

Abstract
The polyketides are a diverse group of natural products with great significance as human and veterinary pharmaceuticals. A significant barrier to the production of novel genetically engineered polyketides has been the lack of available heterologous expression systems for functional polyketide synthases (PKSs). Herein, we report the expression of an intact functional PKS in Escherichia coli and Saccharomyces cerevisiae. The fungal gene encoding 6-methylsalicylic acid synthase from Penicillium patulum was expressed in E. coli and S. cerevisiae and the polyketide 6-methylsalicylic acid (6-MSA) was produced. In both bacterial and yeast hosts, polyketide production required coexpression of 6-methylsalicylic acid synthase and a heterologous phosphopantetheinyl transferase that was required to convert the expressed apo-PKS to its holo form. Production of 6-MSA in E. coli was both temperature- and glycerol-dependent and levels of production were lower than those of P. patulum, the native host. In yeast, however, 6-MSA levels greater than 2-fold higher than the native host were observed. The heterologous expression systems described will facilitate the manipulation of PKS genes and consequent production of novel engineered polyketides and polyketide libraries.
Polyketide synthases (PKSs) are large multifunctional enzymes that catalyze the synthesis of the carbon backbones of a class of natural products known as polyketides (for reviews, see refs. 1 and 2). There are several thousand known polyketides that share basic mechanisms of biosynthesis. PKSs, like fatty acid synthases, catalyze repeated condensations of acyl CoA esters to build a linear polyketide chain. At all stages of biosynthesis, the polyketide chain is tethered as a thiol ester to a Cys residue of a ketosynthase domain or to a phosphopantetheine moiety of an acyl carrier protein-like domain of the PKS; ultimately, the polyketide chain is released from the PKS by cleavage of the thiol ester, usually accompanied by cyclization. By varying chain length, acyl CoA substrates, reductive modifications, and stereochemistry at each condensation step, nature has achieved an enormous structural diversity in evolving the polyketide natural products.
There are three basic types of PKSs. Modular PKSs contain a separate active site or catalytic domain for every reaction catalyzed in the biosynthetic pathway, and the multiple domains are contained on only a few polypeptide chains. In contrast, bacterial aromatic PKSs have only a few active sites on separate polypeptide chains, and the active sites are used iteratively. Finally, fungal PKSs are of the iterative type, but the active sites are found on a single polypeptide chain.
Because of the clinical significance of this class of compounds, there is considerable interest in developing systems for the production of novel designed polyketides, sometimes referred to as “unnatural” natural products (3, 4). Such systems involve (i) the exchange of sequences encoding one or more activities from one PKS gene to another to produce a hybrid gene and (ii) expression of the hybrid gene to form a PKS and its polyketide product. Thus far heterologous production of polyketides has been limited to organisms closely related to natural hosts, and in Streptomyces spp., especially Streptomyces coelicolor. Both modular and aromatic PKS genes have been functionally expressed in S. coelicolor (5, 6), and recently the expression of the fungal PKS 6-methylsalicylic acid synthase (6-MSAS) has been reported in this organism (7). Although the S. coelicolor expression system permits manipulation of the polyketide biosynthetic pathway, certain methodologies such as transformation, genetic manipulation, and cell culture are cumbersome and have unpredictable outcomes, making this organism a less than ideal host.
Clearly, heterologous expression of functional PKSs in organisms such as Escherichia coli or Saccharomyces cerevisiae would be advantageous because it would enable use of the advanced knowledge and technology available with these organisms. Thus far, only fragments of PKS genes have been expressed in E. coli (8, 9), and the acyl carrier protein domains were only partially modified, or in some cases not modified at all, with the essential 4′-phosphopantetheine cofactor (Scheme 1 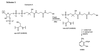 ). Presumably, the modification was not efficiently catalyzed by the endogenous E. coli holo-acyl carrier protein synthase. A similar lack of posttranslational modification prevented heterologous expression of functional nonribosomal peptide synthases (10). Recently, a number of phosphopantetheinyl (P-pant) transferases of various specificity have been reported (11), and it has been shown that coexpression of an appropriate P-pant transferase with a nonribosomal peptide synthase module in E. coli yields the modified protein (12). It seemed reasonable, therefore, to coexpress PKSs with one or more of the recently reported P-pant transferases to produce a holo-PKS capable of polyketide production.
). Presumably, the modification was not efficiently catalyzed by the endogenous E. coli holo-acyl carrier protein synthase. A similar lack of posttranslational modification prevented heterologous expression of functional nonribosomal peptide synthases (10). Recently, a number of phosphopantetheinyl (P-pant) transferases of various specificity have been reported (11), and it has been shown that coexpression of an appropriate P-pant transferase with a nonribosomal peptide synthase module in E. coli yields the modified protein (12). It seemed reasonable, therefore, to coexpress PKSs with one or more of the recently reported P-pant transferases to produce a holo-PKS capable of polyketide production.
In the present work, we describe the coexpression of the fungal PKS 6-MSAS from Penicillium patulum, with the surfactin P-pant transferase (Sfp) from Bacillus subtilis, in E. coli and S. cerevisiae. The transformed bacterial and yeast cells produced large amounts of the polyketide product 6- methylsalicylic acid (6-MSA, Scheme 1).
EXPERIMENTAL PROCEDURES
Materials.
Plasmids pQE60 and pET22(+) were from Qiagen (Chatsworth, CA) and Novagen, respectively. S. cerevisiae InvSc1(MATα his3Δ1 leu2 trp1–289 ura3–52) was from Invitrogen and made competent by lithium acetate treatment, by using a kit purchased from Invitrogen. The yeast–E. coli shuttle plasmid pYT was a gift from S. Hawkes (University of California, San Francisco). YepFLAG-1 was obtained from Kodak. pUC8-sfp, carrying the P-pant transferase gene for surfactin biosynthesis (sfp), has been described (13). E. coli SJ16 was obtained from the E. coli Genetic Stock Center (New Haven, CT.) Unless otherwise specified, procedures involving yeast growth and manipulation were performed as described (14).
Construction of S. cerevisiae Shuttle Vectors.
The alcohol dehydrogenase 2 (ADH2) promoter was amplified from yeast genomic DNA by PCR. The forward primer 5′-GGGAGCTCGGATCCATTTAGCGGCCGCAAAACGTAGGGGC contained 15 bases complementary to the 5′ ADH2 sequence (boldface type) and introduced SacI (nucleotides 3–8), BamHI (nucleotides 9–14), and NotI (nucleotides 20–27) restriction sites. The reverse primer 5′ CCGAATTCTAGAGGTTTCATATGGTATTACGATATAGTTAATAG contained 21 bases complementary to the 3′ ADH2 sequence (boldface type) and introduced NdeI (nucleotides 18–23), XbaI (nucleotides 7–12), and EcoRI (nucleotides 3–8) sites. The forward primer for the terminator 5′-GGGAATTCATAGTCGACCGGACCGATGCCTTCACGATTTATAG introduced EcoRI (nucleotides 3–8), SalI (nucleotides 12–17), and RsrII (nucleotides 17–24) restriction sites and the reverse primer 5′-TTTTCTATTATAAGATGAAAAACGAGGGGAGCTCCCATGGCC introduced XhoI (nucleotides 29–34) and Asp718 (nucleotides 35–40) sites. Nucleotides complementary to the ADH2 terminator region are shown in boldface type.
The PCR-amplified ADH2 promoter and terminator were introduced into pBluescript SK(−) by a three-fragment ligation of the SacI/EcoRI promoter, the EcoRI/Asp718 terminator and the 2.8-kbp large fragment of SacI/Asp718-digested p-Bluescript. The resulting plasmid pKOS12–43d2 (Fig. 1) contained cloning sites (L2) for 6-MSAS and sfp genes, sites (L1 and L3) for transferring the promoter/terminator cassette into yeast shuttle vectors, and sites (L1 and L2) for moving the promoter/gene cassettes from the intermediate Bluescript vector to the yeast shuttle vector.
Figure 1.

Construction of yeast expression vectors. L1, L2, and L3 contain the following restriction sites: BamHI and NotI (L1); NdeI, XbaI, EcoRI, SalI, and RsrII (L2); and XhoI (L3).
For construction of the yeast vector pKOS12–101c, the ADH2 promoter/terminator cassette was moved as a BamHI–XhoI fragment from pKOS12–43d2 to the BamHI/SalI-digested pYT. To construct the Trp vector, a DNA linker containing BamHI, NotI, NcoI, RsrII, XhoI, and SalI sites was first introduced into the PacI/NotI-digested YepFLAG-1 yeast shuttle vector to give pKOS12–48a; next, the ADH2 promoter/terminator was moved as a BamHI/XhoI cassette from pKOS12–43d2 to the BamHI–SalI large fragment of pKOS12–48a to give pKOS12–126b.
Construction of S. cerevisiae Expression Vectors.
The ORF encoding 6-MSAS (7) was cloned into the Bluescript vector pKOS12–43d2 as a NdeI–XbaI fragment to give pKOS12–71d and moved into the modified pYT vector pKOS12–101c as a NotI/RsrII cassette to produce pKOS12–102d (see Fig. 1). The sfp gene was amplified by PCR from the plasmid pUC8-sfp, by using the forward primer 5′-TAGACACATATGAAGATTTACGGAATTTATATG, which introduced a NdeI restriction site (nucleotides 7–12), and the reverse primer 5′-TACATTCTAGAAATTATAAAAGCTCTTCG, which introduced an XbaI site (nucleotides 6–11); sequences complementary to the sfp gene are shown in boldface type. The amplified sfp gene was cloned into pKOS12–43d2 as a NdeI–XbaI fragment to give pKOS12–109c, and the ADH2 promoter/sfp cassette was moved as a BamHI–SalI fragment to the yeast shuttle vector pKOS12–126b to give pKOS12–128a.
Expression of 6-MSAS and Sfp in S. cerevisiae.
Competent yeast cells were transformed with the appropriate plasmids and plated on minimal medium lacking Ura, Trp, or both (14). Transformants appeared after 48–72 hr at 30°C. To prepare the 6-MSAS/sfp double transformant, competent yeast cells harboring the sfp vector pKOS12–128a were transformed with pKOS12–102d harboring the 6-MSAS gene. Transformants were selected on minimal medium lacking Trp and Ura.
Minimal medium lacking the appropriate nutritional requirement (Ura, Trp, or Ura and Trp) was inoculated with single yeast colonies harboring appropriate plasmids. Cultures were grown for 24–48 hr at 30°C, after which 100 μl was used to inoculate 10 ml of YPD medium. Cultures were grown for 18 hr at 30°C in an orbital shaker at 225 rpm. YPD medium (50 ml) was inoculated with 0.5 ml of the overnight cultures and incubated at 30°C for 142 hr. One-milliliter aliquots were removed at various times, and cells were collected by centrifugation. The cells were suspended in SDS/PAGE loading buffer, boiled for 2 min, and subjected to SDS/PAGE. Supernatants from each aliquot were assayed for glucose and for 6-MSA.
Expression of 6-MSAS in E. coli.
The 1.1-kbp XhoI–XbaI fragment of pQE60 was ligated with the 5-kbp XhoI–XbaI fragment of pET22b(+) to give pKOS007–90, containing the T5 promoter, two lac operators, and lacIq. A linker containing NdeI and SpeI restriction sites was inserted into the EcoRI/HindIII-digested pKOS007–90 to give pKOS007–95 in which the NdeI sequence replaced the NcoI of pKOS007–90. The 6-MSAS expression vector pKOS007–109 was constructed by ligating a NdeI–XbaI fragment containing the 6-MSAS ORF (7) to the large NdeI–SpeI fragment of pKOS007–95, leaving about 1 kbp of the linker between the SpeI and HindIII sites of the vector.
The sfp gene and the lac promoter were excised as a 1.1-kbp EcoRI–PvuII fragment from pUC8-sfp, filled in with DNA polymerase I, and cloned into the EcoRV site of pACYC-184 to form pKOS007–108. The orientation of the sfp gene with respect to the promoter was verified by HindIII digestion.
E. coli C2435 was cotransformed with both pKOS007–108 and pKOS007–109. ATCC medium 765 (15) supplemented with 10% glycerol was inoculated with a single colony, and the culture was grown until the OD600 reached 1.0. Gene expression was induced by addition of isopropyl β-d-thiogalactoside to 0.5 mM (final concentration), and the culture was incubated for 24 hr at 30°C. Protein expression was monitored by SDS/PAGE (10% gels) of total cellular protein, followed by Coomassie staining.
Identification and Characterization of 6-MSA.
S. cerevisiae and E. coli cultures were monitored for production of 6-MSA as follows. One-milliliter aliquots of cultures were cleared of cells by centrifugation, and supernatants were analyzed by HPLC. HPLC was performed on a Beckman System Gold Chromatograph fitted with a C18 reverse-phase column (Beckman Ultrasphere, 2 × 150 mm). The liquid chromatography parameters were as follows: solvent A, 1% acetic acid in water; solvent B, 1% acetic acid in acetonitrile; gradient, 20% B to 80% B in 30 min, then to 100% in 2 min; flow rate, 0.5 ml/min; detection by diode-array UV spectroscopy (Beckman noveau Software for Windows). In some experiments, the effluent from the HPLC column was directed to an on-line mass spectrometer (PE Sciex API 100 LC), equipped with multiview software for the Macintosh. The concentration of 6-MSA in cell-free medium was determined by comparing the area of the HPLC peak at 306 nm to that of authentic 6-MSA.
For large-scale preparation of 6-MSA, a 500-ml yeast culture harboring 6-MSAS and sfp expression vectors was grown for 120 hr, and the cells were collected by centrifugation. The supernatant (280 ml) was acidified with 28 ml of glacial acetic acid and extracted with 280 ml of ethyl acetate. The extract was concentrated to dryness under reduced pressure, the residue was recrystallized from water (16), and the crystals were dried under vacuum over KOH. 1H and 13C NMR were performed on a Varian XL-400 spectrometer. The liquid chromatography, MS, and NMR characteristics of 6-MSA produced in S. cerevisiae (all characteristics) and E. coli (liquid chromatography and MS) were compared with those of authentic 6-MSA.
RESULTS
Production of 6-MSA in S. cerevisiae.
Growth of S. cerevisiae harboring the 6-MSAS expression plasmid pKOS12–102d for ≥48 hr resulted in expression of a protein that migrated on SDS/PAGE gels at ∼190 kDa, the predicted size of 6-MSAS (17) (Fig. 2A, lanes 2–5). The 190-kDa protein was not expressed in yeast containing the control plasmid pKOS12–101c (Fig. 2A, lane 1). Likewise, yeast harboring the sfp expression plasmid, pKOS12–128a, produced an ∼30-kDa protein (Fig. 2B, lane 2), consistent with the predicted size of Sfp (13), which was not present in yeast harboring the control plasmid pKOS12–126b (Fig. 2B, lane 1). Yeast transformed with both 6-MSAS and sfp expression plasmids produced both ∼190-kDa and ∼30-kDa protein products (Fig. 2C).
Figure 2.

(A) Expression of 6-MSAS in S. cerevisiae. Yeast harboring control plasmid pKOS12–101c (lane 1) or the 6-MSAS expression plasmid pKOS12–102d were grown for 48 (lane 5), 72 (lane 4), 96 (lane 3), or 116 (lane 2) hr. (B) Expression of Sfp in S. cerevisiae. Yeast harboring control plasmid pKOS12–126b (lane 1) or the sfp expression plasmid pKOS12–128a were grown for 92 hr (lane 2). (C) Expression of Sfp and 6-MSAS in S. cerevisiae. Yeast harboring the sfp expression plasmid pKOS12–126b and the control plasmid pKOS12–101c (lane 1) or pKOS12–126b and the 6-MSAS expression plasmid pKOS12–102d (lane 2) were boiled in loading buffer and subjected to SDS/PAGE. Proteins were stained with Coomassie blue. Arrows indicate the positions of Sfp and 6-MSAS. Molecular mass markers are indicated in kDa.
The production of 6-MSA by yeast harboring both 6-MSAS and sfp expression plasmids was monitored by HPLC of cell-free supernatants. Yeast harboring the control plasmid pKOS12–101c or the control plasmid and the sfp expression plasmid pKOS12–128a produced no 6-MSA (Fig. 3, traces b and d). Yeast expressing 6-MSAS produced a barely detectable amount of 6-MSA (Fig. 3, trace c). Yeast expressing both 6-MSAS and Sfp produced as much as 1.7 g of 6-MSA per liter of culture (Fig. 3, trace a).
Figure 3.

HPLC of 6-MSA. Traces: a, from 5 μl of cell-free extract from yeast harboring both the 6-MSAS (pKOS12–102d) and sfp (pKOS12–128a) expression plasmids; b, from 20 μl of cell-free extract from yeast harboring the control plasmid pKOS12–101c; c, pKOS12–102d; d, pKOS12–128a and pKOS12–101c. AU = absorbance units at 306 nm.
The kinetics of yeast growth and 6-MSA production for the double transformant (6-MSAS and sfp) are shown in Fig. 4A. The commencement of 6-MSAS/Sfp expression and 6-MSA production coincided with glucose depletion, consistent with derepression of the ADH2 promoter. The amount of 6-MSA produced reached a plateau after approximately 60 hr of growth and remained constant up to 150 hr.
Figure 4.

Kinetics of 6-MSA production in S. cerevisiae and E. coli. (A) Growth of S. cerevisiae (□, OD600) and 6-MSA production (♦, 6-MSA in g/liter of cell-free medium). Glucose was depleted after ∼16 hr of growth. (B) 6-MSA production (6-MSA in g/liter of cell-free medium) in E. coli.
The 6-MSA produced by S. cerevisiae comigrated on HPLC with authentic 6-MSA (retention time = 20.1 min). Mass spectral analysis of the product gave a parent ion of 153 atomic mass units (M + H+), consistent with the molecular weight of 6-MSA. After extraction with ethyl acetate and recrystallization from water, 280 ml of cell-free yeast culture medium yielded 240 mg of 6-MSA as crystalline needles. 1H and 13C NMR analyses confirmed the presence of 6-MSA: (1H NMR [DMSO-d6 400 Mhz] δ [ppm] 2.33 [s, 3H], 6.70 [d, J = 7.6 Hz, 1H], 6.73 [d, J = 8.24 Hz, 1H], 7.18 [dd J = 7.6 Hz, 7.6 Hz, 1H], with tetramethylsilane as the internal reference). (13C NMR [DMSO-d6, 100 Mhz] δ [ppm] 20.9, 113.9, 119.0, 121.2, 131.4, 137.7, 157.5 and 171.0, with DMSO-d6 as the internal reference, where DMSO is dimethyl sulfoxide). The NMR data were consistent with data reported for 6-MSA (7).
Production of 6-MSA in E. coli.
Isopropyl β-d-thiogalactoside induction of E. coli transformed with the 6-MSAS expression vector pKOS007–109, followed by incubation at 37°C for 4 hr, resulted in the expression of the ∼190-kDa 6-MSAS at ∼5% of the total protein. However, most of the protein was insoluble, and 6-MSA was not detected by HPLC of the medium. When the β-Ala auxotroph E. coli SJ16 (18) containing pKOS007–109 was incubated with β-[3H]Ala before and after induction, no radioactivity was found in the 6-MSAS band by SDS/PAGE (data not shown). These data suggest that the protein was not modified with the essential 4′-phosphopantetheine cofactor by the endogenous P-pant transferase. In a similar experiment in which the sfp expression vector was also present, a detectable amount of radioactivity migrated as the 190-kDa 6-MSAS band in SDS/PAGE gels, indicating that Sfp catalyzed P-pant transfer to 6-MSAS. However, the low levels of soluble protein may have not been sufficient to produce detectable 6-MSA. The temperature of incubation was lowered to enhance proper protein folding, and glycerol was included in the medium to increase levels of the intracellular malonyl CoA substrate (19). When cells were grown at 30°C in the absence of glycerol or at 37°C in the presence of 10% glycerol, no 6-MSA was produced. Thus, neither of these parameters was solely responsible for the lack of 6-MSA production. However, at 30°C and in the presence of 10% glycerol, 6-MSA was produced at 75 mg/liter after about 24 hr of incubation (Fig. 4B).
DISCUSSION
One of the challenges facing the efficient generation of “unnatural” polyketides by manipulation of PKS genes is the development of heterologous high-level expression systems for fully functional PKSs. We investigated polyketide expression in E. coli and S. cerevisiae, well-established hosts for genetic transformation, protein expression, and high-density growth in liquid medium. Expression vectors were designed to contain the T5 RNA polymerase promoter in E. coli and the ADH2 promoter in yeast so that potentially toxic expression could be tightly regulated. The fungal PKS gene for 6-MSAS was chosen as a paradigm for heterologous PKS expression because it is available as a single ORF, its substrates—acetyl CoA, malonyl CoA, and NADPH—are present in the intended hosts, and its polyketide product, 6-MSA, is easily detectable. Furthermore, 6-MSAS purified to homogeneity catalyzes the production of 6-MSA in vitro, indicating that additional unknown host factors are not required for polyketide production (17).
In the present work, when E. coli harboring an expression vector containing the 6-MSAS gene was induced, the 190-kDa 6-MSAS was observed on SDS/PAGE gels but no 6-MSA was detectable in the medium. It has been reported that the endogenous E. coli fatty acid holo-acyl carrier protein synthase does not appear to efficiently modify PKSs (9), which provides one explanation for the lack of polyketide production by PKSs expressed in E. coli. We tested whether a relatively nonspecific P-pant transferase (i.e., Sfp, the B. subtilis P-pant transferase involved in the synthesis of surfactin; ref. 11) might recognize and appropriately modify PKSs. Indeed, we demonstrated that Sfp was capable of in vitro and in vivo phosphopantetheinylation of a fragment of the erythromycin PKS expressed in E. coli (unpublished data). We also demonstrated that appropriate posttranslational modification of the 6-MSAS PKS was achieved with Sfp in E. coli (data not shown). Nevertheless, upon analysis of the medium we could not detect the polyketide product MSA.
The production of an appropriately modified 6-MSAS that was nonfunctional seemed incongruent with the fact that purified 6-MSAS is catalytically active in vitro (17). Therefore, we sought to identify other factors that might be responsible for the lack of polyketide production. One possibility was that the host cells might not have sufficient levels of substrates—acyl CoA esters, and in particular malonyl CoA—to support 6-MSA synthesis. Intracellular levels of CoA esters in E. coli have been shown to increase significantly when cells are grown in glycerol-containing medium (19). Another possibility was that there was an insufficient amount of properly folded 6-MSAS to effect detectable 6-MSA production. Indeed, we found that the production of 6-MSA by E. coli occurred upon inclusion of glycerol in the medium and by lowering the temperature during protein expression. When cells were grown at 30°C and in 10% glycerol, 6-MSA was optimally produced at 75 mg/liter, which is some 10-fold lower than that observed in the natural host (17) and similar to that produced in the heterologous host S. coelicolor (7).
Concurrent with the above studies, we attempted expression of 6-MSAS and production of 6-MSA in S. cerevisiae. Induction of S. cerevisiae transformed with an expression vector containing the 6-MSAS gene resulted in the expression of 6-MSAS, but only a small amount (<0.75 μg/liter) of 6-MSA was observed in the medium. We assumed that, as with E. coli, PKSs are not efficient substrates for the endogenous yeast holo-acyl carrier protein synthase. When S. cerevisiae was transformed with both the 6-MSAS and sfp genes, coexpression resulted in the high-level production of 6-MSA in the medium. Up to 1.7 g of 6-MSA per liter was produced by the recombinant yeast, some 2-fold greater than that produced by the natural host P. patulum (17) and 25-fold greater than that by the heterologous host S. coelicolor harboring the 6-MSAS gene (7).
In summary, we have demonstrated expression of the 6-MSAS gene and high-level production of the polyketide 6-MSA in both E. coli and S. cerevisiae. The results show that 6-MSA synthesis requires only the presence of a properly folded PKS modified with the phosphopantetheine cofactor and adequate levels of acyl CoA substrates. These heterologous expression systems will greatly facilitate both genetic engineering of PKS genes to produce novel polyketides and the high-level production of polyketides. The results also raise the possibility of expressing polyketides in other organisms, such as plants and mammalian cells. Accordingly, experiments are in progress to evaluate E. coli and S. cerevisiae as hosts for the production of other polyketides and to assess other organisms as hosts for polyketide expression.
Acknowledgments
We thank Dr. Hong Fu for assistance with liquid chromatography/MS and NMR, and Melanie Betlach for assistance with figures. We thank Dr. Chaitan Khosla for providing the plasmid containing the 6-MSAS ORF and Dr. Anthony J. Brake for yeast genomic DNA and advice.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: PKS, polyketide synthase; 6-MSAS, 6-methylsalicylic acid synthase; 6-MSA, 6-methylsalicylic acid; P-pant, phosphopantetheinyl.
References
- 1.Hutchinson C R, Fujii I. Annu Rev Microbiol. 1995;49:201–238. doi: 10.1146/annurev.mi.49.100195.001221. [DOI] [PubMed] [Google Scholar]
- 2.Tsoi C J, Khosla C. Chem Biol. 1995;2:355–362. doi: 10.1016/1074-5521(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 3.Hutchinson C R. Bio/Technology. 1994;12:375–380. doi: 10.1038/nbt0494-375. [DOI] [PubMed] [Google Scholar]
- 4.Verdine G L. Nature (London) 1996;384:11–13. doi: 10.1038/384011a0. [DOI] [PubMed] [Google Scholar]
- 5.Kao C M, Katz L, Khosla C. Science. 1994;265:509–512. doi: 10.1126/science.8036492. [DOI] [PubMed] [Google Scholar]
- 6.McDaniel R, Ebert-Khosla S, Hopwood D A, Khosla C. Science. 1993;262:1546–1550. doi: 10.1126/science.8248802. [DOI] [PubMed] [Google Scholar]
- 7.Bedford D J, Schweizer E, Hopwood D A, Khosla C. J Bacteriol. 1995;177:4544–4548. doi: 10.1128/jb.177.15.4544-4548.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crosby J, Sherman D H, Bibb M J, Revill W P, Hopwood D A, Simpson T J. Biochim Biophys Acta. 1995;1251:32–42. doi: 10.1016/0167-4838(95)00053-w. [DOI] [PubMed] [Google Scholar]
- 9.Roberts G A, Staunton J, Leadlay P F. Eur J Biochem. 1993;214:305–311. doi: 10.1111/j.1432-1033.1993.tb17925.x. [DOI] [PubMed] [Google Scholar]
- 10.Pfeifer E, Pavela-Vrancic M, von Dohren H, Kleinkauf H. Biochemistry. 1995;34:7450–7459. doi: 10.1021/bi00022a019. [DOI] [PubMed] [Google Scholar]
- 11.Lambalot R H, Gehring A M, Flugel R S, Zuber P, LaCelle M, Marahiel M A, Reid R, Khosla C, Walsh C T. Chem Biol. 1996;3:923–936. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 12.Ku J, Mirmira R G, Liu L, Santi D V. Chem Biol. 1997;4:203–207. doi: 10.1016/s1074-5521(97)90289-1. [DOI] [PubMed] [Google Scholar]
- 13.Nakano M M, Corbell N, Besson J, Zuber P. Mol Gen Genet. 1992;232:313–321. doi: 10.1007/BF00280011. [DOI] [PubMed] [Google Scholar]
- 14.Ausubel F M. Current Protocols in Molecular Biology. New York: Greene & Wiley; 1987. [Google Scholar]
- 15.Atlas R M, Parks L C. Handbook of Microbiological Media. Boca Raton, FL: CRC; 1993. [Google Scholar]
- 16.Seidel J L, Epstein W W, Davidson D W. J Chem Ecol. 1990;16:1791–1816. doi: 10.1007/BF01020495. [DOI] [PubMed] [Google Scholar]
- 17.Spencer J B, Jordan P M. Biochem J. 1992;288:839–846. doi: 10.1042/bj2880839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackowski S, Rock C O. J Bacteriol. 1981;148:926–932. doi: 10.1128/jb.148.3.926-932.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vallari D S, Jackowski S, Rock C O. J Biol Chem. 1987;262:2468–2471. [PubMed] [Google Scholar]