

Abstract
The time of origin of the animal phyla is controversial. Abundant fossils from the major animal phyla are found in the Cambrian, starting 544 million years ago. Many paleontologists hold that these phyla originated in the late Neoproterozoic, during the 160 million years preceding the Cambrian fossil explosion. We have analyzed 18 protein-coding gene loci and estimated that protostomes (arthropods, annelids, and mollusks) diverged from deuterostomes (echinoderms and chordates) about 670 million years ago, and chordates from echinoderms about 600 million years ago. Both estimates are consistent with paleontological estimates. A published analysis of seven gene loci that concludes that the corresponding divergence times are 1,200 and 1,000 million years ago is shown to be flawed because it extrapolates from slow-evolving vertebrate rates to faster-evolving invertebrate rates, as well as in other ways.
The time of origin of the metazoan phyla is controversial. A common view is that the first coelomates appeared in the late Neoproterozoic, some 700 million years (My) ago, and the divergence between protostomes and deuterostomes occurred about 600 My ago. The divergence between the deuterostome phyla (echinoderms and chordates) may have occurred during the Vendian, before the beginning of the Cambrian 544 My ago (1–8). Fossil remains of nearly all readily fossilizable animal phyla have been recovered from Cambrian rocks (4).
This interpretation has been challenged on the grounds that it relies on negative evidence, namely the scarcity of fossil remains preceding the Cambrian, followed by the relatively sudden appearance during the Cambrian of many diverse phyla, classes, and orders. This “Cambrian explosion” might simply reflect the difficulty of preservation and discovery of soft-bodied and perhaps tiny animals (9–11). Resolution of this controversy has been sought in DNA sequence data and the theory of the molecular clock. An early study of cytochrome c sequences yielded results consistent with the Cambrian explosion view, although with slightly earlier dates, placing the origin of two protostome phyla, the annelids and arthropods, at 750 My ago, and the divergence between protostomes and deuterostomes at 720 My ago (12–16). Wray et al. (17) have, however, recently concluded from the analysis of seven genes that the divergence of protostomes and deuterostomes occurred nearly twice as early as the Cambrian—i.e., about 1,200 My ago—and that chordates diverged from the echinoderms about 1,000 My ago.
Crucial to these conclusions and others that rely on molecular data is the hypothesis of the molecular clock that molecular evolutionary rates for a particular gene are constant through time and across taxa. Yet we know that genes can evolve at disparate rates at different times or in different taxa (18–21). In this paper, we examine 18 different genes. Our results are consistent with the Cambrian explosion view—namely, they suggest that the divergence of protostomes and deuterostomes occurred in the late Neoproterozoic, around 544–700 My ago, and that the divergence between echinoderms and chordates preceded the Cambrian, but not very much. The genes that we have analyzed include six that were also studied by Wray et al. (17), but we have eliminated by a statistical procedure those branches of the evolutionary tree that are evolving at rates significantly different from the average. Examination of ref. 17 manifests a variety of methodological problems and indicates that the data depart importantly from the assumption of constant evolutionary rates.
MATERIALS AND METHODS
Genes and DNA Sequences.
The 18 protein-coding genes are listed in Tables 1 and 2. The α- and β-globin genes result from a duplication that occurred during the evolution of higher fishes (gnathostomes); a previous gene duplication, which may have slightly preceded the origin of chordates, yielded the vertebrate hemoglobin and myoglobin genes. The α- and β-globin genes are listed separately in Table 1, but only one globin has been analyzed in animals other than the gnathostome vertebrates. The genes listed in Table 1 and the DNA sequences analyzed are the same used by Wray et al. (ref. 17; and http://life.bio.sunysb.edu/ee/precambrian). We have omitted the 18S rRNA gene, which they included, because we were unable to obtain reliable alignment of the sequences across the diverse taxa. Table 2 lists 12 additional loci for which sequences are available in the electronic databases for several vertebrate taxa and at least one invertebrate taxon, and for which linear trees could be constructed (see below; list of sequences, accession numbers, and alignments are available from the first author upon request).
Table 1.
Divergence times between chordates and invertebrate phyla, derived from six protein-encoding gene loci
| Locus | No. of taxa | Method | Chordate–invertebrate divergence time, My
|
|||
|---|---|---|---|---|---|---|
| Echinoderms | Arthropods | Annelids | Molluscs | |||
| ATPase 6 | 15 | Poisson | 514.4 ± 47.4 | 496.3 ± 52.2 | NA | NA |
| Gamma | 523.4 ± 47.8 | 584.0 ± 53.0 | ||||
| Cytochrome c | 22 | Poisson | 502.2 ± 133.1 | 513.3 ± 118.0 | 677.8 ± 174.8 | 518.1 ± 137.0 |
| Gamma | 454.0 ± 108.1 | 473.8 ± 96.9 | 613.9 ± 143.5 | 481.7 ± 114.2 | ||
| Cytochrome oxidase I | 21 | Poisson | 721.1 ± 87.8 | 773.2 ± 86.3 | NA | NA |
| Gamma | 635.3 ± 68.7 | 675.5 ± 61.0 | ||||
| Cytochrome oxidase II | 38 | Poisson | NA | 365.4 ± 46.0 | 355.7 ± 49.5 | NA |
| Gamma | 427.3 ± 51.7 | 410.7 ± 53.9 | ||||
| α-Hemoglobin | 29 | Poisson | 541.9 ± 51.4 | 572.3 ± 53.9 | 551.0 ± 53.2 | 599.4 ± 51.5 |
| Gamma | 899.2 ± 79.9 | 960.5 ± 84.7 | 941.4 ± 83.2 | 1,308.6 ± 111.6 | ||
| β-Hemoglobin | 33 | Poisson | NA | NA | NA | 897.1 ± 125.2 |
| Gamma | 1,283.7 ± 141.8 | |||||
| NADH 1 | 13 | Poisson | 533.5 ± 49.1 | 697.0 ± 66.8 | 660.9 ± 65.8 | 688.1 ± 68.5 |
| Gamma | 625.2 ± 52.6 | 806.6 ± 69.6 | 794.2 ± 70.8 | 856.2 ± 76.1 | ||
| Average | Poisson | 563 ± 40 | 570 ± 60 | 561 ± 74 | 676 ± 82 | |
| Gamma | 628 ± 76 | 655 ± 83 | 690 ± 115 | 983 ± 197 | ||
α-Hemoglobin and β-hemoglobin are calibrated separately in vertebrates but are represented by only one locus in invertebrates. NA indicates that sequences from the taxonomic groups being compared are not included in the linear tree. Divergence times are mean ± SE; SE for the average is calculated from the variance among the loci means.
Table 2.
Divergence times between chordates and arthropods, derived from 12 protein-encoding genes
| Locus | No. of taxa* | Divergence time, My
|
|
|---|---|---|---|
| Poisson | Gamma | ||
| Aldehyde dehydrogenase† | 8 (9) | 352.1 ± 41.0 | 393.9 ± 42.1 |
| Adenine phosphoribosyltransferase | 7 (11) | 479.1 ± 82.9 | 568.5 ± 96.4 |
| Carboxylesterase | 7 (8) | 457.4 ± 28.0 | 603.8 ± 35.6 |
| Cathepsin B | 7 (7) | 264.2 ± 29.8 | 288.1 ± 31.3 |
| Glyceraldehyde-3-phosphate dehydrogenase | 12 (12) | 571.9 ± 77.2 | 586.9 ± 77.4 |
| Glutamine sythetase | 9 (10) | 712.6 ± 90.9 | 787.7 ± 99.8 |
| Histone H2b | 6 (8) | 838.9 ± 302.7 | 873.0 ± 314.7 |
| Heat-shock protein | 7 (8) | 1,042.7 ± 260.7 | 1,098.2 ± 273.7 |
| Lysozyme | 6 (26) | 228.9 ± 29.6 | 261.6 ± 32.1 |
| Mn-Superoxide dismutase | 7 (7) | 484.9 ± 83.5 | 544.1 ± 92.2 |
| Ornithine decarboxylase | 8 (9) | 1,314.7 ± 224.0 | 1,610.3 ± 271.4 |
| Urate oxidase | 6 (7) | 756.6 ± 137.6 | 909.6 ± 161.6 |
| Average | 625 ± 94 | 710 ± 110 | |
Results are mean ± SE; SE for the average is calculated from the variance among the loci means.
Number of taxa in the linear trees; in parentheses is the number of taxa in the original data set that includes those eliminated from the linear tree.
The invertebrate taxa compared for aldehyde dehydrogenase are annelids rather than arthropods.
Sequence Alignment and Genetic Distances.
Sequences were aligned by using the clustal w computer program (22) with adjustments made by eye. Genetic distances were calculated in two ways: with the gamma correction for multiple amino acid replacements; and with the Poisson correction (23). We set the gamma parameter to 2, and thus the gamma method yields results virtually identical to those obtained with Dayhoff’s (24) PAM (accepted point mutation) matrix, used by ref. 17. The Poisson model assumes equal rates of substitution across all sites within a given sequence, whereas the gamma model allows for violations of this assumption in the calculation of genetic distances (25–28). Reference times from the fossil record for the divergence between vertebrate groups used in the rate calibrations are the same as in ref. 17.
Statistical Methods.
We use the Z statistic of the “two-cluster test” (29) to ascertain whether the average amino acid substitution rates are statistically similar between the invertebrate and the vertebrate sets of sequences. In our test, the groups compared are not required to be monophyletic. To map divergence times onto our phylogenetic tree, we compute the height of the ancestral node as half the genetic distance between two reference sequence clusters, and determine the ratio of divergence time to node height. The substitution rate is the average of this ratio for all reference nodes. We multiply this average by the estimated heights of deeper nodes to obtain estimates of divergence times.
We apply the “branch-length” test of ref. 29 to a phylogenetic tree of the gene sequences, reconstructed by using the neighbor joining method of ref. 30 and the Poisson correction for multiple replacements (31). This test calculates the genetic distance from root to tip for each lineage, and it determines for each taxon whether or not it has evolved at a rate significantly different from the average. A χ2 statistic is computed that evaluates the extent to which the entire tree (excluding in this case the outgroup nonmetazoan sequences) conforms to a molecular clock, and the aberrant sequences (P < 0.05) are removed from the tree. The test is then reapplied to the remaining sequences, and the process is repeated until the remaining “linearized tree” consists only of lineages that are all evolving at a uniform rate.
Estimation of Divergence Time in Linear Trees.
Calibration of substitution rates is performed by mapping divergence times from the fossil record onto specific nodes of the linear tree, allowing a direct extrapolation to deeper divergence times. Under the assumptions of the molecular clock model, the number of amino acid substitutions separating two protein sequences is on average proportional to the time elapsed since the divergence of these sequences. If at least one time estimate can be assigned to a node of a phylogenetic tree of a set of protein sequences, then time estimates can be obtained for the remaining nodes. By using the phylogenetic tree to reconstruct divergence times, the covariance between pairwise genetic distances can be directly measured as the variance of the estimated branch lengths shared by the pair of distances (23).
Denote by tu the value of an unknown divergence time and an estimate of this value by t̂u. Given a reconstructed phylogenetic tree, which we assume is correct, binary, and obeys a molecular clock, an estimate of the unknown divergence time can be computed as
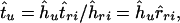 |
1 |
where ĥu estimates the height of the interior node (measured in terms of the number of amino acid substitutions per site) that corresponds to time tu; ĥri, t̂ri, and r̂ri denote the estimated height, divergence time and their ratio, respectively, for the ith reference node that can be assigned a known estimate of the absolute time.
With data sets for which more than one reference node is available, multiple r̂ri values are estimated; under the assumptions of the molecular-clock theory, all r̂ris have the same expected value. Although the error associated with the estimate of the interior-node height, ĥri, can be easily calculated, the corresponding error associated with absolute time estimate, t̂ri, is not known. An unweighted average of the different estimates of r is calculated as
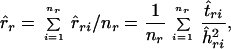 |
2 |
where nr is the total number of available reference nodes. By using the ordinary least-squares method, the node heights hu and hr can be estimated from the pairwise distances, dijs, between protein sequences as
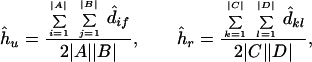 |
3 |
where A and B are two sequence clusters joined together by node u, and C and D are the two corresponding clusters for node r; |A| represents the number of sequences in cluster A. Using the delta-technique (32), we can express the variance of the time estimate tu as
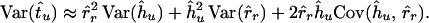 |
4 |
In recent studies involving phylogenetic estimation of the divergence time, the variance of the reference paleontological time estimate, Var(t̂r) is implicitly assumed to be zero (e.g., refs. 17 and 33). Although a rigorous estimate for Var(t̂r) is not currently available, it is likely to be nonzero and may considerably inflate the resulting value of Var(t̂u).
The values of Var(r̂r) and Cov(ĥu, r̂r) can be approximated with the delta-technique as
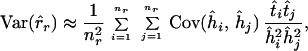 |
5 |
and
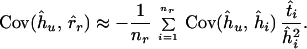 |
6 |
Estimates of the variances and covariances of ĥi and ĥj are calculated by the method of ref. 23, and the covariance between two evolutionary distances is calculated as the variance of the longest path shared by the distances in the true tree. With this approach and the first-order approximation Var(d̂ij) ≈ dij/s, where s is the number of sequence sites used for the calculation of distances,
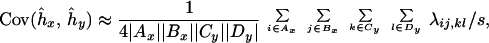 |
7 |
where λij,kl is the length of the longest route in the true tree that includes both the path from sequence i to sequence j and the path from sequence k to sequence l, and Ax and Bx, Cy and Dy are pairs of unique clusters corresponding to the interior nodes x and y, respectively.
RESULTS
Table 1 gives the time estimates (in millions of years) for the divergence between the chordates and various invertebrate phyla. Chordates and echinoderms are deuterostomes, more closely related to each other than they are to the protostomes, represented in the table by three phyla: Arthropoda, Annelida, and Mollusca. The order of branching among these three phyla is uncertain, although annelids and mollusks appear to be closer to each other than either is to arthropods (3, 34). For the present purposes, we shall assume that the three protostome phyla are equally divergent from the vertebrates.
The time estimates in Table 1 are derived from genetic distances estimated with two different methods for correcting for multiple substitutions. The Poisson method assumes that the rate of substitution for a particular sequence is identical for all sites, whereas the gamma method does not make this assumption. The six genes (the α- and β-globin genes present in the vertebrates are represented by only one gene in the invertebrates) in Table 1 are the same genes analyzed by Wray et al. (17). The 18S rRNA gene analyzed by these authors is not included in our analysis because we could not obtain alignments that would be unambiguous and robust—i.e., that could be extended from one to another set of taxa comparisons. The taxa represented in Table 1 are a subset of the taxa analyzed by Wray et al. (17). We eliminated by the branch-length method those lineages showing rates statistically different from the average for the particular gene locus. Wray et al. also eliminated “invertebrate sequences that showed consistently faster rates” (ref. 17, p. 571), although they do not say whether statistical significance was used for this elimination.
The average time estimates derived from the loci shown in Table 1 are consistent with the common view that the animal phyla that appear in the Cambrian fossil record, but not earlier, diverged before the Cambrian (≈700–540 My ago) but not much earlier as proposed by Wray et al. (17). Table 2 gives time estimates for the protostome–deuterostome divergence obtained by analysis of 12 additional gene loci. The estimated time of divergence is again somewhat greater with the gamma than with the Poisson correction but consistent with the hypothesis that it occurred during the late Neoproterozoic. The combined results from all 18 genes are summarized in Table 3. Fig. 1 shows the phylogeny of the phyla on geological and time scales, using the average of the Poisson and gamma estimates for the two divergence points (protostomes–deuterostomes and echinoderms–chordates) with shading indicating the range between the means obtained by the two methods. The time estimates displayed in Fig. 1 are consistent with commonly accepted paleontological interpretations.
Table 3.
Estimated divergence times between echinoderms and chordates and between them and three deuterostome phyla (arthropods, annelids, and mollusks)
Figure 1.

Estimated divergence times for selected animal phyla. Mean divergence times are the averages based on 18 gene loci: 673 million years for the protostome–deuterostome divergence and 595 million years for the echinoderm–chordate divergence. Shading extends to the estimates obtained with two different methods of correcting for multiple substitutions. The boundary between Vendian and Cambrian is at 544 My (8).
DISCUSSION
The theory of the molecular clock has provided useful, sometimes definitive, information toward settling matters of phylogenetic topology and the time of remote evolutionary events. The theory takes into account that each particular gene or protein evolves at a distinct rate and thus may serve as an independent molecular clock. Yet, heterogeneity across taxa and/or through time is often the case, and particular molecular clocks may have very erratic behavior (e.g., see refs. 18–21, 35).
The time of origin of animal phyla remains unsettled. The abundant appearance of most readily fossilizable animal phyla in the Cambrian fossil record, but not earlier, is frequently taken as indication that most animal phyla evolved shortly before the Cambrian (1–7, 36). Others, however, think it likely that animal phyla may have originated much earlier and that absence of their fossil remains before the Cambrian is due to one or more of the following conditions: smallness of the early metazoa, lack of hard body parts, and unsuitable geological circumstances for fossilization and preservation. Thus, it has been proposed that the protostomes and deuterostomes diverged around 1,200 My ago and the two deuterostome phyla, echinoderms and chordates, around 1,000 My ago (17).
We have sought evidence on this matter by analyzing 18 gene loci coding for proteins that have been sequenced in numerous relevant taxa. Extrapolation from known divergence times determined by the fossil record depends on “linear” trees—i.e., phylogenies in which the lineages are all evolving at the same rate, which rate is to be extrapolated to determine the unknown dates. We have, therefore, tested our trees for linearity and excluded all branches that could be shown to evolve at rates significantly different from the average for the tree.
The branch-length test (29) provides a statistical method to determine whether a particular taxon has evolved at a significantly faster or slower rate than the average, as determined by a χ2 statistic that evaluates the extent to which the entire tree conforms to a molecular clock. Calibration of substitution rates is performed by mapping directly all available divergence times from the fossil record onto specific nodes of the linear tree.
The variances of the time estimates are often quite large (see Tables 1 and 2) because of the reduced number of taxa, but also because our calculations take into account the nonindependence of the phylogenetically correlated protein sequences, rather than treating distance estimates as independent observations, as done in ref. 17. The gamma method gives somewhat greater time estimates of divergence than the Poisson method, but the two sets of estimates are fairly similar, with the notable exception of the globins. Higher vertebrates possess α- and β-globin gene families, each with several members. Numerous duplications have occurred that become entangled through a “birth-and-death” process, by which a gene in one species but not in others is lost and replaced by a paralogous one within the same genome (37–39). If paralogous and orthologous genes are intermingled, the mean and variance will increase because the divergence time of paralogous proteins corresponds to the time of gene duplication rather than to the time of speciation. It seems likely that the particular history of the globin genes may account for the large variances and discrepancies obtained for them. We could have eliminated the globins from our analysis, but we have left them, with the understanding that they may be inflating the overall average estimates of divergence time. Removing the globins virtually does not change the average estimates obtained with the Poisson method: 568 ± 52 and 602 ± 54 for the vertebrate–echinoderm and protostome–deuterostome divergences, respectively. But it reduces the averages obtained with the gamma method: 560 ± 43 and 666 ± 64 (628 ± 76 and 736 ± 65 in Table 3) and brings them closer to the Poisson estimates.
The results summarized in Table 3 and Fig. 1 are consistent with the Cambrian explosion theory proposing that the animal phyla originated not much before the Cambrian, during the late Neoproterozoic, some 544–700 My ago. This conclusion contrasts with the estimates obtained by Wray et al. (17), who propose that the protostome–deuterostome divergence is twice or more as old as the Cambrian, having occurred about 1,200 My ago, and that the echinoderm–chordate divergence is about 1,000 My old. They investigated the same set of six protein-encoding genes shown in Table 1 plus the 18S rRNA gene. The taxa in their study include the taxa used in Table 1 as well as taxa that we excluded because of lineages evolving significantly faster or slower than the average. We obtained the gene sequences from their web site. On the whole, they include (see Table 4) about twice as many taxa as used in Table 1.
Table 4.
Tests for homogeneity of sequence divergence rates in the data of Wray et al. (17)
| Locus | No. of taxa | Vertebrates–invertebrates
|
Among lineages*
|
||
|---|---|---|---|---|---|
| Z† | P | χ2 | P | ||
| ATPase 6 | 25 | −2.58 | <0.01 | 70 | <0.001 |
| Cytochrome c | 44 | −0.08 | NS | 2,073 | <0.001 |
| Cytochrome oxidase I | 31 | −1.19 | NS | 157 | <0.001 |
| Cytochrome oxidase II | 57 | −0.78 | NS | 104 | <0.001 |
| α-Hemoglobin | 85 | 0.23 | NS | 242 | <0.001 |
| β-Hemoglobin | 75 | 0.33 | NS | 353 | <0.001 |
| NADH 1 | 34 | −3.00 | <0.01 | 112 | <0.001 |
NS, not significant.
Goodness of fit of the metazoan sequences to a lineanized tree.
Two-tailed normal deviate statistic from the “two-cluster test”: negative values indicate that the average substitution rate is faster for the invertebrates than for the vertebrates.
Wray et al. (17) argue that their results are robust because (i) genetic distances and divergence times are highly correlated (p. 570 and their table 1) and (ii) the relative rate test indicates low rate variation, with standard errors ranging from 0.5% to 2.0% of the mean (p. 571 and their table 3). However, the relative rate test has low statistical power, and it has long ago been shown that apparently insignificant variation in average rates may hide differences in rate of evolution along the branches of the star phylogeny by 200% and more (e.g., ref. 1, see figure 9-23). Statistically more powerful tests have been developed, such as the branch-length test. High correlation and significant regression between distances and times are not, either, convincing evidence of uniform rates. To put it plainly, a time-dependent process such as molecular evolution will provide positive correlation with, and significant regression on, time without implication that the rate of change is constant. If we record the average time taken by travelers between Los Angeles and each of San Diego, San Francisco, and New York, we would likely find strong correlation between distance and time, even though different travelers may be going by car, rail, or plane. It would be folly to extrapolate the average time–distance rate observed between Los Angeles and these three cities and use it for estimating the distance between Los Angeles and London. The following sources of evidence further manifest that the results of Wray et al. (17) are not robust.
First, their regression methods introduce a host of statistical problems. Genetic distances between taxa are very highly correlated, yet all possible pairwise combinations of genetic distance measurements are computed and treated as independent observations in the regression calculations. Wray et al. (17) attempt to account for this nonindependence by assuming that the total number of degrees of freedom in the data is equal to the number of nodes in the corresponding binary tree. Unfortunately, while their approach, including the bootstrap and Mantel test, can detect the stochastic component of the total error, it is powerless to correct for the systematic bias in the computation of the mean slope caused by the dominance of distances that involve large clusters of nearly identical sequences. As a result, differences in substitution rates between vertebrates and invertebrates are amplified by the nonindependence of distance measurements and by the disproportionate representation of the vertebrate taxa. A statistically rigorous method of extrapolating distance times and estimating variances must take into account the phylogenetic relationships underlying the molecular sequences.
Second, we have applied the “two-cluster test” (29) to test for each locus in the data set of ref. 17 whether the average substitution rates differ between the vertebrate and invertebrate groups. The Z statistic indicates that the vertebrate rate of evolution is significantly slower than the invertebrate rate at two loci (Table 4). A slower rate in the vertebrate sequences (indicated by the negative sign of Z; see below about hemoglobin), which were used for calibrating divergence times, introduces a systematic bias in the extrapolation, exaggerating the time divergence estimates between the vertebrate and invertebrate phyla.
Third, we have applied the “branch-length test” (29) to the phylogenetic tree reconstructed for each locus with the neighbor-joining method (30) using the Poisson correction for multiple replacements (23). This test calculates the genetic distance from root to tip for each lineage and determines for each taxon whether it has evolved at a rate significantly different from the average. A χ2 statistic evaluates the extent to which the entire tree (excluding the outgroup nonmetazoan sequences) conforms to a molecular clock. Table 4 shows a significant departure from a molecular clock at P < 0.001 for every locus in the data set of ref. 17.
Fourth, we notice in figure 1 of Wray et al. (17) that the data points at each of four loci consist of two discrete sets, approximately 0–150 My and 300–450 My. We have calculated the rate of evolution by the regression of genetic distance on known divergence time separately for the two sets of data points (without imposing the restriction that the regression line pass through the origin, similarly as done by Wray et al. in ref. 17). If the rate of evolution is approximately constant through time, one would expect that the rates obtained for the two sets of data points should be fairly similar. The two rates are quite disparate and significantly so (Fig. 2).
Figure 2.

Rates of molecular evolution (genetic distance versus time) obtained with the data points of Wray et al. (17), but separately for those 0–150 and 300–450 My old. The regression slopes for each of the four loci follow (given in parentheses are F value for the test of equal regression slopes, degrees of freedom, and P; see ref. 40, p. 499). ATPase 6, 0.00279 and −0.00053 (F = 8.79; df = 1, 101; P < 0.005); NADH 1 = 0.00224 and 0.00019 (F = 33.04; df = 1, 101; P < 0.001); cytochrome oxidase I, 0.00052 and −0.00006 (F = 14.31; df = 1, 116; P < 0.001); and cytochrome oxidase II, 0.00070 and −0.00026 (F = 19.85; df = 1, 322; P < 0.001).
Fifth, we notice that only vertebrate data are used to calibrate the protein-coding genes, but echinoderms and mollusks are also used for calibrating 18S rRNA (ref. 17, legend for table 1). We ask whether the 18S rRNA rate would remain the same if only vertebrate data are used, as for the other genes. The regression slope obtained for the vertebrate data alone is 0.77 × 10−4, but it is twice as large, 1.5 × 10−4 when mollusks and echinoderms are added (ref. 17, table 1). If only vertebrates had been used for calibrating the clock, as done by Wray et al. for the other genes, the protostome–deuterostome divergence times would be 2,600–3,200 My, surely much too ancient. In any case, we see that the vertebrates are evolving slower than the invertebrates for the 18S rRNA gene, as noted above for protein-coding genes.
Sixth, we notice in table 3 of Wray et al. (17) that the mean genetic distances between animals and other multicellular kingdoms do not seem conspicuously different from the distances between bacteria and animals. We show in Table 5 the mean distances obtained by averaging over loci the distances provided by Wray et al. (17). The average distance between the eubacteria and animals is 1.51 ± 0.50, not significantly different from the distance between plants and animals of 1.40 ± 0.62, or between fungi (protist for hemoglobin) and animals, 1.68 ± 0.81. We have used these data to estimate a and b in Fig. 3. Using the mean genetic distances that Wray et al. give in their table 3, we conclude that b ≈ 0, even though the time span encompasses the evolution from the prokaryote to the eukaryote cell, the proliferation of the protist phyla, and the origin of multicellularity. If we exclude the hemoglobin gene, which shows the most heterogeneous rates in Wray et al. (ref. 17, table 3), the mean genetic distances from metazoa become 0.792 ± 0.217 for plant, 0.892 ± 0.235 for fungus/yeast, and 1.044 ± 0.211 for the bacterium; and the b/a ratio becomes 0.24. If we accept Wray et al.’s value of a = 1,200 My, b would be 288 My rather than a typical estimate of ≈2,000 My.
Table 5.
Mean genetic distances between metazoa and nonmetazoa calculated as the averages between the values given by Wray et al. (ref. 17, table 3) for six gene loci, and time parameters derived from the distances
| All metazoans | Vertebrates | Invertebrates | |
|---|---|---|---|
| Genetic distance | |||
| Metaphyte | 1.40 ± 0.62 | 1.44 ± 0.66 | 1.22 ± 0.44 |
| Yeast | 1.68 ± 0.81 | 1.63 ± 0.76 | 1.79 ± 0.92 |
| Eubacterium | 1.51 ± 0.50 | 1.49 ± 0.50 | 1.53 ± 0.50 |
| Phylogenetic time parameters* | |||
| a1 (metaphyte) | 0.698 | 0.719 | 0.610 |
| a2 (yeast) | 0.841 | 0.813 | 0.897 |
| a (average) | 0.770 | 0.766 | 0.753 |
| b1 (metaphyte) | 0.057 | 0.024 | 0.156 |
| b2 (yeast) | −0.087 | −0.069 | −0.131 |
| b (average) | −0.015 | −0.023 | 0.013 |
| b/a ratio | <0 | <0 | 0.017 |
Data for 18S rRNA are not included, because no eubacterium comparison is given by Wray et al. (17).
Time parameters are given as lineage change—i.e., as half the genetic distances: a reflects the lineage change from the origin of the multicellular kingdoms to the present; b is for the lineage change from the divergence between bacteria and eukaryotes to the origin of the multicellular kingdoms (see Fig. 3).
Figure 3.

Phylogeny of eubacteria and multicellular kingdoms, with branch lengths arbitrary. The averages for the genetic distances given in table 3 of ref. 17 are 1.50 ± 0.50 between bacteria and animals, which is not greater than 1.40 ± 0.62 between plant and animals or 1.68 ± 0.81 between yeast/mold (protist for one gene) and animals. Assuming that the lineage change is half the genetic distance between taxa, the value of b is about 0, even though it encompasses the evolution from prokaryote to eukaryote and from protist to multicellularity.
It seems warranted to conclude that the estimates of invertebrate–vertebrate divergence times obtained by Wray et al. (17) are invalid, owing to methodological problems and violations of the molecular clock. Extrapolations to distant times from molecular evolutionary rates estimated within confined data-sets are fraught with danger (18–21). Nevertheless, our time estimates, obtained by systematic elimination of erratic rates, are consistent with the common interpretation that protostomes and deuterostomes originated in the late Neoproterozoic, during the 160 My preceding the Cambrian.
Acknowledgments
We thank W. M. Fitch, R. R. Hudson, R. K. Selander, and J. W. Valentine for discussions. This research was supported by National Institutes of Health Grant GM42397.
ABBREVIATION
- My
million years
References
- 1.Dobzhansky T, Ayala F J, Stebbins G L, Valentine J W. Evolution. San Francisco: Freeman; 1977. [Google Scholar]
- 2.Valentine J W. Syst Zool. 1973;22:97–102. [Google Scholar]
- 3.Valentine J W. Proc Natl Acad Sci USA. 1989;86:2272–2275. doi: 10.1073/pnas.86.7.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valentine J W, Awramik S M, Signor P W, Sadler P M. Evol Biol. 1991;25:279–356. [Google Scholar]
- 5.Lipps J H, Signor P W, editors. Origin and Early Evolution of Metazoa. New York: Plenum; 1992. [Google Scholar]
- 6.Gould S J. Wonderful Life. New York: Norton; 1989. [Google Scholar]
- 7.Schopf J W, Klein C, editors. The Proterozoic Biosphere. A Multidisciplinary Study. Cambridge, U.K.: Cambridge Univ. Press; 1992. [Google Scholar]
- 8.Grotzinger J P, Bowring S A, Saylor B Z, Kaufman A J. Science. 1995;270:598–604. [Google Scholar]
- 9.Boaden P J S. Zool J Linn Soc. 1989;96:217–227. [Google Scholar]
- 10.Conway Morris S. Nature (London) 1993;361:219–225. [Google Scholar]
- 11.Glaessner M F. The Dawn of Animal Life. Cambridge, U.K.: Cambridge Univ. Press; 1984. [Google Scholar]
- 12.Brown R H, Richardson M, Boulter D, Ramshaw J A M, Jeffries R P S. Biochem J. 1972;128:971–974. doi: 10.1042/bj1280971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phillipe H, Chenuil A, Adoutte A. Development (Cambridge, UK) Suppl. 1994;1994:15–25. [Google Scholar]
- 14.Runnegar B. Lethaia. 1982;15:199–205. [Google Scholar]
- 15.Runnegar B. Paleontology. 1986;29:1–24. [Google Scholar]
- 16.Erwin D H. Lethaia. 1989;22:251–257. [Google Scholar]
- 17.Wray G A, Levinton J S, Shapiro L H. Science. 1996;274:568–573. [Google Scholar]
- 18.Ayala F J. Proc Natl Acad Sci USA. 1997;94:7776–7783. doi: 10.1073/pnas.94.15.7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ayala F J, Barrio E, Kwiatowski J. Proc Natl Acad Sci USA. 1996;93:11729–11734. doi: 10.1073/pnas.93.21.11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ayala F J. J Heredity. 1986;77:226–235. doi: 10.1093/oxfordjournals.jhered.a110227. [DOI] [PubMed] [Google Scholar]
- 21.Gillespie J H. The Causes of Molecular Evolution. New York: Oxford Univ. Press; 1991. [Google Scholar]
- 22.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nei M, Stephens J C, Saitou N. Mol Biol Evol. 1985;2:66–85. doi: 10.1093/oxfordjournals.molbev.a040333. [DOI] [PubMed] [Google Scholar]
- 24.Dayhoff M O. Atlas of Protein Sequence and Structure. Vol. 5. Washington, DC: National Biomedical Research Foundation; 1978. , Suppl. 3. [Google Scholar]
- 25.Golding G B. Mol Biol Evol. 1983;1:125–142. doi: 10.1093/oxfordjournals.molbev.a040303. [DOI] [PubMed] [Google Scholar]
- 26.Jin L, Nei M. Mol Biol Evol. 1990;10:1396–1402. doi: 10.1093/oxfordjournals.molbev.a040588. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z. Mol Biol Evol. 1993;10:1396–1402. doi: 10.1093/oxfordjournals.molbev.a040082. [DOI] [PubMed] [Google Scholar]
- 28.Takahata N. Proc R Soc London Ser B. 1991;243:13–18. doi: 10.1098/rspb.1991.0003. [DOI] [PubMed] [Google Scholar]
- 29.Takezaki N, Rzhetsky A, Nei M. Mol Biol Evol. 1995;12:823–833. doi: 10.1093/oxfordjournals.molbev.a040259. [DOI] [PubMed] [Google Scholar]
- 30.Saitou N, Nei M. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 31.Zuckerkandl E, Pauling L. In: Evolving Genes and Proteins. Bryson V, Vogel H J, editors. New York: Academic; 1965. pp. 97–166. [DOI] [PubMed] [Google Scholar]
- 32.Kendall M B. The Advanced Theory of Statistics. New York: Hafner; 1956. [Google Scholar]
- 33.Hedges S B, Parker P H, Sibley C G, Kumar S. Nature (London) 1996;381:226–229. doi: 10.1038/381226a0. [DOI] [PubMed] [Google Scholar]
- 34.Eernisse F J, Albert J S, Anderson F E. Syst Biol. 1992;41:305–330. [Google Scholar]
- 35.Li W-H. Molecular Evolution. Sunderland, MA: Sinauer; 1997. [Google Scholar]
- 36.Valentine J W. In: Patterns of Evolution. Hallam A, editor. Amsterdam: Elsevier; 1977. pp. 27–58. [Google Scholar]
- 37.Ohita T, Nei M. Mol Biol Evol. 1994;11:469–482. doi: 10.1093/oxfordjournals.molbev.a040127. [DOI] [PubMed] [Google Scholar]
- 38.Koonin E V, Mushegian A R. Curr Opin Genet Dev. 1996;6:757–762. doi: 10.1016/s0959-437x(96)80032-3. [DOI] [PubMed] [Google Scholar]
- 39.Nei M, Gu X, Sitnikova T. Proc Natl Acad Sci USA. 1997;94:7799–7806. doi: 10.1073/pnas.94.15.7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sokal R R, Rohlf F J. Biometry. 2nd Ed. New York: Freeman; 1981. [Google Scholar]