

Abstract
The fixation of advantageous mutations by natural selection has a profound impact on patterns of linked neutral variation. While it has long been appreciated that such selective sweeps influence the frequency spectrum of nearby polymorphism, it has only recently become clear that they also have dramatic effects on local linkage disequilibrium. By extending previous results on the relationship between genealogical structure and linkage disequilibrium, I obtain simple expressions for the influence of a selective sweep on patterns of allelic association. I show that sweeps can increase, decrease, or even eliminate linkage disequilibrium (LD) entirely depending on the relative position of the selected and neutral loci. I also show the importance of the age of the neutral mutations in predicting their degree of association and describe the consequences of such results for the interpretation of empirical data. In particular, I demonstrate that while selective sweeps can eliminate LD, they generate patterns of genetic variation very different from those expected from recombination hotspots.
SELECTIVE sweeps, in which a beneficial mutation is swept to fixation in a population by natural selection, have a profound impact on patterns of linked genetic variation through what is known as the hitchhiking effect (Maynard Smith and Haigh 1974). Although simple in concept, studies of the process continue to uncover novel and unusual properties that have direct implications for the detection of such events from empirical data. For example, the realization that the interaction of hitchhiking with recombination can lead to an excess of high-frequency-derived mutations (Fay and Wu 2000) gave novel insights into the well-known fact that hitchhiking can lead to a bias toward low-frequency polymorphism (Fu and Li 1993; Braverman et al. 1995). Recently, studies of the effects of selective sweeps on patterns of linkage disequilibrium (LD) have also identified characteristic, and perhaps surprising, patterns (Kim and Stephan 2002; Przeworski 2002; Kim and Nielsen 2004; Reed and Tishkoff 2005; Stephan et al. 2006). For example, while sweeps can lead to an increase in LD while they are still in progress (Hudson et al. 1994; Sabeti et al. 2002), when the beneficial mutation has reached fixation, LD across the selected site is eliminated (Kim and Nielsen 2004; Stephan et al. 2006). Interpreting empirical patterns of genetic variation in the light of such observations is therefore potentially confusing and raises important questions. For example, are positions at which LD is observed to break down rapidly the result of selective sweeps or recombination hotspots? Indeed, it has been demonstrated that for certain population genetic methods selective sweeps may be falsely interpreted as hotspots of recombination (Reed and Tishkoff 2005).
The aim of this article is to provide an intuitive interpretation of the effects of selective sweeps on patterns of LD, through considering the relationship between LD and the structure of the underlying genealogical history. Previous work has shown that there is a direct quantitative relationship between the magnitude of LD observed between a pair of neutral mutations and the correlation structure of the underlying genealogy (McVean 2002). By using the conventional approximation that strong selective sweeps lead to short, star-like genealogies at the selected site, this theory is extended to examine the correlation structure between the genealogies of neutral loci either separated by or adjacent to the selected site. Comparison with the results of stochastic simulation demonstrates that this theory predicts the qualitative and, to some extent, quantitative, behavior of LD around a selective sweep. In addition, the theory identifies the importance of the age of neutral mutations (relative to the selected one) in determining patterns of LD and predicts large differences in the nature of the breakdown of LD around a selective sweep and a recombination hotspot.
TWO-LOCUS IDENTITIES AND A GENEALOGICAL INTERPRETATION OF LD
Informally, LD between neutral alleles at two loci arises because of correlations in the genealogical history of the two loci. Put another way, if the time to the MRCA (most recent common ancestor) for a pair of chromosomes at a given position, x, on the genome is informative about the time to the MRCA for the same pair of chromosomes at another genomic position, y (relative to any other pair of chromosomes), the alleles at the two loci are expected to show significant LD. However, different statistical measures of LD focus on different aspects of such correlation. Here we focus on one widely used two-locus measure of LD for biallelic loci, the square of the correlation coefficient in allelic state or 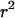 (Hill and Robertson 1968). For a pair of biallelic loci, with alleles 0 and 1 at locus x and also 0 and 1 at locus y, the statistic is defined as
(Hill and Robertson 1968). For a pair of biallelic loci, with alleles 0 and 1 at locus x and also 0 and 1 at locus y, the statistic is defined as
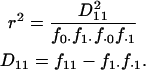 |
(1) |
Here,  is the sample frequency of the 11 haplotype and
is the sample frequency of the 11 haplotype and 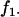 is the marginal sample frequency of the “1” allele at locus x. Note that for biallelic loci the value of
is the marginal sample frequency of the “1” allele at locus x. Note that for biallelic loci the value of  does not depend on which allele is assigned the value 1. Consequently, in what follows the subscript for D is omitted.
does not depend on which allele is assigned the value 1. Consequently, in what follows the subscript for D is omitted.
Ideally, we wish to calculate the expected value of 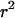 between alleles at the two loci, conditioning on observing at least one of each allele at each of the two loci in a sample of size n sequences:
between alleles at the two loci, conditioning on observing at least one of each allele at each of the two loci in a sample of size n sequences:
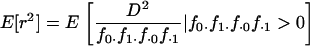 |
(2) |
There is, unfortunately, no simple expression for this expectation, although recent advances have been made in its numerical evaluation (Song and Song 2007). However, it is possible to derive expressions for a related quantity, called 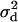 :
:
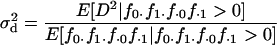 |
(3) |
(Ohta and Kimura 1971). After this point the conditioning on segregation at the two loci will be implicit. It can be shown through Monte Carlo simulation (Hudson 1985; McVean 2002) that Equation 3 is a good approximation to the expectation of 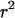 (i.e., Equation 2) for large sample sizes and when rare variants are excluded.
(i.e., Equation 2) for large sample sizes and when rare variants are excluded.
Previous work (Strobeck and Morgan 1978; Hudson 1985) showed that the statistic 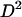 can be rewritten in terms of two-locus identity coefficients:
can be rewritten in terms of two-locus identity coefficients:
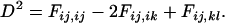 |
(4) |
To understand the two-locus identity coefficients, consider sampling four chromosomes at random with replacement from a population and labeling them i, j, k, and l. The three terms on the right-hand side of Equation 4 are, respectively, the probability that sequences i and j are identical in state at both sites x and y, the probability that sequences i and j are identical at locus x and that sequences i and k are identical at site y, and the probability that sequences i and j are identical at site x and sequences k and l are identical at site y. These three configurations, which are referred to as A, B, and C, respectively, are central to the following discussion and are represented in Figure 1A. A similar expression applies to the sample statistic where the chromosomes are drawn (with replacement) from the sample (Hudson 1985). In small samples it is therefore possible that i, j, etc., are not distinct.
Figure 1.—

(A) Two-locus configurations relating to Equation 4 showing the three ways in which two chromosomes at each of two loci can be sampled. (B) The model of a selective sweep. Chromosomes (bottom bars) at a locus where there has been a recent and complete selective sweep (shaded triangle) are related to each through a star-like genealogy. However, recombination events (dotted arrow) during the selected phase allow lineages to escape to the ancestral background. In the model, neutral mutations (circles) occur on the portion of the genealogy older than the selected mutation, the neutral phase. In the example shown, only a single lineage recombines, such that the two neutral mutations are in perfect association; r2 = 1.
The key point about Equation 4 is that the expectation of 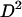 can be written in terms of the expectation of these two-locus identity coefficients. Under the infinite-sites model, in which each polymorphism observed is the result of a single mutation event within the sample's history, it is possible to relate the two-locus identities to the expectations of genealogical properties at the two loci (McVean 2002). For example,
can be written in terms of the expectation of these two-locus identity coefficients. Under the infinite-sites model, in which each polymorphism observed is the result of a single mutation event within the sample's history, it is possible to relate the two-locus identities to the expectations of genealogical properties at the two loci (McVean 2002). For example,
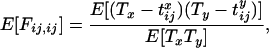 |
(5) |
where Tx is the total time in the genealogy (i.e., the sum of the branch lengths) at locus x and 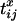 is the coalescence time for sequences i and j at locus x. By obtaining similar expressions for the other two-locus identities and also the denominator of Equation 3, it was shown that
is the coalescence time for sequences i and j at locus x. By obtaining similar expressions for the other two-locus identities and also the denominator of Equation 3, it was shown that
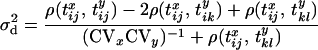 |
(6) |
(McVean 2002), where 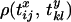 is the Pearson correlation coefficient between the coalescence time for sequences i and j at locus x and the coalescence time for sequences k and l at locus y and CVx is the coefficient of variation in the time to the most recent common ancestor (MRCA) for a pair of randomly sampled chromosomes at locus x,
is the Pearson correlation coefficient between the coalescence time for sequences i and j at locus x and the coalescence time for sequences k and l at locus y and CVx is the coefficient of variation in the time to the most recent common ancestor (MRCA) for a pair of randomly sampled chromosomes at locus x,  . Note that there are three correlations in Equation 6, relating to the three sample configurations (see Equation 4 and Figure 1A).
. Note that there are three correlations in Equation 6, relating to the three sample configurations (see Equation 4 and Figure 1A).
The most important implication of Equation 6 is that it provides a quantitative approach for relating patterns of LD to features of the underlying genealogical history. For example, demographic histories in which the population has increased, decreased, or remained constant in size influence LD both through their effects on the correlation structure of genealogies and through their effects on the coefficient of variation in time to the MRCA. For example, population growth reduces the coefficient of variation thus reducing LD, while population bottlenecks increase the coefficient of variation, increasing LD. The theory can also be extended to consider more complex situations, for example, the case of a series of island populations connected by migration (Wakeley and Lessard 2003). In the next section, the theory is extended to the case of a pair of neutral loci linked to a site that has undergone a complete selective sweep in which the beneficial mutation has just reached fixation in the population.
MODELING GENEALOGIES UNDER A SELECTIVE SWEEP
Looking back in time, a neutral locus on a single lineage at some genetic distance  from a selected site (where r is the genetic map distance in Morgans and Ne is the effective population size, assumed to be diploid) can either recombine away from the selected mutation before its removal from the population, with probability p, or not, with probability q = 1 − p. The probability of “escape” is a function of the recombination rate and the frequency trajectory of the selected mutation, itself a random variable determined by the scaled selection coefficient
from a selected site (where r is the genetic map distance in Morgans and Ne is the effective population size, assumed to be diploid) can either recombine away from the selected mutation before its removal from the population, with probability p, or not, with probability q = 1 − p. The probability of “escape” is a function of the recombination rate and the frequency trajectory of the selected mutation, itself a random variable determined by the scaled selection coefficient 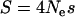 . By approximating the trajectory of the selected mutation by that of the deterministic expectation, it has been previously shown that
. By approximating the trajectory of the selected mutation by that of the deterministic expectation, it has been previously shown that
 |
(7) |
(Maynard Smith and Haigh 1974; Kaplan et al. 1989; Stephan et al. 1992; Durrett and Schweinsberg 2004). Implicit within this formula is an expression for the age of the selected mutation:
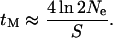 |
(8) |
As for all expressions relating to age, this is expressed in units of 2Ne generations. When there is more than a single lineage to consider (i.e., a sample of size n > 1), the shape of genealogy under the selected mutation has to be considered. However, if a selective sweep is sufficiently strong, this genealogy can be approximated as a star phylogeny (Maynard Smith and Haigh 1974; Kim and Stephan 2002) with the age of the common ancestor, 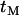 , taken from Equation 8 (Figure 1B). Although this approximation can be criticized (Barton 1998; Durrett and Schweinsberg 2004; Etheridge et al. 2006), it nevertheless has proved very useful in analytical treatments of hitchhiking, because of the resulting independence between lineages in whether they recombine away from the selected mutation.
, taken from Equation 8 (Figure 1B). Although this approximation can be criticized (Barton 1998; Durrett and Schweinsberg 2004; Etheridge et al. 2006), it nevertheless has proved very useful in analytical treatments of hitchhiking, because of the resulting independence between lineages in whether they recombine away from the selected mutation.
A further simplifying assumption, 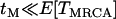 , is also made, where
, is also made, where 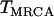 is the time until the MRCA for a sample of n chromosomes. Under the standard neutral model,
is the time until the MRCA for a sample of n chromosomes. Under the standard neutral model, 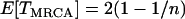 . Looking back in time, the history of the sample can therefore be divided into two phases (Figure 1B). During the first “selection phase” the only events that can occur are recombination events that move neutral loci from the background of the selected allele to that of the ancestral, wild-type allele. The end of the selection phase is marked by the origin of the selected mutation at which point all chromosomes carrying the selected allele coalesce immediately, and the selected allele is removed. Subsequently, in the “neutral phase,” the history of the remaining lineages follows that of the standard neutral model. In the extreme, the selection phase can be considered instantaneous with respect to the timescale of the neutral coalescent process (i.e.,
. Looking back in time, the history of the sample can therefore be divided into two phases (Figure 1B). During the first “selection phase” the only events that can occur are recombination events that move neutral loci from the background of the selected allele to that of the ancestral, wild-type allele. The end of the selection phase is marked by the origin of the selected mutation at which point all chromosomes carrying the selected allele coalesce immediately, and the selected allele is removed. Subsequently, in the “neutral phase,” the history of the remaining lineages follows that of the standard neutral model. In the extreme, the selection phase can be considered instantaneous with respect to the timescale of the neutral coalescent process (i.e., 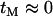 ) and therefore any mutations segregating must have occurred on the portion of the genealogy that predates the origin of selected mutation. Under this assumption if no lineages have recombined to the ancestral background at a given distance from the selected site, there will be no polymorphism in the sample.
) and therefore any mutations segregating must have occurred on the portion of the genealogy that predates the origin of selected mutation. Under this assumption if no lineages have recombined to the ancestral background at a given distance from the selected site, there will be no polymorphism in the sample.
By dividing the history of the sample into these two phases it can be seen that the effect of the selective sweep on patterns of LD is determined by how it influences the configuration of chromosomes found at the start of the neutral phase (just further back in time than the origin of the selected mutation). In particular, we need to calculate the transition probabilities that describe how each of the initial configurations, A, B, and C, is distributed at the start of the neutral phase. For example, consider configuration A where the selected site separates the two neutral loci (Figure 2). Depending on the distribution of recombination events that move a neutral locus from the selected to the ancestral background, this initial configuration can be transformed into any of 10 possible states at the end of the selected phase. The removal of the selected mutation subsequently transforms these 10 configurations, through coalescence of those still carrying the selected mutation, to any of configurations A, B, and C or to ones where one or both of the neutral loci coalesce (indicated by O in Figure 2). Details of the probabilities of each transition are given in appendixes a and b.
Figure 2.—

Transition probabilities for the two-stage model of a selective sweep. The initial configuration (type A), where the selected mutation (solid circle) separates the two neutral loci (triangles), can be transformed into one of 10 different configurations at the end of the selection phase (the open circle indicates the ancestral, unselected mutation). Probabilities for each transition are shown in terms of pi and qi, respectively, the probability of a recombination event occurring in interval i during the selective sweep and the probability of no recombination ( ). The removal of the selected mutation induces coalescence between any chromosomes still carrying the selected mutation. The configurations at the start of the neutral phase can be classified into types A, B, or C, corresponding to the three configurations in Equation 4, or into type O in which at least one of the two loci has coalesced.
). The removal of the selected mutation induces coalescence between any chromosomes still carrying the selected mutation. The configurations at the start of the neutral phase can be classified into types A, B, or C, corresponding to the three configurations in Equation 4, or into type O in which at least one of the two loci has coalesced.
APPENDIX A.
Transition probabilities when the selected mutation separates the neutral loci
| Configuration at end of selection phase | Probability given starting configuration
|
Configuration at start of neutral phase | ||
|---|---|---|---|---|
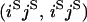 |
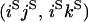 |
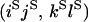 |
||
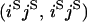 |
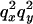 |
0 | 0 | O |
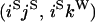 |
 |
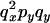 |
0 | O |
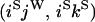 |
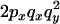 |
 |
0 | O |
 |
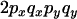 |
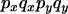 |
0 | B |
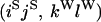 |
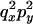 |
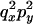 |
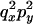 |
O |
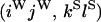 |
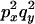 |
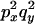 |
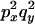 |
O |
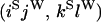 |
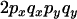 |
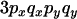 |
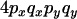 |
B |
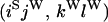 |
 |
 |
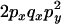 |
C |
 |
 |
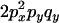 |
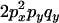 |
C |
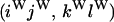 |
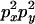 |
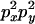 |
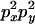 |
C |
 |
0 |  |
0 | O |
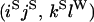 |
0 | 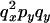 |
 |
O |
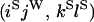 |
0 | 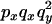 |
 |
O |
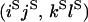 |
0 | 0 | 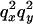 |
O |
An example of the transition probabilities for the changes in configuration that occur during the selection is given in Figure 2. Here we give the transition probabilities for the different starting configurations. For notation, let  represent the configuration where at locus x (to the left of the selected site) the two chromosomes i and j have been sampled and both carry the selected allele and at locus y the same two chromosomes have been sampled and again, both carry the selected allele. Using this notation,
represent the configuration where at locus x (to the left of the selected site) the two chromosomes i and j have been sampled and both carry the selected allele and at locus y the same two chromosomes have been sampled and again, both carry the selected allele. Using this notation, 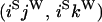 is, for example, the configuration where at locus x chromosome i carries the selected allele and chromosome j carries the wild type, while at locus y chromosome i has again been sampled (and therefore by necessity carries the selected allele), while a third chromosome, k, carries the wild-type allele. The transition probabilities during the selection phase for each of the three starting configurations are given below. Note that
is, for example, the configuration where at locus x chromosome i carries the selected allele and chromosome j carries the wild type, while at locus y chromosome i has again been sampled (and therefore by necessity carries the selected allele), while a third chromosome, k, carries the wild-type allele. The transition probabilities during the selection phase for each of the three starting configurations are given below. Note that 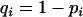 and that the labels i, j, k, and l are arbitrary identifiers for chromosomes drawn at random (with replacement) from the sample. For example, the configurations
and that the labels i, j, k, and l are arbitrary identifiers for chromosomes drawn at random (with replacement) from the sample. For example, the configurations  and
and 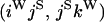 would be equivalent.
would be equivalent.
APPENDIX B.
Transition probabilities when the selected mutation is adjacent to the neutral loci
| Configuration at end of selection phase | Probability given starting configuration
|
Configuration at start of neutral phase | ||
|---|---|---|---|---|
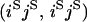 |
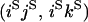 |
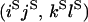 |
||
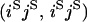 |
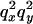 |
0 | 0 | O |
 |
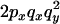 |
0 | 0 | A |
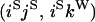 |
 |
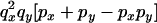 |
0 | O |
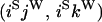 |
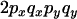 |
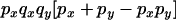 |
0 | B |
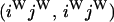 |
 |
0 | 0 | A |
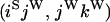 |
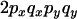 |
 |
0 | B |
 |
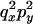 |
 |
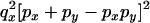 |
O |
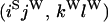 |
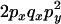 |
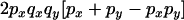 |
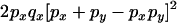 |
C |
 |
 |
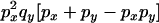 |
0 | B |
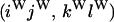 |
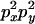 |
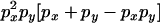 |
 |
C |
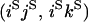 |
0 | 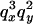 |
0 | O |
 |
0 |  |
0 | O |
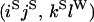 |
0 |  |
 |
O |
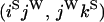 |
0 | 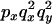 |
0 | A |
 |
0 | 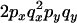 |
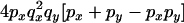 |
B |
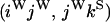 |
0 |  |
0 | B |
 |
0 | 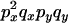 |
 |
C |
 |
0 | 0 |  |
O |
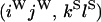 |
0 | 0 |  |
O |
 |
0 | 0 | 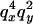 |
O |
The transition probabilities during the selection phase for each of the three starting configurations are given. Notation is as in appendix a. Note that px refers to the probability of recombination to the wild-type background for the proximal locus, while py refers to the probability of a recombination event between the proximal and the distal locus that moves the distal locus to the wild-type background.
Once the transition probabilities to each possible state at the start of the neutral phase have been calculated, it is a simple matter to obtain expressions for the necessary genealogical statistics. In particular, for each starting configuration we can write the expectation of the product of the coalescence time at the two neutral loci as a function of these transition probabilities. For example,
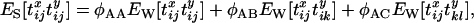 |
(9) |
where 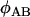 is the probability that configuration A in the sampled chromosomes (all of which carry the selected mutation) results in configuration B at the start of the neutral phase. The subscript S on the left-hand side indicates that the expectation refers to the selected allele, while the subscript W on the right-hand side indicates that these expectations refer to the wild-type allele (i.e., the standard neutral expectations). Under the standard neutral model these quantities are known for different configurations of chromosomes. In particular,
is the probability that configuration A in the sampled chromosomes (all of which carry the selected mutation) results in configuration B at the start of the neutral phase. The subscript S on the left-hand side indicates that the expectation refers to the selected allele, while the subscript W on the right-hand side indicates that these expectations refer to the wild-type allele (i.e., the standard neutral expectations). Under the standard neutral model these quantities are known for different configurations of chromosomes. In particular,
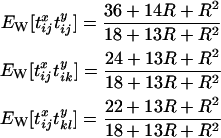 |
(10) |
(Pluzhnikov and Donnelly 1996; McVean 2002). Expressions similar to Equation 9 can be obtained for the other initial configurations B and C. Note that it is not necessary to include in Equation 9 a term for transitions to state O, as the expected product of coalescence times for this state is zero under the assumption 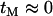 .
.
Finally, because the configurations can be thought of as relating to subsamples (with replacement) from a sample of n sequences, there is a possibility that sequences i, j, k, and l may not be distinct (the same sequence could be picked twice). A simple correction has to be made to the expectations,
 |
(11) |
where n is the sample size (Hudson 1985; McVean 2002).
NEUTRAL LOCI SEPARATED BY THE SELECTED SITE
First, consider the case of two loci separated by the selected site and distant from it by recombination distances of 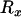 and
and 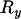 , respectively, such that the probabilities of a lineage escaping the selective sweep are
, respectively, such that the probabilities of a lineage escaping the selective sweep are 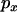 and
and 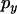 , respectively. By considering the probability of recombination in each interval it can be shown that
, respectively. By considering the probability of recombination in each interval it can be shown that
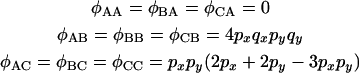 |
(12) |
(see appendix a). Consequently
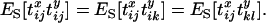 |
(13) |
It follows that whatever the values of  and
and 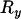
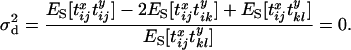 |
(14) |
In other words, LD across the selected site (as measured by 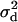 ) is zero or at least no greater than background levels caused by finite sample size. This result agrees with previous findings (Kim and Nielsen 2004; Stephan et al. 2006) obtained by simulation and analysis of deterministic models of selection. It is worth noting that a deterministic model (in which drift during the selection phase is ignored) is equivalent to assuming that no coalescent events occur during this period, the same assumption as is made here.
) is zero or at least no greater than background levels caused by finite sample size. This result agrees with previous findings (Kim and Nielsen 2004; Stephan et al. 2006) obtained by simulation and analysis of deterministic models of selection. It is worth noting that a deterministic model (in which drift during the selection phase is ignored) is equivalent to assuming that no coalescent events occur during this period, the same assumption as is made here.
However, it is also worth noting that while LD may be zero, there is actually nonzero correlation in coalescence time. For example, if 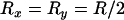 and
and 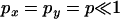 , it can be shown that
, it can be shown that
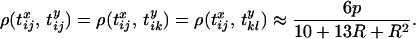 |
(15) |
It is perhaps surprising that there should be nonzero correlation in the time to the MRCA at the two neutral loci, but yet no LD. The nonzero correlation arises because lineages that escape the sweep will have low, though nonzero, correlations in the time to the MRCA resulting from the neutral part of their ancestry. For example, Equation 15 is derived by noting that when the recombination rate is low, the most probable configuration that arises in which both neutral loci escape the sweep is configuration B (this is true for all initial configurations). However, each initial configuration requires exactly the same set of recombination events to occur to reach configurations B and C at the start of the neutral phase, so the resulting correlation structure is the same for each initial configuration, and there is no LD.
NEUTRAL LOCI ON THE SAME SIDE OF THE SELECTED LOCUS
Now consider a pair of loci that are both on the same side of the selected site, with the nearer (or proximal), x, being at recombination distance  and the more distant (or distal), y, being at a recombination distance
and the more distant (or distal), y, being at a recombination distance 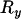 from x. In this situation the different initial configurations have different probabilities of resulting in each configuration at the start of the neutral phase. For example, configuration A can escape the sweep through a single recombination, while configuration C requires a minimum of two recombination events to escape the sweep. By considering the effect of recombination events occurring in each part of each chromosome during the selection phase (see appendix b) it follows that for configuration A
from x. In this situation the different initial configurations have different probabilities of resulting in each configuration at the start of the neutral phase. For example, configuration A can escape the sweep through a single recombination, while configuration C requires a minimum of two recombination events to escape the sweep. By considering the effect of recombination events occurring in each part of each chromosome during the selection phase (see appendix b) it follows that for configuration A
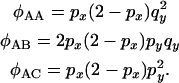 |
(16) |
For configuration B
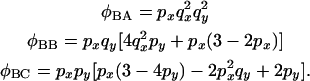 |
(17) |
While for configuration C
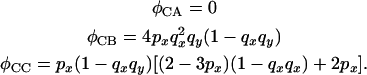 |
(18) |
The mean and variance of the time to coalescence at each locus are
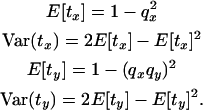 |
(19) |
These results can be used to derive numerical expressions for Equation 6 for various parameter values (Figure 3). However, several important features of the results can be identified. First, when 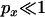 it follows that
it follows that
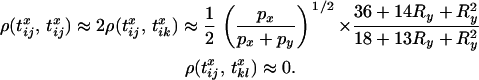 |
(20) |
Under this approximation, Equation 6 evaluates at zero. However, when  , such that
, such that  , it is also critical to account for the finite sample size, such that i, j, k, and l are not necessarily distinct. Under these conditions a good approximation for the expected LD is
, it is also critical to account for the finite sample size, such that i, j, k, and l are not necessarily distinct. Under these conditions a good approximation for the expected LD is
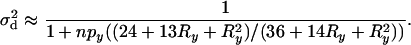 |
(21) |
Equation 21 predicts that conditional on observing polymorphism at the linked neutral loci there will be perfect correlation (i.e., 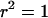 ) between the alleles if there is no recombination between them (Figure 3). This result can be understood by noting that the most probable way in which polymorphism will be observed if
) between the alleles if there is no recombination between them (Figure 3). This result can be understood by noting that the most probable way in which polymorphism will be observed if 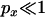 is if a single lineage escapes the selective sweep. Any neutral mutations must occur during the neutral phase, in which only two lineages will be present (the lineage leading to the MRCA of the selected mutation and the escaped lineage), leading to perfect association (in effect the mutations will occur on the same branch of the unrooted genealogy, as in Figure 1B). Another prediction of Equation 21 is that the magnitude of LD decreases rapidly as the recombination rate between the neutral loci increases. Indeed for moderate to large sample sizes it should decrease below that expected for an identical pair of neutral sites unaffected by a sweep (Figure 3). From a genealogical perspective, any recombination events occurring between the two neutral loci will rapidly lead to a breakdown in the correlation of the genealogies at the two positions. Informally, the effect can also be understood in terms of allele frequency. When
is if a single lineage escapes the selective sweep. Any neutral mutations must occur during the neutral phase, in which only two lineages will be present (the lineage leading to the MRCA of the selected mutation and the escaped lineage), leading to perfect association (in effect the mutations will occur on the same branch of the unrooted genealogy, as in Figure 1B). Another prediction of Equation 21 is that the magnitude of LD decreases rapidly as the recombination rate between the neutral loci increases. Indeed for moderate to large sample sizes it should decrease below that expected for an identical pair of neutral sites unaffected by a sweep (Figure 3). From a genealogical perspective, any recombination events occurring between the two neutral loci will rapidly lead to a breakdown in the correlation of the genealogies at the two positions. Informally, the effect can also be understood in terms of allele frequency. When 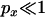 , polymorphism at the proximal locus is most likely to be in the form of a singleton (i.e., one chromosome differs from all the others). Recombination between the proximal and the distal loci will allow nonsingleton polymorphism at the distal locus and this is likely to show weak LD with the singleton allele at the proximal locus.
, polymorphism at the proximal locus is most likely to be in the form of a singleton (i.e., one chromosome differs from all the others). Recombination between the proximal and the distal loci will allow nonsingleton polymorphism at the distal locus and this is likely to show weak LD with the singleton allele at the proximal locus.
Figure 3.—

The effect of a nearby selective sweep on LD between a pair of linked neutral loci. Numerical evaluation of Equation 6 is shown with the correction for finite sample size in the case where both neutral loci are on the same side of the selected site. 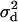 is shown as a function of the recombination rate, R = 4Ner, between the neutral loci (x-axis) and the probability of escape for the proximal locus being 0.001 (solid line), 0.1 (dark-shaded line), and 0.5 (light-shaded line). The dotted line shows the evaluation of Equation 6 under the neutral model. The probability of escape for the distal locus is given by
is shown as a function of the recombination rate, R = 4Ner, between the neutral loci (x-axis) and the probability of escape for the proximal locus being 0.001 (solid line), 0.1 (dark-shaded line), and 0.5 (light-shaded line). The dotted line shows the evaluation of Equation 6 under the neutral model. The probability of escape for the distal locus is given by 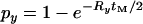 with
with 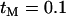 .
.
As the recombination rate between the proximal neutral locus and the selected site increases, the impact of the selective sweep diminishes and the LD between the neutral loci approaches that expected under the neutral model. However, the two key features of the pattern remain. First, if the neutral loci are very closely linked, LD is generally increased relative to the neutral expectation. Second, weakly linked neutral loci show a small decrease in LD relative to the neutral case (Figure 3). Both features can be explained by the above reasoning.
INCORPORATING NEUTRAL MUTATIONS YOUNGER THAN THE SELECTED MUTATION
So far, it has been assumed that the time to the origin of the beneficial mutation is approximately zero, such that any polymorphism found in the sample has to be older than the selected mutation. However, when the probability of a lineage escaping the selective sweep by recombination is low the expected time in genealogies in which no recombination occurs is considerable relative to the total expected time in the genealogy. Consequently, when 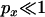 it is relatively likely that polymorphism observed in a sample that has experienced a selective sweep may be more recent than the selected mutation. From the genealogical perspective, considering such recent mutations is equivalent to setting
it is relatively likely that polymorphism observed in a sample that has experienced a selective sweep may be more recent than the selected mutation. From the genealogical perspective, considering such recent mutations is equivalent to setting 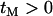 . Because no coalescent events occur during the selected phase, the only influence of a nonzero value of
. Because no coalescent events occur during the selected phase, the only influence of a nonzero value of 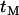 is to increase the expected coalescence time (it has no effect on the correlations in coalescence time or variance) and consequently decrease the coefficient of variation in coalescence time, thus reducing LD. When the neutral loci are either side of the selected site LD is low anyway, so inclusion of recent mutation has little or no impact on LD. However, when the two neutral loci are on the same side of the selected mutation recent mutation can have a considerable impact on LD, because neutral mutations older than the selected one will typically show strong LD if they are themselves tightly linked (as described above). To get an idea for the importance of including recent mutations, note that when
is to increase the expected coalescence time (it has no effect on the correlations in coalescence time or variance) and consequently decrease the coefficient of variation in coalescence time, thus reducing LD. When the neutral loci are either side of the selected site LD is low anyway, so inclusion of recent mutation has little or no impact on LD. However, when the two neutral loci are on the same side of the selected mutation recent mutation can have a considerable impact on LD, because neutral mutations older than the selected one will typically show strong LD if they are themselves tightly linked (as described above). To get an idea for the importance of including recent mutations, note that when 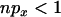 , typically at most one lineage will escape the sweep and the contribution of the neutral phase to the expected time in the genealogy of the sample is ∼
, typically at most one lineage will escape the sweep and the contribution of the neutral phase to the expected time in the genealogy of the sample is ∼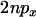 . Under these same conditions the total length of the genealogy within the selected phase is
. Under these same conditions the total length of the genealogy within the selected phase is 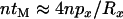 . Consequently, the probability that an observed neutral mutation at the proximal locus is older than the selected mutation is ∼
. Consequently, the probability that an observed neutral mutation at the proximal locus is older than the selected mutation is ∼ . In humans the average recombination rate is
. In humans the average recombination rate is 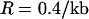 in European populations (Myers et al. 2005), so that a polymorphism 5 kb from the selected site will have only a 50% probability of being older than the selected mutation.
in European populations (Myers et al. 2005), so that a polymorphism 5 kb from the selected site will have only a 50% probability of being older than the selected mutation.
Figure 4 shows that inclusion of recent mutations has a marked effect on 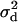 . When the recombination rate between the neutral loci is zero, mutations older than the selected one are predicted to show (and do show) monotonically decreasing LD as a function of increasing
. When the recombination rate between the neutral loci is zero, mutations older than the selected one are predicted to show (and do show) monotonically decreasing LD as a function of increasing 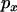 . However, when recent mutations are considered, LD very close to the selected site is near zero when
. However, when recent mutations are considered, LD very close to the selected site is near zero when 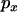 is small. LD increases as
is small. LD increases as 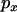 increases, exceeding the neutral expectation at intermediate values of
increases, exceeding the neutral expectation at intermediate values of 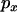 . Finally, as
. Finally, as  approaches one, the expected LD decreases toward neutral expectation. The nonmonotonic relationship between the distance of the neutral loci from the selected site and the strength of LD is actually more marked in the simulations (see below) than in the theoretical predictions. Qualitatively similar patterns are predicted when the neutral loci are only partially linked (data not shown).
approaches one, the expected LD decreases toward neutral expectation. The nonmonotonic relationship between the distance of the neutral loci from the selected site and the strength of LD is actually more marked in the simulations (see below) than in the theoretical predictions. Qualitatively similar patterns are predicted when the neutral loci are only partially linked (data not shown).
Figure 4.—

The effects of a nearby selective sweep on a pair of completely linked neutral mutations (A) when only neutral mutations within the neutral phase are considered and (B) when neutral mutations can also occur during the selected phase. In each plot the solid line and shaded circles indicate numerical evaluation of Equation 6 and the dotted line and solid circles indicate the results of stochastic simulation carried out under the assumed model (i.e., no coalescence is allowed during the selective phase). Note the dramatic effect of including recent neutral mutations; there is a monotonic decline of LD between old neutral mutations as they get further from the selected site. However, because recent mutations have little or no LD, their inclusion results in LD between the neutral loci maximizing at some distance from the selected site. For each point, 105 simulations were carried with a sample size of 20 and with a recombination rate between the neutral loci of zero. (A) 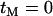 and (B)
and (B) 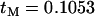 (note that under the simulation scheme the two series do not agree for
(note that under the simulation scheme the two series do not agree for 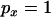 because of the additional time in the tips of the trees in B).
because of the additional time in the tips of the trees in B).
STOCHASTIC SIMULATION
To examine the accuracy of the results obtained here, Monte Carlo simulations were performed under two different models for the selective sweep. In series A, the effects of a selective sweep were simulated under the approximate model used as the basis of the analytical results. Specifically the genealogical history is divided into two phases: a phase of duration 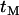 during which the only events that can occur are recombination events that move lineages from the selected to the wild-type background, a point of instant coalescence between all lineages still carrying the selected allele, and a neutral phase. In series B, fully stochastic models of selective sweeps were simulated using the program SelSim (Spencer and Coop 2004). Briefly, the method first simulates a stochastic trajectory for the selected mutation backward in time using a diffusion approximation (Coop and Griffiths 2004) and then subsequently performs a structured coalescent simulation conditional on the trajectory. By performing the two series of simulations it is possible to examine both the accuracy of Equation 6 as an approximation to the expectation of
during which the only events that can occur are recombination events that move lineages from the selected to the wild-type background, a point of instant coalescence between all lineages still carrying the selected allele, and a neutral phase. In series B, fully stochastic models of selective sweeps were simulated using the program SelSim (Spencer and Coop 2004). Briefly, the method first simulates a stochastic trajectory for the selected mutation backward in time using a diffusion approximation (Coop and Griffiths 2004) and then subsequently performs a structured coalescent simulation conditional on the trajectory. By performing the two series of simulations it is possible to examine both the accuracy of Equation 6 as an approximation to the expectation of 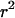 and the accuracy of the approximate model for selective sweeps. For efficiency simulations were carried out by placing mutations uniformly on the simulated genealogies at loci x and y and the ith simulation was assigned a weight given by the product of the total branch lengths at each site,
and the accuracy of the approximate model for selective sweeps. For efficiency simulations were carried out by placing mutations uniformly on the simulated genealogies at loci x and y and the ith simulation was assigned a weight given by the product of the total branch lengths at each site, 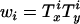 . Expected values of
. Expected values of 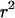 are estimated from the weighted average over ≥105 simulations for each parameter combination.
are estimated from the weighted average over ≥105 simulations for each parameter combination.
Where the selected site separates the two neutral loci the extent of association between the neutral loci in the series A simulations was, as predicted, no higher than background (data not shown). When the selected site does not separate the neutral loci the results are highly sensitive to assumptions about the duration of the selective phase (Figure 4, A and B; note that there is no recombination between the neutral loci). In Figure 4A it was assumed that the age of the selected mutation was negligible compared to the age of the neutral genealogy, 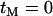 . In Figure 4B, the age of the selected mutation was fixed at
. In Figure 4B, the age of the selected mutation was fixed at 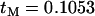 , the average obtained by fully stochastic simulation with S = 400 and a sample size of 20. There are two key features of these results. First, in both cases Equation 6 typically overestimates the expected value of
, the average obtained by fully stochastic simulation with S = 400 and a sample size of 20. There are two key features of these results. First, in both cases Equation 6 typically overestimates the expected value of 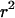 , although the expression is accurate when the probability of escape is low. The second key point is the difference the inclusion of recent neutral mutations makes. As predicted, mutations older than the selected one do typically show very strong LD. However, when the probability of escaping the selective sweep is very low, recent neutral mutations make the majority contribution to LD, such that the average value of
, although the expression is accurate when the probability of escape is low. The second key point is the difference the inclusion of recent neutral mutations makes. As predicted, mutations older than the selected one do typically show very strong LD. However, when the probability of escaping the selective sweep is very low, recent neutral mutations make the majority contribution to LD, such that the average value of 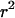 is very low.
is very low.
Figure 5 shows the comparison between the analytical results and the average value of 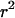 calculated from the fully stochastic simulations. These give qualitatively the same results as those obtained under the approximate model of a selective sweep. When the selected mutation separates the neutral mutations there is no LD between them (Figure 5A), irrespective of the level of diversity observed. When the selected site does not separate the neutral loci the LD between linked neutral loci is zero when the proximal locus is very close to the selected site, increases beyond its neutral expectation as the probability of escape increases, and then decreases back to the neutral expectation. This feature is seen both when the neutral loci are completely linked (Figure 5B) and when they are only partially linked (Figure 5C). The most notable difference between the two series of simulations is that in series A the approximation was a considerable overestimate of the true LD, whereas in series B it is typically a slight underestimate. In the absence of a selective sweep Equation 6 is typically an overestimate of
calculated from the fully stochastic simulations. These give qualitatively the same results as those obtained under the approximate model of a selective sweep. When the selected mutation separates the neutral mutations there is no LD between them (Figure 5A), irrespective of the level of diversity observed. When the selected site does not separate the neutral loci the LD between linked neutral loci is zero when the proximal locus is very close to the selected site, increases beyond its neutral expectation as the probability of escape increases, and then decreases back to the neutral expectation. This feature is seen both when the neutral loci are completely linked (Figure 5B) and when they are only partially linked (Figure 5C). The most notable difference between the two series of simulations is that in series A the approximation was a considerable overestimate of the true LD, whereas in series B it is typically a slight underestimate. In the absence of a selective sweep Equation 6 is typically an overestimate of 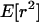 , as it also is when the approximate model is used as the basis of stochastic simulation (Figure 4). The most likely explanation for the underestimate in Figure 5 is that the genealogy under the selected mutation is not star shaped, and hence there can be significant LD between neutral mutations that occur during the selective phase. Indeed, as the sample size increases, the approximation of a star-like genealogy in the selective phase becomes progressively worse (Durrett and Schweinsberg 2004).
, as it also is when the approximate model is used as the basis of stochastic simulation (Figure 4). The most likely explanation for the underestimate in Figure 5 is that the genealogy under the selected mutation is not star shaped, and hence there can be significant LD between neutral mutations that occur during the selective phase. Indeed, as the sample size increases, the approximation of a star-like genealogy in the selective phase becomes progressively worse (Durrett and Schweinsberg 2004).
Figure 5.—

The effects of a selective sweep on patterns of LD if the selected site is either separating (A) or adjacent to (B and C) the neutral loci. In each plot the shaded line indicates the prediction of Equation 6 allowing for finite sample size but not for recent mutations, the solid line indicates the prediction of Equation 6 allowing for finite sample size and for recent mutations, and the solid circles and dotted line show the values obtained by fully stochastic simulation. The configuration relating to each plot is shown in the top right corner (triangles, neutral loci; circles, selected loci). (A) The selected site is at the midpoint between the two selected loci, which are separated by the recombination fraction shown. (B) The selected site is adjacent to the neutral loci at the recombination fraction indicated beyond the proximal locus and the two neutral loci are completely linked. (C) The same as B except the two neutral loci are separated by a recombination fraction of 4Ner = 5. In all cases 106 simulations were performed with a sample size of 20 and a scaled selection coefficient of 4Ns = 400 using the SelSim package (Spencer and Coop 2004).
In summary, the stochastic simulations demonstrate that the combination of Equation 6 and the approximate model of a selective sweep provides a reasonably accurate quantitative prediction of the effects of selective sweeps on the average value of r2. They do not, of course, predict the full distribution and the approximation gets progressively worse for weaker selection coefficients (data not shown). Informally, the approximation appears to be valuable for S > 100.
DISCUSSION
The results presented here provide a detailed understanding of the effects of selective sweeps on patterns of linkage disequilibrium, particularly for the case where a mutation of large effect has recently reached fixation in the population. Although previous theoretical and simulation-based studies have demonstrated some of the patterns described, the genealogical perspective taken provides an intuitive approach to understanding key features of the process. In particular, two key features can be identified.
Selective sweeps can eliminate LD:
If a selective sweep is sufficiently strong and recent, such that the genealogy of the sample at the selected site can be approximated as a star (i.e., all lineages coalesce at the same time), all LD between neutral loci separated by the selected site is eliminated. As previously noted (Kim and Nielsen 2004), there is a simple genealogical explanation for this observation. In effect, the genealogical interpretation of LD implies that significant LD will occur when the coalescent time for a pair of chromosomes at one position on a chromosome is informative about the coalescent time for the same pair of chromosomes at another position (relative to the coalescent time of all other pairs of chromosomes). Within a star-like genealogy all pairs of chromosomes coalesce at the same time. Consequently the coalescent time for a given pair at one point is uninformative about the coalescent time at any other point for the same pair (i.e., there is no variance in coalescence time within the star), and there is no LD. Moving away from the selected site recombination events will allow linked neutral sites to revert to the neutral distribution of genealogies. However, such “recovery” from the star-like genealogy happens independently on the two sides of the selected site. Consequently, the coalescent time for a pair of chromosomes on one side of the selected site will always be uninformative about the coalescent time for the same pair of chromosomes on the other side.
What is the implication of this result for understanding patterns of variation? The most obvious issue is that selective sweeps, through abolishing LD, may create patterns that look like recombination hotspots. Indeed, it has been shown that one statistical test for hotspots does have an elevated false positive rate at selective sweeps (Reed and Tishkoff 2005). However, it should be noted that the patterns of genetic variation (and underlying genealogies) associated with a hotspot and those associated with a selective sweep are strikingly different. In humans, hotspots are typically short (1–2 kb) regions where there is a very rapid breakdown in LD, and there are many “detectable” recombination events and no distortion to the distribution of marginal genealogies (i.e., no distortion to the frequency distribution of neutral variation) (Jeffreys et al. 2001). In contrast, a selective sweep of considerable strength will affect the density and frequency distribution of polymorphism over considerable distances. For example, a scaled selection coefficient of 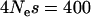 (a selection coefficient of ∼1% in humans) will affect the frequency distribution of polymorphism up to a genetic distance of at least
(a selection coefficient of ∼1% in humans) will affect the frequency distribution of polymorphism up to a genetic distance of at least  on either side (this is the distance at which there is a 50% chance of lineage escaping the sweep). In humans, the average recombination rate is ∼
on either side (this is the distance at which there is a 50% chance of lineage escaping the sweep). In humans, the average recombination rate is ∼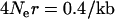 in European populations (Myers et al. 2005), such that a region some 140 kb in size should be strongly affected. In short, even if a sweep does influence LD in such a way as to resemble a hotspot, the sweep is also likely to lead to unusual patterns of variation that are indicative of a selective sweep.
in European populations (Myers et al. 2005), such that a region some 140 kb in size should be strongly affected. In short, even if a sweep does influence LD in such a way as to resemble a hotspot, the sweep is also likely to lead to unusual patterns of variation that are indicative of a selective sweep.
One way to ask the question of whether selective sweeps can create false hotspots is to ask whether, conditioning on seeing polymorphism at given genetic distances on either side of the selected mutation, the evidence for historical recombination is greater or less than under the neutral model. Table 1 shows how selective sweeps influence the probability of seeing all four possible haplotypes relative to the neutral case. Under the infinite-sites model such data sets are direct evidence for recombination (Hudson and Kaplan 1985). The patterns are quite striking: sweeps lead to a dramatic decrease in the probability of observing all four haplotypes relative to the neutral model. This is true whether all mutations are considered or just those >10% in frequency. In short, selective sweeps do not lead to any increase in the evidence for recombination. The reported bias to one method for detecting hotspots (Reed and Tishkoff 2005) therefore is likely to result from the fact that this method uses a nongenealogical model for patterns of variation. Analysis of data sets simulated with selective sweeps indicates that coalescent-based estimators of the recombination rate show no such local increase in estimated rate. Rather, the depression in the opportunity for recombination at such sites also leads to a slight decrease in average estimated rate (Figure 6).
TABLE 1.
Probability of observing an incompatibility across a selective sweep
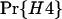 a a
|
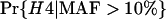
|
|||
|---|---|---|---|---|
| 4Nerb | 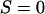 |
 |
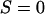 |
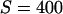 |
| 0.1 | 0.0019 | 0.00007 | 0.0044 | 0.00062 |
| 0.2 | 0.0038 | 0.00016 | 0.0094 | 0.0012 |
| 0.4 | 0.0086 | 0.00031 | 0.020 | 0.0028 |
| 1 | 0.024 | 0.0011 | 0.053 | 0.0095 |
| 2 | 0.051 | 0.0030 | 0.11 | 0.024 |
| 4 | 0.098 | 0.0087 | 0.21 | 0.055 |
| 10 | 0.18 | 0.035 | 0.36 | 0.15 |
| 20 | 0.23 | 0.086 | 0.46 | 0.29 |
| 40 | 0.26 | 0.17 | 0.53 | 0.44 |
| 100 | 0.29 | 0.28 | 0.59 | 0.58 |
Estimated from 106 simulations with n = 20, conditioning on segregation at both neutral loci.
The selected locus is at the midpoint of the neutral loci, which are separated by the recombination distance shown. Note that neutral simulations are conditioned on fixation of a neutral mutation.
Figure 6.—

The effect of selective sweeps on estimates of the recombination rate. For data sets previously simulated with a selective sweep (the position of which is indicated by the vertical bar) and constant recombination rate (R = 10; indicated by the dotted line) (Reed and Tishkoff 2005), a model of variable rate recombination was fitted using the reversible-jump MCMC method of McVean et al. (2004), using a block penalty of 5. Four series of data sets were analyzed, each of 100 replicates, with S = 4Nes = 10, 100, 177.8, and 562.3, respectively (θ = 10 and n = 100 for each). Each chart shows the median (solid line) and quartiles (shaded lines) of the distribution of the estimated rate. In no case is there a tendency to overestimate the recombination rate.
Selective sweeps can increase (and decrease) LD:
While LD between neutral loci is eliminated by a selective sweep at an intervening site, if the selected site does not separate the neutral loci LD can be increased or decreased depending on their proximity to the selected site. A further complication is that the age of the neutral mutations relative to the selected one has critical consequences for the magnitude of LD. If both neutral loci are closely linked to the selected site, mutations older than the selected one will typically show strong LD and younger mutations will typically have little or no LD. When both features are combined the result is a nonmonotonic relationship between the proximity of a pair of neutral loci to a selected one and the strength of LD.
What are the implications of these results for the interpretation of empirical patterns of genetic variation? Previous work has suggested that incorporating information on LD does not greatly improve the power of statistical approaches to identifying selective sweeps (Kim and Nielsen 2004). This result is understandable given the complexity of the patterns described. One possibility is that incorporating information about the age of linked neutral polymorphism (for example, by comparison with related populations in which no sweep is thought to have occurred) may increase the power to detect selection. In particular, sweeps will lead to series of old SNPs at low frequency and in strong LD interleaved with series of young SNPs at low frequency and in very low LD. Of course, inferences about the age of a mutation within the population that has experienced selection will be confounded by the effect of the sweep.
One argument against using patterns of LD directly to make inferences about selective sweeps is that their effects on LD can all be understood in terms of the generation of a star-like genealogy at the selected site. Consequently, the most powerful methods for detecting selective sweeps will be those that are most powerful at detecting local star-like genealogies with short times to the MRCA (Kim and Stephan 2002; Kim and Nielsen 2004; Nielsen et al. 2005). For example, of existing methods to detect recent, complete selective sweeps, perhaps the most powerful is one that compares models with and without a local star-like genealogy at a putatively selected site using only the allele-frequency distribution (Nielsen et al. 2005). However, what the results presented here show is that selective sweeps can induce unusual patterns of association between neutral mutations near selected sites, a feature that is currently not considered in this method. In effect, the results suggest that there may be additional information about selective sweeps in the way genetic variation recovers around a selected locus; however, it remains to be seen whether such recovery differs systematically from cases where star-like genealogies have occurred by chance or through population bottlenecks.
Acknowledgments
I thank Nick Barton, Alison Etheridge, Rasmus Nielsen, Jay Taylor, and two anonymous reviewers for discussion and comments on the manuscript and Wolfgang Stephan for providing the original inspiration for this work.
References
- Barton, N. H., 1998. The effect of hitch-hiking on neutral genealogies. Genet. Res. 72: 123–133. [Google Scholar]
- Braverman, J. M., R. R. Hudson, N. L. Kaplan, C. H. Langley and W. Stephan, 1995. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics 140: 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop, G., and R. C. Griffiths, 2004. Ancestral inference on gene trees under selection. Theor. Popul. Biol. 66: 219–232. [DOI] [PubMed] [Google Scholar]
- Durrett, R., and J. Schweinsberg, 2004. Approximating selective sweeps. Theor. Popul. Biol. 66: 129–138. [DOI] [PubMed] [Google Scholar]
- Etheridge, A. M., P. Pfaffelhuber and A. Wakolbinger, 2006. An approximate sampling formula under genetic hitchhiking. Ann. Appl. Probab. 16: 685–729. [Google Scholar]
- Fay, J. C., and C. I. Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., and W. H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., and A. Robertson, 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38: 226–231. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1985. The sampling distribution of linkage disequilibrium under an infinite allele model without selection. Genetics 109: 611–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111: 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., K. Bailey, D. Skarecky, J. Kwiatowski and F. J. Ayala, 1994. Evidence for positive selection in the superoxide dismutase (Sod) region of Drosophila melanogaster. Genetics 136: 1329–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys, A. J., L. Kauppi and R. Neumann, 2001. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat. Genet. 29: 217–222. [DOI] [PubMed] [Google Scholar]
- Kaplan, N. L., R. R. Hudson and C. H. Langley, 1989. The “hitchhiking effect” revisited. Genetics 123: 887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and R. Nielsen, 2004. Linkage disequilibrium as a signature of selective sweeps. Genetics 167: 1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160: 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- McVean, G. A., 2002. A genealogical interpretation of linkage disequilibrium. Genetics 162: 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean, G. A., S. R. Myers, S. Hunt, P. Deloukas, D. R. Bentley et al., 2004. The fine-scale structure of recombination rate variation in the human genome. Science 304: 581–584. [DOI] [PubMed] [Google Scholar]
- Myers, S., L. Bottolo, C. Freeman, G. McVean and P. Donnelly, 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., S. Williamson, Y. Kim, M. J. Hubisz, A. G. Clark et al., 2005. Genomic scans for selective sweeps using SNP data. Genome Res. 15: 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., and M. Kimura, 1971. Linkage disequilibrium between two segregating nucleotide sites under the steady flux of mutations in a finite population. Genetics 68: 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluzhnikov, A., and P. Donnelly, 1996. Optimal sequencing strategies for surveying molecular genetic diversity. Genetics 144: 1247–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., 2002. The signature of positive selection at randomly chosen loci. Genetics 160: 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, F. A., and S. A. Tishkoff, 2005. Positive selection can create false hotspots of recombination. Genetics 172: 2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti, P. C., D. E. Reich, J. M. Higgins, H. Z. Levine, D. J. Richter et al., 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837. [DOI] [PubMed] [Google Scholar]
- Song, Y. S., and J. S. Song, 2007. Analytic computation of the expectation of the linkage disequilibrium coefficient r2. Theor. Popul. Biol. 71: 49–60. [DOI] [PubMed] [Google Scholar]
- Spencer, C. C., and G. Coop, 2004. SelSim: a program to simulate population genetic data with natural selection and recombination. Bioinformatics 20: 3673–3675. [DOI] [PubMed] [Google Scholar]
- Stephan, W., T. Wiehe and M. W. Lenz, 1992. The effect of strongly selected substitutions on neutral polymorphism: analytical results based on diffusion theory. Theor. Popul. Biol. 41: 237–254. [Google Scholar]
- Stephan, W., Y. S. Song and C. H. Langley, 2006. The hitchhiking effect on linkage disequilibrium between linked neutral loci. Genetics 172: 2647–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobeck, C., and K. Morgan, 1978. The effect of intragenic recombination on the number of alleles in a finite population. Genetics 88: 829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., and S. Lessard, 2003. Theory of the effects of population structure and sampling on patterns of linkage disequilibrium applied to genomic data from humans. Genetics 164: 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]