 8.25, p < 0.01) (Figure 5D).
Fig. 5.
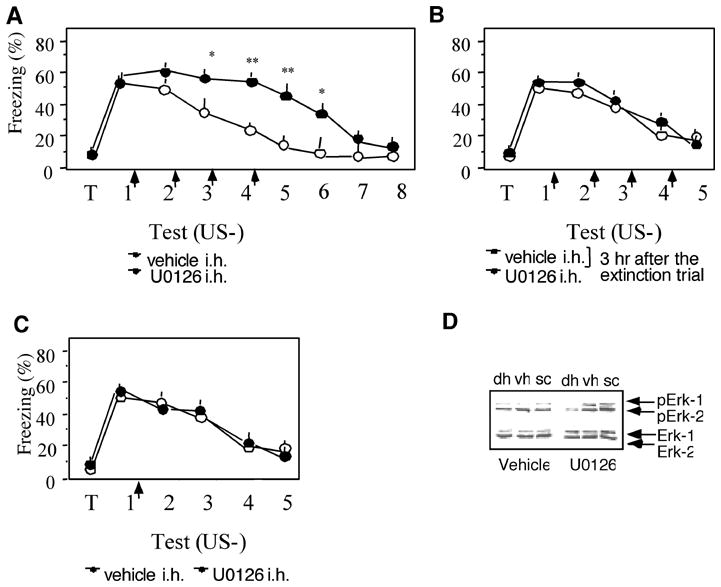
The effects of U0126 are reversible, specific and localized. (A) After cessation of i.h. treatment with U0126, mice rapidly extinguish the fear response, as revealed by similarly low freezing levels of U0126- and vehicle-injected mice. (B) Injection of U0126 3 hr after the extinction trial did not affect extinction when compared to the vehicle-injected control. (C) A single injection of U0126 after the first nonreinforced trial did not affect extinction. No. of mice/group was 8–11.(D) U0126 selectively reduced the levels of pErk-1/2 in total lysates of the dorsal hippocampus (DH), but not ventral hippocampus (VH) or somatosensory cortex (SC). The number of mice per group was 5.
These results demonstrated that inhibition of Mek in the dorsal hippocampus disrupts the activation of Erk-1/2 and prevents the extinction of contextual fear.
4. Discussion
We have demonstrated that Erk-1/2 phosphorylation in the dorsal hippocampus parallels and contributes to the extinction of conditioned contextual freezing. Up-regulation of pErk-1/2 was observed both after contextual fear conditioning and its extinction however, the activation and localization patterns of these proteins within hippocampal neurons significantly differed between these experimental conditions.
The specificity of hippocampal Erk-1/2 up-regulation for fear conditioning when compared to unpaired immediate shock presentations has been demonstrated by Atkins et al. (1998) and our previous data (Sananbenesi et al., 2002) however reports on Erk-1/2 alterations during extinction have been less consistent. An overall increase of pErk-1/2 has been previously observed in total hippocampal extracts under experimental conditions associated with enhanced (Cannich et al. 2003; Chen et al., 2005) or impaired (Wang et al., 2004) contextual fear extinction. Therefore, the localization pattern and physiological role of this signaling pathway remained elusive. Separate screening of the cytoplasmic and nuclear hippocampal proteome pointed to significant alterations of two major components of the Erk cascade, Erk-1 and Erk-2. Although the employed conditions enabled to identify only a subset of proteins, the employed pH was the most suitable to detect concomitantly phosphorylation shifts as well as changes of protein levels (Stannard et al., 2003). These initial screens, indicating enhanced Erk-2 phosphorylation and Erk-1 nuclear accumulation, were verified by immunoblot and immunohistochemical approaches.
The increase of pErk-2 in the cytoplasmic hippocampal extracts of fear conditioned mice was in agreement with earlier studies demonstrating that pErk-2 is the main isoform activated during the consolidation of contextual fear memory (Atkins et al., 1998; Sananbenesi et al., 2002; Giovannini et al., 2003; Buckley & Caldwell, 2004). In contrast to other brain areas, such as the cortex, central and basolateral nuclei of the amygdala, where activated Erk-1/2 localize within the nucleus ( Bhat et al., 1998; Schafe et al., 2000) acting as a transcriptional regulators, a predominantly cytoplasmic activation pattern of Erk-1/2 in the hippocampus is commonly observed in vivo during fear conditioning (Sananbenesi et al., 2002), pharmacological manipulations (Winder et al., 1999; Refojo et al., 2005) or seizures (Bhat et al., 1998). On this basis, nuclear up-regulation of pErk-1, and less consistently pErk-2, observed prior to and during initial trials revealing reduced freezing behavior, may be specific for processes underlying extinction. Actin-dependent Erk-1/2 activation and nuclear translocation (Zhao et al., 2005) is a possible mechanism that is consistent with our previous studies demonstrating a significant role of hippocampal actin rearrangement in this process (Fischer et al., 2005). However, on the basis of recent evidence demonstrating that the cytoplasm may not be an exclusive site of Erk phosphorylation (Mandl et al., 2005), a compartmentalized activation in the nucleus cannot be excluded. Further analysis will reveal the upstream mechanisms causing the differences in Erk activation and localization and elucidate whether a subset of downstream nuclear targets of Erk-1/2 might specifically regulate gene expression associated with extinction when compared to conditioning.
The significantly accelerated activation of Erk-1/2 observed after nonreinforced when compared to reinforced trials was in line with recent observations (Chen et al., 2005). In the latter study, however, these alterations subsided within one hour after one nonreinforced trial, whereas we observed elevated levels of pErk-1/2 at this time point after several consecutive trials. Some procedural differences, such as somewhat longer contextual exposures in our experiments, may account for this discrepancy.
By employing two inhibitors of Mek, we demonstrated that disruption of dorsohippocampal Erk-1/2 phosphorylation impaired contextual fear extinction. A series of control experiments showed that these inhibitors produced neuroanatomically and temporally specific effects, and did not exert nonspecific actions on freezing behavior. It has been reported that post-trial administration of Mek inhibitors may affect memory retrieval (Szapiro et al., 2003) or reconsolidation (Kelly et al., 2003; Cestari et al., 2006). In contextual fear conditoning, however, neither post-trial inhibition of Mek/Erk employed here, or PI3/Akt shown earlier (Chen et al., 2005), affected retrieval or reconsolidation. This conclusion is drawn from the observation that injections after the initial nonreinforced trials did not impair contextual freezing during subsequent memory tests. Taking into account that in addition to Mek, PI3 also contributes to the activation of Erk-1/2, at least after the first nonreinforced contextual presentation (Chen et al., 2005), it remains to be determined whether these pathways converge on Erk as the main mediator, or Erk and Akt synergistically act on downstream processes associated with fear reduction. Thus, some of the numerous PI3/Akt substrates belonging to the Wnt, NF kappaB or other protein kinase pathways (Workman et al., 2006) may act in concert with Erk-1/2 to enable extinction.
A role of hippocampal Mek/Erk signaling in extinction was first demonstrated with the passive avoidance paradigm (Szapiro et al., 2003). Thus, the observation that Mek/Erk also contributes to the extinction of contextual freezing suggests that this pathway may exert a broader role in extinction processes involving the hippocampal brain area. Nevertheless, some differences unique for each paradigm need to be mentioned. In our study, a single injection of a Mek inhibitor did not prevent extinction and did not result in transient impairments of retrieval. In addition, differences in the time course of activation and distribution of pErk-1/2 between acquisition and extinction of contextual fear strongly suggest that the upstream activators and probably downstream effectors of pErk-1/2 are distinctively involved in these processes. Thus, it is not likely that the molecular mechanisms underlying extinction are similar to those for acquisition of contextual freezing, as it has been previously suggested for inhibitory avoidance (Szapiro et al., 2003). Because both behaviors similarly depend on contextual memory retrieval, some of the observed discrepancies may be attributable to differences in the intensity of contextual fear (Rachman & Hodgson, 1974) and stress hormone levels (Korte et al., 1992) accompanying avoidance versus immobility.
It has been of an increasing interest to understand and dissociate the molecular mechanisms triggered by nonreinforced trials, taking into account that several processes such as memory retrieval, reconsolidation and extinction could spatio-temporally overlap (Nader, 2003; Dudai & Eisenberg, 2004). Additionally, the hippocampal network has been implicated in anticipatory anxiety states associated with conflictual (McNaughton & Gray, 2000) or aversive (Ploghaus et al., 2001) situations. Thus, at least four conditions (retrieval, fear state, reconsolidation and extinction) may trigger the observed Erk-1/2 activation patterns and result in one outcome, in this case, conditioned freezing behavior. The day-by-day analyses of data obtained from the molecular and pharmacological experiments allowed excluding some of the processes interfering with extinction. For example, retrieval of the conditioning (Chen et al., 2005) or extinction memories may activate pErk-1/2. Under this scenario however, nonreinforced trials would be expected to always trigger Erk-1/2 phosphorylation. Such possibility was not experimentally supported because once extinction of freezing was stable over several trials pErk-1/2 levels were low despite of the retrieval of the extinction memory. A second possibility is that the fear state up-regulates pErk-1/2. Although pErk-1/2 were activated by trials showing high freezing and even stronger by initial trials showing reduction of freezing, it cannot be completely ruled out that some residual fear could account for signaling alterations without eliciting overt freezing behavior. Alternatively, Erk-1/2 might be triggered with some specificity to contribute to extinction processes. This option is supported by pharmacological experiments demonstrating a significant long-term role of this pathway in extinction of contextual freezing, but not memory retrieval, reconsolidation or fear expression. Although several mechanisms by which hippocampal processes mediate extinction can be considered (Lattal et al., 2006), it may be hypothesized that the Erk-1/2 pathway either contributes to the formation of an inhibitory extinction memory or, alternatively, by modifying the conditioning contextual memory and “updating” the new information obtained after extinction trials into the amygdalar circuits consolidating extinction memory (Chhatwal et al., 2006) or lateral septum regulating anticipatory anxiety (McNaughton and Gray, 2000). The later view is more consistent with the proposed role of the hippocampus in contextual processing (Huff and Rudy, 2004).
A final consideration concerns the employed extinction paradigm. There is evidence that short and long exposures leading to within- and between-session extinction may engage different molecular mechanisms (Pedreira & Maldonado, 2003). We have selected multiple short exposures resulting in gradual between-session extinction in order to better analyze the individual processes accompanying nonrenforced trials, and interfere with Mek/Erk signaling in the immediate post-exposure period. Similar approaches were employed in most experiments discussed above (Szapiro et al., 2003; Wang et al., 2004; Chen et al., 2005). This paradigm may be particularly relevant for human studies showing that one-time interventions may impede extinction, whereas interventions delivered in more than one session are effective in preventing the development of anxiety disorders (Rothbaum and Davis, 2003). It remains to be established whether within-session extinction of contextual fear also involves the Mek/Erk signaling pathway.
In conclusion, the findings obtained in this study support an accumulating evidence on the contribution of hippocampal mechanisms to the neuronal circuitry mediating extinction of contextual freezing (Szapiro et al., 2003; Fischer et al., 2004; Chen et al., 2005), however it remains to be established whether these alterations reflect the consolidation of extinction memory or other regulatory processes (Corcoran et al., 2005) leading to a reduction of freezing behavior. Importantly, Mek/Erk activation in other brain areas, such as the basolateral amygdala (Lu et al., 2001) and prefrontal cortex (Hugues et al., 2004), contributes to the extinction of freezing to explicit cues, suggesting that Erk-1/2 may be important targets for therapeutic approaches designed to relieve persistent fear symptoms accompanying anxiety disorders.
Acknowledgments
Supported by the National Institute of Mental Health, MH073669 to J.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nature Neuroscience. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Engber TM, Finn JP, Koury EJ, Contreras PC, Miller MS, Dionne CA, Walton KM. Region-specific targets of p42/p44MAPK signaling in rat brain. Journal of Neurochemistry. 1998;70:558–571. doi: 10.1046/j.1471-4159.1998.70020558.x. [DOI] [PubMed] [Google Scholar]
- Buckley CT, Caldwell KK. Fear conditioning is associated with altered integration of PLC and ERK signaling in the hippocampus. Pharmacology Biochemistry and Behavior. 2004;79:633–640. doi: 10.1016/j.pbb.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Buss KA, Davidson RJ, Kalin NH, Goldsmith HH. Context-specific freezing and associated physiological reactivity as a dysregulated fear response. Developmental Psychology. 2004;40:583–594. doi: 10.1037/0012-1649.40.4.583. [DOI] [PubMed] [Google Scholar]
- Cannich A, Wotjak CT, Kamprath K, Hermann H, Lutz B, Marsicano G. CB1 cannabinoid receptors modulate kinase and phosphatase activity during extinction of conditioned fear in mice. Learning and Memory. 2004;11:625–632. doi: 10.1101/lm.77904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestari V, Costanzi M, Castellano C, Rossi-Arnaud C. A role for ERK2 in reconsolidation of fear memories in mice. Neurobiology of Learning and Memory. 2006 doi: 10.1016/j.nlm.2006.01.003. in press. [DOI] [PubMed] [Google Scholar]
- Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nature Neuroscience. 2006;9:870–872. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nature Neuroscience. 2005;8:925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Desmond TJ, Frey KA, Maren S. Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. Journal of Neuroscience. 2005;25:8978–8987. doi: 10.1523/JNEUROSCI.2246-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Rainnie D, Cassell M. Neurotransmission in the rat amygdala related to fear and anxiety. Trends in Neurosciences. 1994;17:208–214. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Dudai Y, Eisenberg M. Rites of passage of the engram: reconsolidation and the lingering consolidation hypothesis. Neuron. 2004;44:93–100. doi: 10.1016/j.neuron.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Distinct roles of hippocampal de novo protein synthesis and actin rearrangement in extinction of contextual fear. Journal of Neuroscience. 2004;24:1962–1966. doi: 10.1523/JNEUROSCI.5112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini MG, Efoudebe M, Passani MB, Baldi E, Bucherelli C, Giachi F, Corradetti R, Blandina P. Improvement in fear memory by histamine-elicited ERK2 activation in hippocampal CA3 cells. Journal of Neuroscience. 2003;23:9016–9023. doi: 10.1523/JNEUROSCI.23-27-09016.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C. Startle reactivity and anxiety disorders: aversive conditioning, context, and neurobiology. Biological Psychiatry. 2002;52:958–975. doi: 10.1016/s0006-3223(02)01665-7. [DOI] [PubMed] [Google Scholar]
- Huff NC, Rudy JW. The amygdala modulates hippocampus-dependent context memory formation and stores cue-shock associations. Behavioral Neuroscience. 2004;118:53–62. doi: 10.1037/0735-7044.118.1.53. [DOI] [PubMed] [Google Scholar]
- Hugues S, Deschaux O, Garcia R. Postextinction infusion of a mitogen-activated protein kinase inhibitor into the medial prefrontal cortex impairs memory of the extinction of conditioned fear. Learning and Memory. 2004;11:540–543. doi: 10.1101/lm.77704. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kelly A, Laroche S, Davis S. Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. Journal of Neuroscience. 2003;23:5354–5360. doi: 10.1523/JNEUROSCI.23-12-05354.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte SM, Bouws GA, Koolhaas JM, Bohus B. Neuroendocrine and behavioral responses during conditioned active and passive behavior in the defensive burying/probe avoidance paradigm: effects of ipsapirone. Physiology and Behavior. 1992;52:355–361. doi: 10.1016/0031-9384(92)90284-9. [DOI] [PubMed] [Google Scholar]
- Lalonde J, Lachance PE, Chaudhuri A. Monocular enucleation induces nuclear localization of calcium/calmodulin-dependent protein kinase IV in cortical interneurons of adult monkey area V1. Journal of Neuroscience. 2004;24:554–564. doi: 10.1523/JNEUROSCI.1668-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KA, Radulovic J, Lukowiak K. Extinction: Does it or doesn’t it? The requirement of altered gene activity and new protein synthesis. Biological Psychiatry. 2006 doi: 10.1016/j.biopsych.2006.05.038. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annual Review of Neuroscience. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Lu KT, Walker DL, Davis M. Mitogen-activated protein kinase cascade in the basolateral nucleus of amygdala is involved in extinction of fear-potentiated startle. Journal of Neuroscience. 2001;21:RC162. doi: 10.1523/JNEUROSCI.21-16-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl M, Slack DN, Keyse SM. Specific inactivation and nuclear anchoring of extracellular signal-regulated kinase 2 by the inducible dual-specificity protein phosphatase DUSP5. Molecular and Cell Biology. 2005;25:1830–1845. doi: 10.1128/MCB.25.5.1830-1845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton N, Gray JA. Anxiolytic action on the behavioural inhibition system implies multiple types of arousal contribute to anxiety. Journal of Affective Disorders. 2000;61:161–176. doi: 10.1016/s0165-0327(00)00344-x. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neuroscience Letters. 1993;163:109–113. doi: 10.1016/0304-3940(93)90241-c. [DOI] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE. Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behavioral Neuroscience. 1995;109:681–688. doi: 10.1037//0735-7044.109.4.681. [DOI] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE. Contribution of ventrolateral prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Neurobiology of Learning and Memory. 1999;72:244–251. doi: 10.1006/nlme.1999.3907. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- Nader K. Memory traces unbound. Trends in Neurosciences. 2003;26:65–72. doi: 10.1016/S0166-2236(02)00042-5. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- Pedreira ME, Maldonado H. Protein synthesis subserves reconsolidation or extinction depending on reminder duration. Neuron. 2003;38:863–869. doi: 10.1016/s0896-6273(03)00352-0. [DOI] [PubMed] [Google Scholar]
- Ploghaus A, Narain C, Beckmann CF, Clare S, Bantick S, Wise R, Matthews PM, Rawlins JN, Tracey I. Exacerbation of pain by anxiety is associated with activity in a hippocampal network. Journal of Neuroscience. 2001;21:9896–9903. doi: 10.1523/JNEUROSCI.21-24-09896.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouyssegur J, Volmat V, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochemical Pharmacology. 2002;64:755–763. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- Rachman S, Hodgson R. I. Synchrony and desynchrony in fear and avoidance. Behaviour Research and Therapy. 1974;12:311–318. doi: 10.1016/0005-7967(74)90005-9. [DOI] [PubMed] [Google Scholar]
- Radulovic J, Kammermeier J, Spiess J. Relationship between fos production and classical fear conditioning: effects of novelty, latent inhibition, and unconditioned stimulus preexposure. Journal of Neuroscience. 1998;18:7452–7461. doi: 10.1523/JNEUROSCI.18-18-07452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refojo D, Echenique C, Muller MB, Reul JM, Deussing JM, Wurst W, et al. Corticotropin-releasing hormone activates ERK1/2 MAPK in specific brain areas. Proceedings of the National Academy of Sciences U S A. 2005;102:6183–6188. doi: 10.1073/pnas.0502070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: interactions between Erk-1/2 and Elk-1. Molecular and Cellular Neuroscience. 2002;21:463–476. doi: 10.1006/mcne.2002.1188. [DOI] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Mitogen-activated protein kinase signaling in the hippocampus and its modulation by corticotropin-releasing factor receptor 2: a possible link between stress and fear memory. Journal of Neuroscience. 2003;23:11436–11443. doi: 10.1523/JNEUROSCI.23-36-11436.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Ge H, Ren K, Pena de Ortiz S, Quirk GJ. Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. Journal of Neuroscience. 2004;24:5704–5710. doi: 10.1523/JNEUROSCI.0786-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. Journal of Neuroscience. 2000;20:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimanski LA, Wahlsten D, Nguyen PV. Selective modification of short-term hippocampal synaptic plasticity and impaired memory extinction in mice with a congenitally reduced hippocampal commissure. Journal of Neuroscience. 2002;22:8277–8286. doi: 10.1523/JNEUROSCI.22-18-08277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Cain CK, Ledoux JE. Brain Mechanisms of Fear Extinction: Historical Perspectives on the Contribution of Prefrontal Cortex. Biological Psychiatry. 2006 doi: 10.1016/j.biopsych.2005.10.012. in press. [DOI] [PubMed] [Google Scholar]
- Stannard C, Soskic V, Godovac-Zimmermann J. Rapid changes in the phosphoproteome show diverse cellular responses following stimulation of human lung fibroblasts with endothelin-1. Biochemistry. 2003;42:13919– 13928. doi: 10.1021/bi035414u. [DOI] [PubMed] [Google Scholar]
- Stiedl O, Radulovic J, Lohmann R, Birkenfeld K, Palve M, Kammermeier J, Sananbenesi F, Spiess J. Strain and substrain differences in context- and tone-dependent fear conditioning of inbred mice. Behavioural Brain Research. 1999;104:1–12. doi: 10.1016/s0166-4328(99)00047-9. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. Journal of Neurochemistry. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- Szapiro G, Vianna MR, McGaugh JL, Medina JH, Izquierdo I. The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus. 2003;13:53–58. doi: 10.1002/hipo.10043. [DOI] [PubMed] [Google Scholar]
- Thiels E, Kanterewicz BI, Norman ED, Trzaskos JM, Klann E. Long-term depression in the adult hippocampus in vivo involves activation of extracellular signal-regulated kinase and phosphorylation of Elk-1. Journal of Neuroscience. 2002;22:2054–2062. doi: 10.1523/JNEUROSCI.22-06-02054.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. Journal of Neuroscience. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR. Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nature Neuroscience. 2004;7:635–642. doi: 10.1038/nn1248. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- Workman P, Clarke PA, Guillard S, Raynaud FI. Drugging the PI3 kinome. Nature Biotechnology. 2006;24:794–796. doi: 10.1038/nbt0706-794. [DOI] [PubMed] [Google Scholar]
- Zhao M, Discipio RG, Wimmer AG, Schraufstatter IU. Regulation of CXCR4-mediated nuclear translocation of ERK1/2. Molecular Pharmacology. 2005;69:66–75. doi: 10.1124/mol.105.016923. [DOI] [PubMed] [Google Scholar]