

Abstract
Superoxide has been shown to be critically involved in several pathological manifestations of aging animals. In contrast, superoxide can act as a signaling molecule to modulate signal transduction cascades required for hippocampal synaptic plasticity. Mitochondrial superoxide dismutase (SOD-2 or Mn-SOD) is a key antioxidant enzyme that scavenges superoxide. Thus, SOD-2 may not only prevent aging-related oxidative stress, but may also regulate redox signaling in young animals. We used transgenic mice overexpressing SOD-2 to study the role of mitochondrial superoxide in aging, synaptic plasticity and memory-associated behavior. We found that overexpression of SOD-2 had no obvious effect on synaptic plasticity and memory formation in young mice, and could not rescue the age-related impairments in either synaptic plasticity or memory in old mice. However, SOD-2 overexpression did decrease mitochondrial superoxide in hippocampal neurons, and extended the lifespan of the mice. These findings increase our knowledge of the role of mitochondrial superoxide in physiological and pathological processes in the brain.
Keywords: reactive oxygen species, antioxidant, learning and memory, synaptic plasticity, aging
1. Introduction
Reactive oxygen species (ROS) include superoxide, hydrogen peroxide, hydroxyl radicals, and unstable intermediates derived from the peroxidation of lipids. ROS have been shown to play important roles as both cellular messenger molecules in physiological events, such as activity-dependent synaptic plasticity and memory (Oury, Card, & Klann, 1999; Droge, 2002; Forman, Torres, & Fukuto, 2002; Martindale, & Holbrook, 2002; Stone, & Yang, 2006), and toxic molecules in pathological events, such as ischemia, brain injury, and age-related cell damage (Fukagawa, 1999; Finkel, & Holbrook, 2000; Pratico, 2002; Esposito, Ammendola, Faraonio, Russo, & Cimino, 2004). Thus, ROS can be both beneficial and deleterious to neuronal function, with the balance between ROS and antioxidants critical for maintaining normal neuronal function.
Superoxide dismutases (SODs) are a class of oxidoreductases that remove superoxide from organisms by catalyzing the dismutation of the superoxide radical to hydrogen peroxide. The resulting hydrogen peroxide is metabolized to molecular oxygen and water by either catalase or glutathione peroxidase (Marklund, 1984; Fridovich, 1995; Petersen, Oury, Valnickova, Thogersen, Hojrup, Crapo, & Enghild, 2003). SODs are a crucial part of the cellular antioxidant defense mechanism (Muscoli, Cuzzocrea, Riley, Zweier, Thiemermann, Wang, & Salvemini, 2003). In mammals, there are three different SOD genes encoding three different enzymes.. These SOD isozymes catalyze the same chemical reaction, but display different enzymatic properties and distinct cellular localizations. SOD-1 (Cu/Zn-SOD) is found mainly in intracellular compartments; SOD-2 (also referred to as Mn-SOD) is localized primarily in the mitochondrial matrix; and extracellular SOD (EC-SOD), which is usually found in the extracellular fluid, but can also be found in intracellular vesicle-like structures and on cell surfaces (Oury et al., 1999).
Mitochondrial respiration is a major source of intracellular ROS production (Liu, Fiskum, & Schubert, 2002). Under physiological conditions, around 0.2% of oxygen consumption is converted to ROS in and around mitochondria (Staniek, & Nohl, 2000; St-Pierre, Buckingham, Roebuck, & Brand, 2002). Under conditions of altered cellular metabolism, mitochondrial generation of ROS may even be considerably higher (Albers, & Beal, 2000). As a consequence, mitochondria are enriched with antioxidants in order to tightly regulate those free radicals (Zeevalk, Bernard, Song, Gluck, & Ehrhart, 2005). Mitochondrial superoxide dismutase, also known as SOD-2 or Mn-SOD, is the major antioxidant that controls the release of mitochondrial superoxide (Weisiger, & Fridovich, 1973). Therefore, SOD-2 is important for mitochondrial superoxide-regulated physiological and pathological events.
It has been reported that two different lines of SOD-2 homozygous knockout mice die shortly after birth (Li, Huang, Carlson, Melov, Ursell, Olson, Noble, Yoshimura, Berger, Chan, Wallace, & Epstein, 1995; Lebovitz, Zhang, Vogel, Cartwright, Dionne, Lu, Huang, & Matzuk, 1996), whereas overexpression of SOD-2 extends the lifespan of flies (Sun, Folk, Bradley, & Tower, 2002; Sun, Molitor, & Tower, 2004) and attenuates drug-induced neurotoxicity in mice (Klivenyi, St Clair, Wermer, Yen, Oberley, Yang, & Flint Beal, 1998; Maragos, Jakel, Chesnut, Pocernich, Butterfield, St Clair, & Cass, 2000; Callio, Oury, & Chu, 2005). However, questions concerning the neurological effects of chronic SOD-2 overexpression over the lifetime of an animal have yet to be addressed.
Our recent work on another SOD isozyme, extracellular SOD (EC-SOD), revealed that EC-SOD overexpression improves hippocampal synaptic plasticity and memory-related behavioral performance in aged mice (Hu, Serrano, Oury, & Klann, 2006). In light of our observations with EC-SOD transgenic mice, we utilized SOD-2 overexpressing mice to examine the role of mitochondrial superoxide on hippocampal function during aging. We found that SOD-2 overexpression decreased mitochondrial superoxide levels, but had no obvious impact on hippocampal LTP and memory in either young or aged mice. However, similar to the previous findings in flies, SOD-2 overexpression extended the lifespan of mice. We conclude that although mitochondrial superoxide may not contribute to age-related impairments in hippocampal synaptic plasticity and memory, decreasing the levels of mitochondrial superoxide in mice increases their longevity.
2. Materials and Methods
2.1 SOD-2 transgenic mice
The construction of SOD-2 transgenic mice has been previously described (Ho, Vincent, Dey, Slot, & Crapo, 1998). All mice were housed in Baylor College of Medicine’s Transgenic Mouse Facility, compliant with the NIH guide for Care and Use of Laboratory Animals. The facility is kept on a 12-hour light-dark cycle, with a regular feeding and cage cleaning schedule. Heterozygote mice expressing human SOD-2, driven by the β-actin promoter, were compared with wild-type mice from the same litter for all experiments.
2.2 Western blot analysis
In order to analyze regional SOD-2 expression in the transgenic mice, the entire hippocampus was dissected fresh on ice from PBS-perfused brains from 14–15 mice per group. Dissected tissue from sets of 3–4 mice were combined for homogenization and subjected to Western blot analysis using a rabbit anti-recombinant human SOD-2 antibody (Chang, Kang, Slot, Vincent, & Crapo, 1995), followed densitometry measurement as previously described (Hanford, Fattman, Shaefer, Enghild, Valnickova, & Oury, 2003). The endogenous SOD-1 and catalase levels in hippocampus were determined by Western blots as previously described (Hu et al., 2006). Densitometric analysis of immunoreactivity for each protein was conducted using Scion image software (Scion Corporation). Each experiment was repeated at least three times to ensure validity of the results.
2.3 Detection of mitochondrial O2 •- in primary neuronal culture
Cultures of dissociated hippocampal neurons were isolated from 1–2 day postnatal mice as described previously (Wu, Deisseroth, & Tsien, 2001). Neurons at 15–18 days in vitro (d.i.v.) were incubated with 300 nM MitoTracker-Green FM (Molecular Probes) for 40 min, and then reacted with 5 mM MitoSOX-Red (Molecular Probes) for 10 min. The treated neurons then were fixed by 4% paraformaldehyde in phosphate buffered saline and evaluated for mitochondria (MitoTracker-Green FM: Ex λ 490 nm, Em λ 516 nm) and superoxide (MitoSOX-Red: Ex λ 488 nm, Em λ >590 nm) with a Zeiss LSM 510 confocal microscope system (Zeiss, Germany).
2.4 In vivo detection of O2 •- using dihydroethidium
Dihydroethidium (DHE) was obtained from Molecular Probes (Eugene, OR). To identify cell-specific superoxide formation in the brain in vivo, we utilized DHE as previously described (Hu et al., 2006). Cover glasses were mounted with Vectashield H-1200 containing DAPI (Vector Laboratories, Burlingame, CA). Slices were evaluated for ethidium fluorescence (Ex λ 488 nm, Em λ >590 nm) and DAPI (Ex λ 405 nm, Em λ between 420 and 480 nm) on Zeiss LSM 510 confocal microscope.
2.5 Electrophysiology
Hippocampi from age-matched littermates were removed and 400 μm slices were prepared using a vibratome. The slices were perfused for 1–2 hours with oxygenated artificial cerebrospinal fluid (ACSF) (in mM: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 D-glucose, 2 CaCl2, and 1 MgCl2) in an interface tissue slice chamber at 30–32 ºC. Basal synaptic transmission (input/output) and PPF were examined in these studies as described in (Hu et al., 2006). LTP was induced by one of the following three protocols: one train, three trains (ITI 20 sec) or four trains (ITI 5 min) of 100 Hz HFS for 1 sec after at least 20 min of stable baseline recordings. fEPSPs were recorded every 20 sec and were presented as the average of four individual traces.
2.6 Behavioral studies
For all behavioral experiments, two groups of SOD-2 transgenic mice and wild-type littermates were tested: 3 months of age group and 2 years of age group. Spatial learning and memory was tested using the Morris water maze. During the acquisition phase, mice were given four trials each day (60-sec maximum, with 1-hr ITI) for eight consecutive days. A probe trial was given at the end of the last training day to determine the type of search strategy developed by the mice. The number of platform crossings and the time spent in each quadrant was recorded in the probe trial. A visual platform task was given to 16 one-year old mice (wild-type, n = 8; SOD-2 transgenic, n = 8). The mice were given four trials (60-sec maximum, 1-hr ITI) each day for two consecutive days with the escape platform marked by a visible cue and moved randomly between four locations. The animals' trajectories were recorded with a video tracking system (Noldus EthoVision System). Associative memory was tested using the delayed fear conditioning paradigm and traced fear conditioning paradigm. The experimental protocol is schematically illustrated in Figure 5. The training sessions consisted of a 2-min exploration period followed by two conditioned stimulus-unconditioned stimulus (CS-US) pairings (foot-shock intensity 0.75 mA, 2-sec duration; tone 90 db white noise, 30-sec duration). Context tests were performed in the same training chamber 24 hrs after training. Cue tests were performed in a distinct chamber 25 hrs after training for 6 min; baseline freezing was monitored (3-min duration) prior to presentation of the tone (90 db white noise, 3-min duration).
Figure 5.
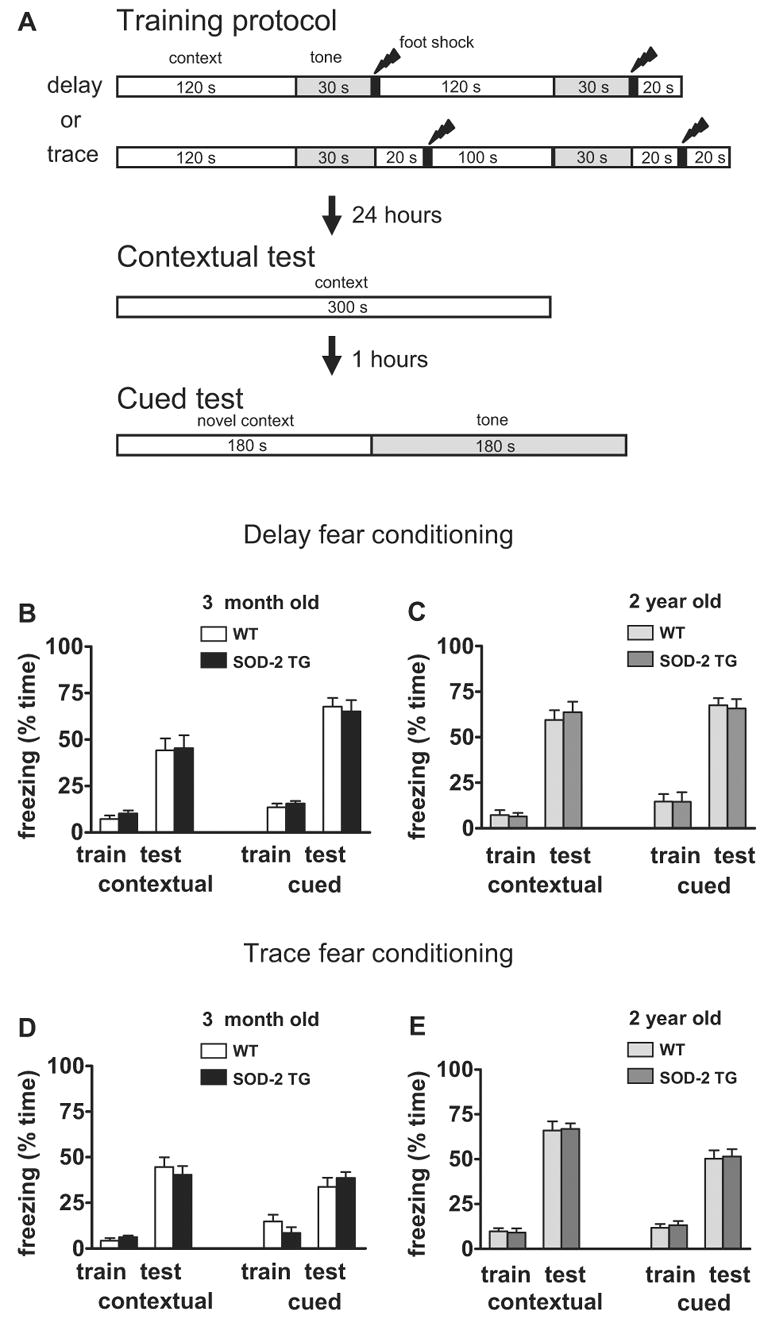
Normal contextual and cued memory in SOD-2 transgenic mice. (A) Illustration of the protocols for the associative fear conditioning experiments. The training sequences for delayed and trace conditioning and the two memory tests are presented. (B, D) Freezing behavior was expressed as the percentage of time during training (train) and testing (test) for 3-month old wild-type (WT) and SOD-2 transgenic mice (SOD-2 TG). Bar graphs grouped on the left show the percentage of time that the mice froze to context 24 hours after training. Bar graphs grouped on the right show the percentage of time that the mice froze to the auditory cue 25 hours after training. (B: WT, n=12; SOD-2 TG, n=12; D: WT, n=13; SOD-2 TG, n=12) (C, E) Similar to B and D, except that 2-year old mice were used for the studies (C: WT, n=12; SOD-2 TG, n=15; E: WT, n=16; SOD-2 TG, n=13).
2.7 Data analysis
All the data are presented as mean ± SEM. For comparisons between two groups, a two-tailed Student’s t-test was used. All time course experiments involving multiple comparisons were analyzed using a two-way ANOVA followed by individual post-tests when applicable. Error probabilities of p<0.05 were considered statistically significant.
3. Results
3.1 Characterization of human SOD-2 expression in mouse hippocampus and its effects on other antioxidant enzymes
Previous analyses of mice overexpressing human SOD-2 using immunogold electron microscopy have confirmed the overexpression of SOD-2 in mitochondria (Oberley, Coursin, Cihla, Oberley, el-Sayyad, & Ho, 1993; Maragos et al., 2000). Enzyme activity assays revealed that heterozygous SOD-2 transgenic mice displayed a 2-fold increase in SOD-2 activity in total brain homogenates (Maragos et al., 2000). We performed Western blot analysis using an anti-human SOD-2 antibody, which reacts with both mouse and human SOD-2, to assess SOD-2 protein expression in different brain regions. SOD-2 transgenic mice showed a 2.6-fold increase in SOD-2 protein expression in the midbrain, a 4.6-fold increase in the striatum (Callio et al., 2005), and a 3.6-fold increase in the hippocampus (Fig. 1A). Overexpression of SOD-2 did not affect the levels of either SOD-1 or catalase (Fig. 1B). These results indicate that SOD-2 transgenic mice exhibit increased expression of SOD-2 and normal expression of SOD-1 and catalase.
Figure 1.
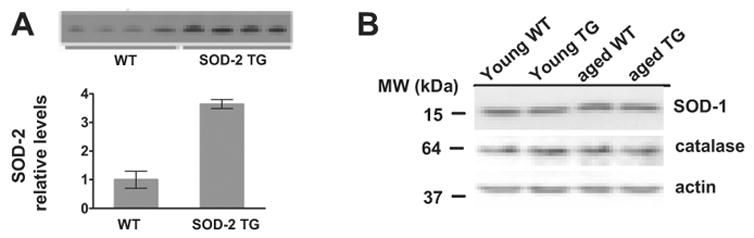
Levels of SOD-2 and other antioxidant enzymes in hippocampal tissue. (A) Western blot analysis of SOD-2 levels in hippocampal homogenates prepared from young adult wild-type (WT) and young adult SOD-2 transgenic mice (SOD-2 TG). (B) Western blot analysis of endogenous SOD-1 and catalase levels in hippocampal tissues prepared from wild-type (WT) and SOD-2 transgenic (SOD-2 TG) at 3 months (young) and 2 years (aged) of age.
3.2 SOD-2 transgenic mice exhibit normal hippocampal LTP
As a prelude to studying hippocampal LTP in young adult and aged SOD-2 transgenic mice, we tested the synaptic input-output relation in hippocampal area CA1 by eliciting synaptic responses with a range of stimulus intensities. We observed no significant differences between wild-type and SOD-2 transgenic mice (Fig. 2A). Second, we examined paired-pulse facilitation (PPF), which is a calcium-dependent form of presynaptic plasticity that is short-lived. Similar to the lack of an effect on the input-output function, we observed no difference in PPF between the two genotypes (Fig. 2B). These results suggest that overexpression of SOD-2 does not exert an adverse effect on general synaptic function or on one type of short-lasting presynaptic plasticity.
Figure 2.
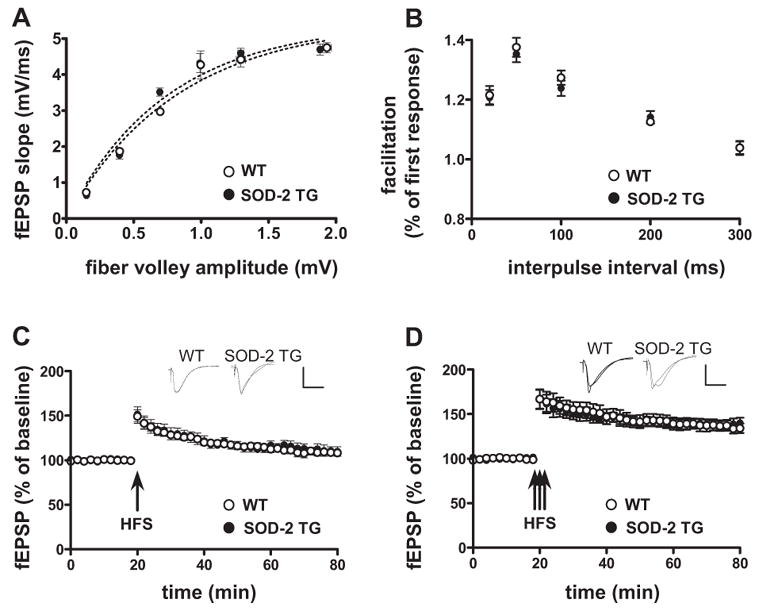
Normal synaptic transmission and plasticity in 3-month old SOD-2 transgenic mice. (A) Baseline synaptic transmission and (B) paired-pulse facilitation in hippocampal area CA1 are normal in SOD-2 transgenic mice. (C) LTP induced by one train of HFS (100 Hz, for 1 sec) did not reveal a difference between young WT and young SOD-2 TG. (D) LTP induced by three trains of HFS (100 Hz, for 1 sec, ITI 20 sec) in area CA1 did not reveal a difference between young WT and young SOD-2 TG. The insets in C and D are sample traces of EPSPs 10 min before and 1 hr after HFS. Calibration: 3 mV, 10 msec. (A: 3 slice/mouse, 5 mice/genotype, n=15; B: 2 slice/mouse, 5 mice/genotype, n=10; C: 2 slice/mouse, 5 mice/genotype, n=10; D: 2 slice/mouse, 5 mice/genotype, n=10).
We proceeded to examine LTP in young SOD-2 transgenic mice and their wild-type littermates. Field excitatory postsynaptic potentials (fEPSPs) were recorded in stratum radiatum in area CA1 of hippocampal slices in response to stimulation of the Schaffer collateral/commissural pathway. Early-phase LTP (E-LTP), induced by high frequency stimulation consisting of either one 100 Hz train (1 sec), three 100 Hz trains (each train 1 sec with an intertrain interval (ITI) of 20 sec), revealed no difference between young wild-type and young SOD-2 transgenic mice (Fig. 2C and 2D). These findings suggest that mitochondrial superoxide does not play a critical role in this type of hippocampal synaptic plasticity.
Because the hippocampus is one of the primary brain regions attacked by age-related neurological disease, we hypothesized that the SOD-2 transgenic mice would be protected against age-related impairments in hippocampal synaptic plasticity. We evaluated LTP in the SOD-2 transgenic mice at 2 years of age. In agreement with the findings from young wild-type mice, overexpression of SOD-2 did not alter either synaptic input-output relation or PPF in the aged mice (Fig. 3A and 3B). Using the same E-LTP induction protocols described above, we found that aged SOD-2 transgenic mice had similar impairments in E-LTP compared to their aged wild-type littermates (Fig. 3C and 3D). It is well known that multiple and spaced stimuli induce a very robust and long-lasting LTP, termed late-phase LTP (L-LTP). We then examined L-LTP induced by four trains of HFS with a 5-min ITI in aged wild-type and SOD-2 transgenic mice. Similar to E-LTP, we observed impaired L-LTP in both wild-type and SOD-2 transgenic mice (Fig. 3E). Taken together, these findings indicate that in contrast to our previous studies with mice that overexpress EC-SOD (Hu et al., 2006), overexpression of SOD-2 does not protect against age-related impairments in hippocampal LTP.
Figure 3.
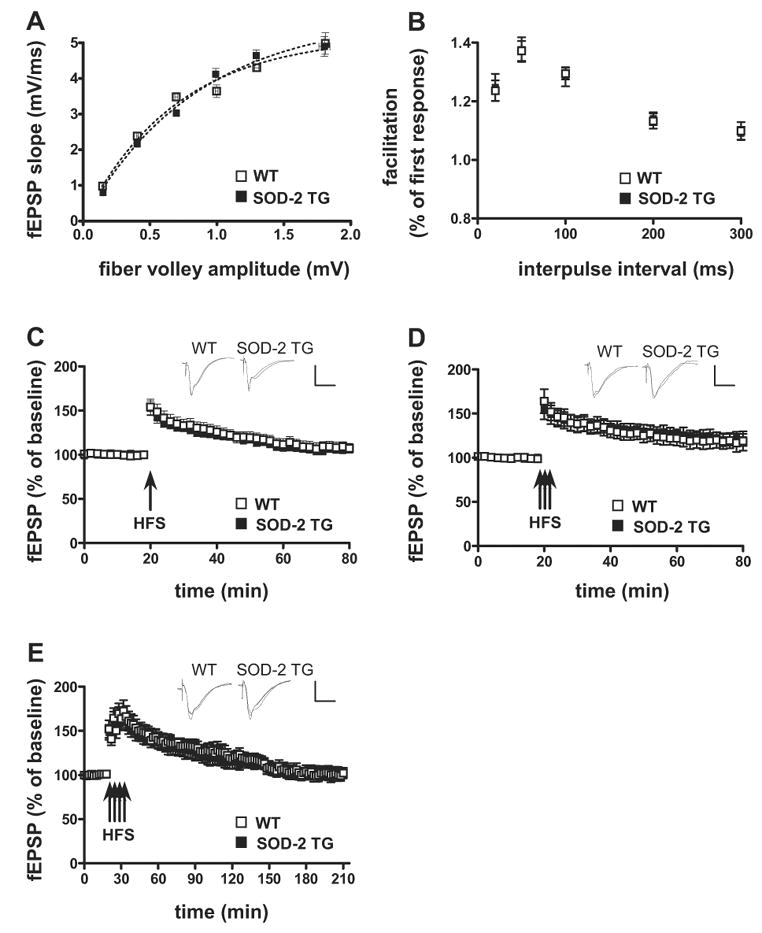
Normal synaptic transmission and plasticity in 2-year old SOD-2 transgenic mice. (A) Baseline synaptic transmission, (B) paired-pulse facilitation, (C) LTP induced by one train of HFS and, (D) LTP induced by three trains of HFS in hippocampal area CA1 of wild-type and SOD-2 transgenic mice were determined as described in Fig. 2, except that 2-year old mice were used for the studies. The insets in C and D are sample traces of EPSPs 10 min before and 1 hr after HFS. (E) Four trains of HFS (100 Hz, for 1 sec, ITI 5 min) induced an impaired L-LTP in area CA1 that was indistinguishable between aged WT and aged SOD-2 TG. The insets in E are sample traces of EPSPs 10 min before and 2.5 h after HFS. Calibration: 3 mV, 10 msec. (A: 3 slice/mouse, 5 mice/genotype, n=15; B: 2 slice/mouse, 5 mice/genotype, n=10; C: 2 slice/mouse, 6 mice/genotype, n=12; D: 2 slice/mouse, 6 mice/genotype, n=12; E: 2 slice/mouse, 6 mice/genotype, n=12).
3.3 SOD2 overexpression does not protect against age-related decline in spatial memory
Because hippocampal LTP is considered to be a potential cellular substrate for hippocampus-dependent memory, our observations of indistinguishable hippocampal LTP in aged SOD-2 transgenic mice lead us to predict that hippocampus-dependent memory function would not be improved in these mice as they age. To test this prediction, we examined the spatial learning and memory of wild-type and SOD-2 transgenic mice in the Morris water maze. We examined mice in two different age groups: 3 months of age and 2 years of age. During acquisition of the spatial memory task, there were no differences with respect to escape latencies (Fig. 4A) and path length (data not shown) between the wild-type and SOD-2 transgenic mice in both age groups, indicating that the wild-type and SOD-2 transgenic mice learn the platform position in a similar fashion. However, we did observe an effect of aging, as the old mice had significantly longer escape latencies than the young mice (Fig. 4A).
Figure 4.
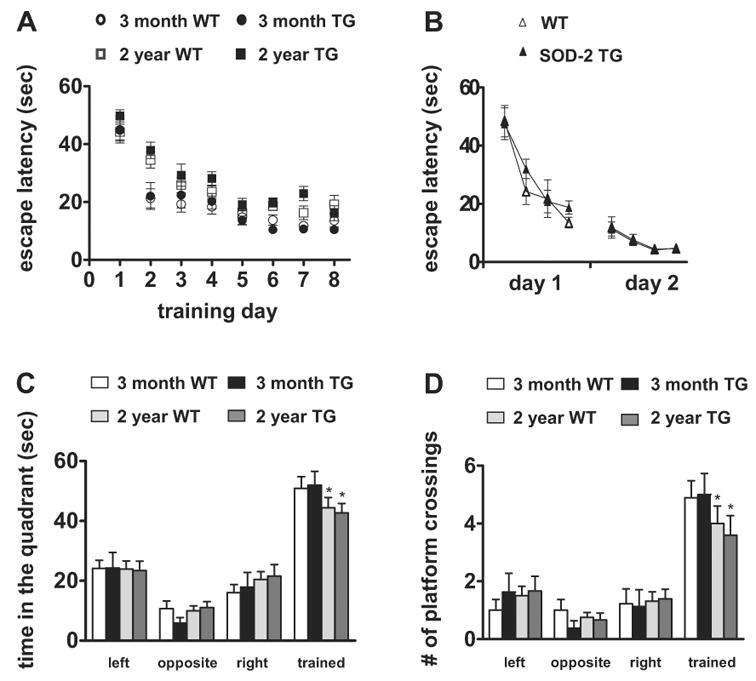
Normal spatial learning and memory in SOD-2 transgenic mice assessed by Morris water maze task. (A) Time spent searching for the platform on each training day. Wild-type (WT) and SOD-2 transgenic (TG) littermates were used for the studies. For each genotype, 3-month group: n=12; 2-year group: n=16. (B) Visible platform search. 12-month old mice were used for this test (WT, n=8; SOD-2 TG, n=8). (C) Time spent in each of the quadrants in the probe trial. The time spent in the target quadrant was not significantly different between wild-type and SOD-2 transgenic in either age group. *p<0.05 compared to the corresponding young mice by Student’s t-test. (D) The number of platform crossings in the probe trial is shown for the target quadrant and the corresponding location in the other quadrants. The number of platform crossings was not significantly different between wild-type and SOD-2 transgenic in either age group. *p<0.05 compared to the corresponding young mice by Student’s t-test.
To exclude the possibility that any differences in spatial memory between the wild-type and SOD-2 transgenic mice was due to poor vision, motivation, or swimming ability, we tested naïve mice (12 months of age) on a visible platform task in which mice learned to associate a distinct visible cue (a black pole) with an escape platform. The position of the platform and swim-start position varied from trial to trial. We found that there were no differences in latencies to find the platform (Fig. 4B), swim speed, or distance traveled (data not shown) in this task. In addition, in open field analysis and the rotating rod task, the wild-type and SOD-2 transgenic mice at all ages exhibited normal locomotor abilities, coordination and exploratory behavior (data not shown).
To assess spatial memory, we tested the mice in a probe trial during which the platform was removed from the pool and the mice were allowed to search for 60 s (1 hr after the completion of the last training trial). We found no significant difference between the wild-type and SOD-2 transgenic mice with respect to the time in the target quadrant (Fig. 4C) and the number of platform crossings (Fig. 4D). All together, our findings suggest that overexpression of SOD-2 does not protect mice against age-related impairments in hippocampus-dependent spatial learning and memory.
3.4 Overexpression of SOD-2 does not alter associative fear memory
Two sets of protocols (illustrated in Fig. 5A) were used to test fear memory in wild-type and SOD-2 transgenic mice: delayed fear conditioning and trace fear conditioning. In each of the training sessions, the wild-type and SOD-2 transgenic mice exhibited comparably low levels of freezing as shown in (Fig. B–E). In delayed fear conditioning, the animals were tested for contextual and cued fear memory 24 hrs following training. There were no apparent differences in freezing to either the context or the tone between the wild-type and SOD-2 transgenic mice in both 3-month age group and the 2-year age group (Fig. 5B, 5C). These findings suggest that overexpression of SOD-2 does not alter fear memory regardless of the age of the mice.
We then determined whether a more hippocampus-dependent training paradigm could reveal a difference in fear memory between the wild-type and SOD-2 mice. Two separate groups of mice 3 months of age and 2 years of age were subjected to a trace fear conditioning paradigm with a trace interval of 20 sec (Misane, Tovote, Meyer, Spiess, Ogren, & Stiedl, 2005). Again, the wild-type and SOD-2 transgenic mice performed comparably on the contextual and cued fear memory test (Fig. 5D, 5E), although the freezing to the tone was less than that observed in delayed fear conditioning because the trace intervals prevented the formation of conditioned tone-dependent fear (Misane et al., 2005). In additional behavioral tests, we found that wild-type and SOD-2 transgenic mice had similar responses in the open field (an exploratory behavior test), the elevated plus maze (an anxiety measurement), acoustic startle response (a hearing ability test) and the hot plate (a pain threshold test), suggesting that there were no gross neurological abnormalities in these animals (data not shown). Collectively, these data indicate that overexpression of SOD-2 has no effects in hippocampus-dependent associative learning in both young and aged mice.
3.5 SOD-2 overexpression reduces mitochondrial superoxide levels in cultured hippocampal neurons
To determine whether SOD-2 overexpression could actually reduce mitochondrial superoxide, we stained primary hippocampal neuronal cultures made from wild-type and SOD-2 transgenic mice with a mitochondria-specific dye, MitoTracker-Green FM, and a cell-permeable mitochondrial superoxide indicator, MitoSOX-Red. In Figure 6A, two classes of principal neurons in mouse hippocampus are shown: pyramidal cells from areas CA1 and CA3, and granule cells from dentate gyrus, which can be identified by their characteristic morphology. A decreased signal of red fluorescence corresponding to levels of mitochondrial superoxide was observed in both pyramidal and granule neurons from SOD-2 transgenic mice compared to the level of signal observed in neurons from wild-type mice (Figs. 6A and 6B). These data demonstrate that overexpression of SOD-2 does reduce the levels of mitochondrial superoxide in hippocampal neurons.
Figure 6.
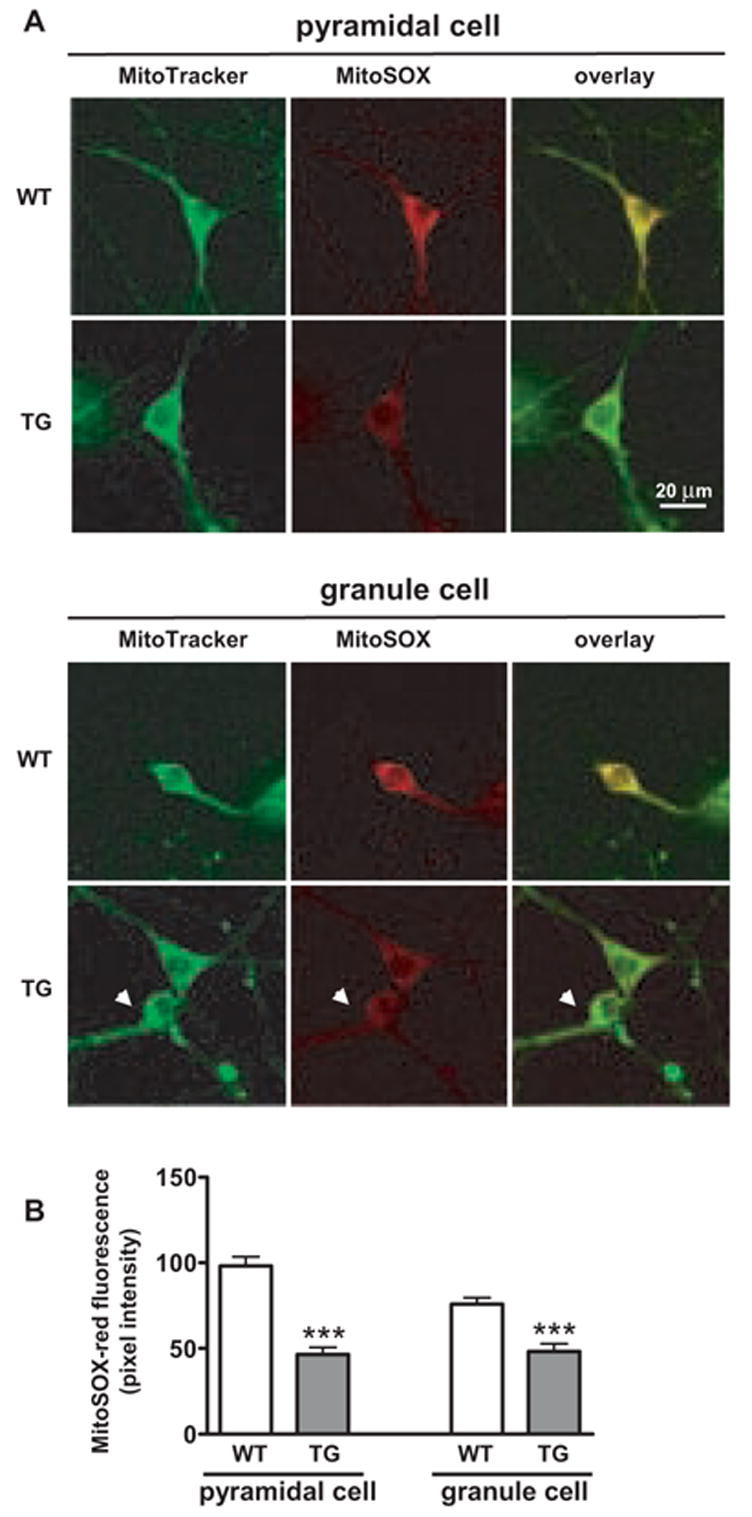
Levels of mitochondrial superoxide in cultured hippocampal neurons from wild-type and SOD-2 transgenic mice. (A) Hippocampal neurons were prepared from wild-type (WT) and SOD-2 transgenic mice (TG). MitoTracker-Green and MitoSOX-Red fluorescence was measured and the images were merged (overlay). Replicate experiments were carried out three times with similar results. Images for pyramidal cells and granule cells are shown. (B) Quantification of DHE fluorescence in hippocampal neurons. DHE fluorescence was measured with Scion software, using one-inch squares randomly selected from the cytosol of hippocampal pyramidal and granule cells. Data were graphed as pixel intensity, mean ± SEM, n > 40. *** denotes statistical significance with a Student’s t-test (***p<0.001).
3.6 SOD-2 overexpression reduces age-related increases in superoxide levels in vivo
To determine whether SOD-2 overexpression could also reduce in vivo superoxide levels in the brain, we utilized dihydroethidium (DHE), a superoxide-sensitive dye that has been used to localize superoxide in tissues (Murakami, Kondo, Kawase, Li, Sato, Chen, & Chan, 1998). Fluorescence reflecting DHE oxidation to ethidium is stable to fixation procedures (Quick, & Dugan, 2001). Mice that received no DHE were used to assess tissue autofluorescence and we found that background tissue fluorescence was low and did not differ between wild-type and SOD-2 transgenic mice (data not shown). We then compared sections prepared from either 3-month old or 2-year old wild-type and SOD-2 transgenic mice that received two intraperitoneal injections of DHE (see Materials and Methods). Representative images for hippocampal area CA1 and area CA3 are shown in Figure 7A. In hippocampal area CA1, superoxide levels were significantly increased in aged mice compared to young mice (Fig. 7B), consistent with our previous findings (Hu et al., 2006). Importantly, aged SOD-2 transgenic mice exhibited decreased superoxide levels compared with aged wild-type mice (Fig. 6B). Examination of other brain regions showed similar patterns of superoxide levels to those measured in hippocampal area CA1 (data not shown). These results suggest that there is an age-related increase in superoxide levels in the brain, and that this increase can be attenuated by overexpression of SOD-2.
Figure 7.
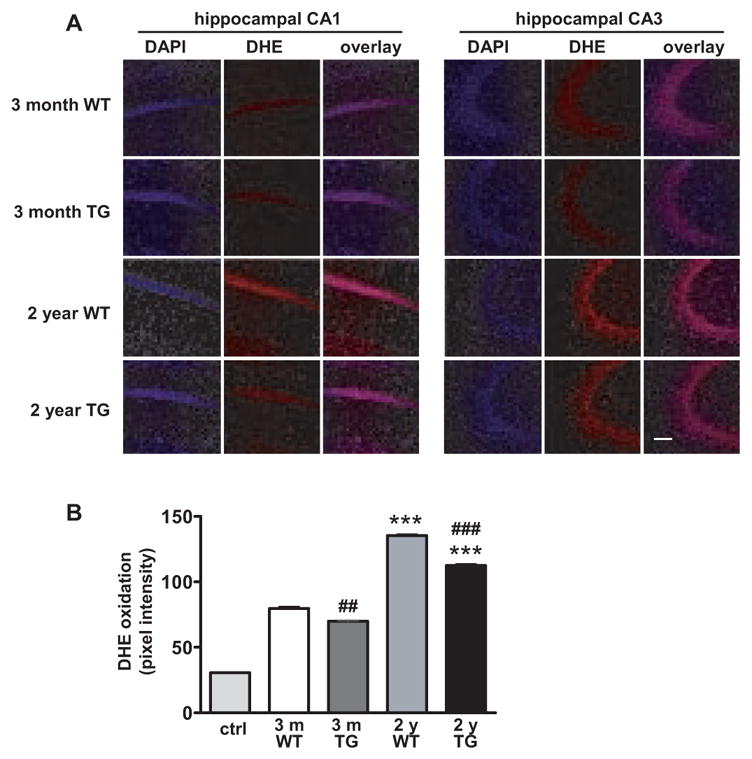
Endogenous levels of superoxide in young and old wild-type mice, and young and old SOD-2 transgenic mice. Brain sections were prepared from four 3-month old (young) and four 2-year old (aged) wild-type (WT) and SOD-2 transgenic (TG) mice that received two intraperitoneal injections of DHE (see Materials and Methods for details). Mice that were not injected with DHE were used as controls for autofluorescence. (A) Coronal sections through area CA1 and CA3 of the hippocampus showing differences in ethidium fluorescence. The images were taken with the same settings and a typical image for each region is shown. The scale bar indicates 75 μm. (B) Quantification of fluorescence in hippocampal area CA1. DHE fluorescence was measured with Scion software, using half inch square throughout the soma of CA1 pyramidal cells from at least 10 sections. Data were graphed as mean pixel intensity ± SEM, n > 60. Tissue autofluorescence (control) from mice that received no DHE is shown by the left-hand bar. Values were compared by one-way ANOVA followed by Tukey’s post hoc tests, indicating significantly enhanced levels in aged mice as compared with the young mice (***p<0.001). Student’s t-test showed a significant difference between young wild-type (young WT) and young SOD-2 transgenic (young TG) mice (##p<0.01), as well as between aged wild-type (aged WT) and aged SOD-2 transgenic (aged TG) mice (###p<0.001).
3.7 SOD-2 overexpression extend mouse lifespan
It has been reported that treatment of SOD-2 homozygous knockout mice with synthetic SOD and catalase mimetics extended their lifespan (Melov, Doctrow, Schneider, Haberson, Patel, Coskun, Huffman, Wallace, & Malfroy, 2001), and that the overexpression of SOD-2 extended the lifespan of flies (Sun et al., 2002; Sun et al., 2004). Therefore, we investigated whether the overexpression of SOD-2 could extend the lifespan of the mouse as well. The natural death of 54 mice which came from 7 litters was counted under normal animal facility conditions. As shown in Figure 8, SOD-2 overexpression increased the lifespan in SOD-2 transgenic mice compared with their wild-type littermates. Key parameters of the survival data obtained were summarized in the table in Figure 8. The was a small, 4% increase in the mean lifespan of the SOD-2 transgenic mice and 18% of the SOD-2 transgenic mice lived longer than 40 months, whereas the longest surviving wild-type mouse was 36 months of age. These results suggest that overexpression of SOD-2 increases the longevity of mice.
Figure 8.
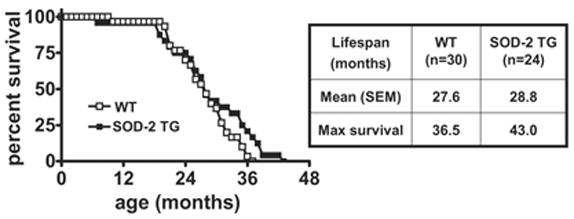
Survival curves for wild-type and SOD-2 transgenic mice. Age at natural death was recorded. Mean life span and maximum survival value are listed in the table on the right.
4. Discussion
Hippocampal LTP is a widely studied form of synaptic plasticity that likely involves cellular and molecular processes relevant to mammalian learning and memory (Bliss, & Collingridge, 1993; Malenka, & Nicoll, 1999). In previous studies we found that superoxide can serve as a cellular messenger during hippocampal LTP. For example, superoxide is able to regulate the autonomous activity of protein kinase C (PKC) during LTP (Knapp, & Klann, 2002), which is essential for the maintenance and expression of LTP (Roberson, English, & Sweatt, 1996). Cell-permeable and cell-impermeable scavengers of superoxide can block/attenuate LTP (Klann, 1998; Klann, Roberson, Knapp, & Sweatt, 1998) and application of superoxide to hippocampal slices can cause a PKC-dependent, LTP-like potentiation (Knapp, & Klann, 2002). In addition, young adult mice that overexpress EC-SOD showed hippocampal LTP deficits (Thiels, Urban, Gonzalez-Burgos, Kanterewicz, Barrionuevo, Chu, Oury, & Klann, 2000). Because mitochondria generate large amounts of superoxide, herein we examined hippocampal LTP, memory-related behavioral performance, and lifespan of the SOD-2 overexpressing mice to investigate the role of superoxide generated by mitochondria in hippocampal function and survival of the animals.
Although several studies indicate that mitochondrial superoxide plays a role in neuronal signal transduction (Werner, & Werb, 2002; Hongpaisan, Winters, & Andrews, 2003, 2004), it is not known whether mitochondrial superoxide contributes to HFS-induced hippocampal LTP. The phospholipid bilayer structure of the mitochondrial membranes does not allow free diffusion of superoxide generated within mitochondria (Balaban, Nemoto, & Finkel, 2005), which would argue against a role of mitochondrial superoxide in hippocampal LTP signaling. Consistent with this idea, we found that SOD-2 transgenic mice exhibit LTP that was indistinguishable from their wild-type littermates (Fig. 2). This result differs from previous observations with either SOD-1 or EC-SOD overexpressing mice, which both showed LTP deficits (Thiels et al., 2000; Kamsler, & Segal, 2003). These findings suggest that mitochondria are likely not the predominant source for superoxide production that is required for HFS-induced hippocampal LTP.
Much of the work elucidating the relationship between ROS and aging has focused on hippocampal function, because decreased hippocampal function is well known to be correlated with biomarkers of aging and is associated with a number of neurodegenerative disorders, such as Alzheimer’s and Parkinson’s disease. ROS, including superoxide, have been implicated in the cell damage observed in each of these conditions (Halliwell, 1989; Berlett, & Stadtman, 1997; Multhaup, Ruppert, Schlicksupp, Hesse, Beher, Masters, & Beyreuther, 1997; Van Remmen, & Richardson, 2001; Harman, 2002; Maier, & Chan, 2002). Thus, increasing antioxidant activity might be of therapeutic benefit for treating the pathologies of normal aging and neurodegenerative disorders. It has been shown that aged SOD-1 or aged EC-SOD overexpressing mice have improved hippocampal LTP compared with that of aged wild-type littermates (Kamsler, & Segal, 2003; Hu et al., 2006), and aged EC-SOD transgenic mice are protected against a decrease in age-dependent impairments in spatial memory (Levin, Christopher, & Crapo, 2005; Hu et al., 2006). Different from the pronounced effects of overexpression of either SOD-1 or EC-SOD on hippocampal function, we found that overexpression of SOD-2, did not impact impairments in either hippocampal synaptic plasticity or hippocampus-dependent memory in aged animals (Fig. 3, Fig. 4, and Fig. 5). Two-year old SOD-2 transgenic mice exhibited a similar age-dependent decline in hippocampal LTP compared to wild-type controls (Fig. 3). In addition, the aged SOD-2 transgenic mice showed marked deficits in spatial learning and memory as well (Fig. 4), which suggests that overexpression of SOD-2 might not protect against the deterioration in hippocampal function in aged animals. The difference in the synaptic plasticity and behavioral phenotypes between the SOD-2 transgenic mice and either SOD-1 or EC-SOD transgenic mice may be attributable to the differences in subcellular compartments in which these isozymes catabolize superoxide.
Previously, it has been shown that motivation can influence the learning and memory in EC-SOD overexpressing mice (Levin, Brucato, & Crapo, 2000). In a low motivational state, young EC-SOD transgenic mice have significantly impaired learning and memory. However, when the EC-SOD transgenic mice were in a high motivational state, the learning and memory impairments were less obvious. Therefore, it is possible that the relatively stressful behavioral tasks used in our study may have masked any subtle differences in the learning and memory capability between the SOD-2 transgenic mice and their wild-type littermates. It is interesting to note that SOD-2 transgenic mice have been found to enhance resistance to a variety of treatments that produce either acute toxicity or oxidative stress; they also showed a decrease in tissue damage in virtually all of these studies (Yen, Oberley, Vichitbandha, Ho, & St Clair, 1996; Chen, Siu, Ho, Vincent, Chua, Hamdy, & Chua, 1998; Ho et al., 1998; Maragos et al., 2000; Callio et al., 2005). For example, it has been reported that, using the same SOD-2 transgenic line as used in our experiments, SOD-2 overexpression protected against methamphetamine-induced brain damage (Maragos et al., 2000) and 6-hydroxydopamine-induced brain injury (Callio et al., 2005). In contrast, a reduction in SOD-2 activity exacerbated neuronal and vascular pathology in hAPP/Aβ expressing mice (Esposito, Raber, Kekonius, Yan, Yu, Bien-Ly, Puolivali, Scearce-Levie, Masliah, & Mucke, 2006). Together with our findings, these results suggest that increasing SOD-2 activity can protect against acute toxicity and stressors in certain brain regions, but SOD-2 overexpression may not impact on hippocampal function during the normal aging process.
Using ex vivo and in vivo methods, we measured mitochondrial superoxide levels in hippocampal neurons prepared from SOD-2 transgenic mice, and overall superoxide levels in the brains of the SOD-2 transgenic mice, respectively. Overexpression of SOD-2 appears to reduce mitochondrial superoxide levels because in cultured hippocampal neurons with a dye specific for mitochondrial superoxide, we found that superoxide levels were substantially decreased compared to wild-type hippocampal neurons (Fig. 6). We also observed that the overall superoxide levels were significantly reduced in SOD-2 transgenic mice (Fig. 7), although this decrease was not as profound as that observed in EC-SOD transgenic mice (Hu et al., 2006). The less effective reduction of superoxide levels in the aged SOD-2 transgenic mice may be the reason we did not observe a reversal of age-related impairments in synaptic plasticity as was the case in EC-SOD transgenic mice (Hu et al., 2006). It would be of interest to determine whether a further reduction in superoxide levels, perhaps by increasing the amount of SOD-2 overexpression, could attenuate the age-related decline in hippocampal function.
Over the past several years, many research groups have used either transgenic or knockout mice to test whether genetic manipulation of SOD can change an animal’s lifespan and the results have been contradictory. For instance, SOD-1 knockout mice have a shortened mouse lifespan, but EC-SOD knockout mice have a normal lifespan (Sentman, Granstrom, Jakobson, Reaume, Basu, & Marklund, 2006). In mice lacking both SOD-1 and EC-SOD, the reduced lifespan of SOD-1 knockouts was not further shortened by EC-SOD deficiency (Sentman et al., 2006). SOD-2 knockout mice died within the first week of life (Li et al., 1995) and treatment of SOD-2 homozygous knockout mice with synthetic SOD and catalase mimetics extended their lifespan (Melov et al., 2001). Overexpression of SOD-1 did not result in an extension of animal lifespan (Huang, Carlson, Gillespie, Shi, & Epstein, 2000), although these transgenic mice showed increased resistance to a variety of oxidative stresses (Cadet, Sheng, Ali, Rothman, Carlson, & Epstein, 1994; Chen, Ying, Simma, Copin, Chan, & Swanson, 2000; Ying, Anderson, Chen, Stein, Fahlman, Copin, Chan, & Swanson, 2000; Huang, Fujimura, Chang, & Chan, 2001). The survival data about mice overexpressing EC-SOD have not been reported, but it has been shown that the aged EC-SOD transgenic mice had decreased oxidative stress and improved cognitive performance compared with their old littermates (Hu et al., 2006). In this study, we investigated whether the overexpression of SOD-2 would extend mouse lifespan. We found that overexpression of SOD-2 did extend the mean and maximum lifespan of the mice compared to their wild-type littermates (Fig. 8). This result is consistent with what has been observed in Drosophila, where transgenic flies with increased SOD-2 activity had increased lifespan (Sun et al., 2002; Sun et al., 2004). Taken together, our findings suggest that the amount of mitochondrial superoxide is an important factor in determining mammalian longevity.
In summary, we have demonstrated that extraordinary scavenging of mitochondrial superoxide by SOD-2 does not appear to be beneficial at either the synaptic level or the cognitive level; however, it extends the lifespan of animals. These results indicate that different SOD isozymes exert different impacts on hippocampal function and aging.
Acknowledgments
This work was supported by National Institutes of Health grants NS034007 and NS047384 (E.K.), HL63700 (T.D.O.) and an Investigator-Initiated Research Grant from the Alzheimer’s Association (E.K.). We thank Laura Villasana and Monica Garcia for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albers DS, Beal MF. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. Journal of Neural Transmission Supplementum. 2000;59:133–154. doi: 10.1007/978-3-7091-6781-6_16. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. Journal of Biological Chemistry. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Sheng P, Ali S, Rothman R, Carlson E, Epstein C. Attenuation of methamphetamine-induced neurotoxicity in copper/zinc superoxide dismutase transgenic mice. Journal of Neurochemistry. 1994;62:380–383. doi: 10.1046/j.1471-4159.1994.62010380.x. [DOI] [PubMed] [Google Scholar]
- Callio J, Oury TD, Chu CT. Manganese superoxide dismutase protects against 6-hydroxydopamine injury in mouse brains. Journal of Biological Chemistry. 2005;280:18536–18542. doi: 10.1074/jbc.M413224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LY, Kang BH, Slot JW, Vincent R, Crapo JD. Immunocytochemical localization of the sites of superoxide dismutase induction by hyperoxia in rat lungs. Laboratory Investigation. 1995;73:29–39. [PubMed] [Google Scholar]
- Chen Y, Ying W, Simma V, Copin JC, Chan PH, Swanson RA. Overexpression of Cu,Zn superoxide dismutase attenuates oxidative inhibition of astrocyte glutamate uptake. Journal of Neurochemistry. 2000;75:939–945. doi: 10.1046/j.1471-4159.2000.0750939.x. [DOI] [PubMed] [Google Scholar]
- Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, Chua BH. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. Journal of Molecular and Cellular Cardiology. 1998;30:2281–2289. doi: 10.1006/jmcc.1998.0789. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiological Reviews. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Esposito F, Ammendola R, Faraonio R, Russo T, Cimino F. Redox control of signal transduction, gene expression and cellular senescence. Neurochemical Research. 2004;29:617–628. doi: 10.1023/b:nere.0000014832.78725.1a. [DOI] [PubMed] [Google Scholar]
- Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, Puolivali J, Scearce-Levie K, Masliah E, Mucke L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. Journal of Neuroscience. 2006;26:5167–5179. doi: 10.1523/JNEUROSCI.0482-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Torres M, Fukuto J. Redox signaling. Molecular and Cellular Biochemistry. 2002;234–235:49–62. [PubMed] [Google Scholar]
- Fridovich I. Superoxide radical and superoxide dismutases. Annual Review of Biochemistry. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- Fukagawa NK. Aging: is oxidative stress a marker or is it causal? Proceedings of the Society for Experimental Biology and Medicine. 1999;222:293–298. doi: 10.1046/j.1525-1373.1999.d01-146.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidants and the central nervous system: some fundamental questions. Is oxidant damage relevant to Parkinson's disease, Alzheimer's disease, traumatic injury or stroke? Acta Neurologica Scandinavica Supplementum. 1989;126:23–33. doi: 10.1111/j.1600-0404.1989.tb01779.x. [DOI] [PubMed] [Google Scholar]
- Hanford LE, Fattman CL, Shaefer LM, Enghild JJ, Valnickova Z, Oury TD. Regulation of receptor for advanced glycation end products during bleomycin-induced lung injury. American Journal of Respiratory Cell and Molecular Biology. 2003;29:S77–81. [PubMed] [Google Scholar]
- Harman D. Alzheimer's disease: role of aging in pathogenesis. Annals of the New York Academy of Sciences. 2002;959:384–395. doi: 10.1111/j.1749-6632.2002.tb02109.x. discussion 463–385. [DOI] [PubMed] [Google Scholar]
- Ho YS, Vincent R, Dey MS, Slot JW, Crapo JD. Transgenic models for the study of lung antioxidant defense: enhanced manganese-containing superoxide dismutase activity gives partial protection to B6C3 hybrid mice exposed to hyperoxia. American Journal of Respiratory Cell and Molecular Biology. 1998;18:538–547. doi: 10.1165/ajrcmb.18.4.2959. [DOI] [PubMed] [Google Scholar]
- Hongpaisan J, Winters CA, Andrews SB. Calcium-dependent mitochondrial superoxide modulates nuclear CREB phosphorylation in hippocampal neurons. Molecular and Cellular Neurosciences. 2003;24:1103–1115. doi: 10.1016/j.mcn.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Hongpaisan J, Winters CA, Andrews SB. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. Journal of Neuroscience. 2004;24:10878–10887. doi: 10.1523/JNEUROSCI.3278-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Serrano F, Oury TD, Klann E. Aging-dependent alterations in synaptic plasticity and memory in mice that overexpress extracellular superoxide dismutase. Journal of Neuroscience. 2006;26:3933–3941. doi: 10.1523/JNEUROSCI.5566-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Fujimura M, Chang YY, Chan PH. Overexpression of copper-zinc superoxide dismutase attenuates acute activation of activator protein-1 after transient focal cerebral ischemia in mice. Stroke. 2001;32:741–747. doi: 10.1161/01.str.32.3.741. [DOI] [PubMed] [Google Scholar]
- Huang TT, Carlson EJ, Gillespie AM, Shi Y, Epstein CJ. Ubiquitous overexpression of CuZn superoxide dismutase does not extend life span in mice. Journals of Gerontology Series A, Biological Sciences and Medical Sciences. 2000;55:B5–9. doi: 10.1093/gerona/55.1.b5. [DOI] [PubMed] [Google Scholar]
- Kamsler A, Segal M. Paradoxical actions of hydrogen peroxide on long-term potentiation in transgenic superoxide dismutase-1 mice. Journal of Neuroscience. 2003;23:10359–10367. doi: 10.1523/JNEUROSCI.23-32-10359.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. Journal of Neurophysiology. 1998;80:452–457. doi: 10.1152/jn.1998.80.1.452. [DOI] [PubMed] [Google Scholar]
- Klann E, Roberson ED, Knapp LT, Sweatt JD. A role for superoxide in protein kinase C activation and induction of long-term potentiation. Journal of Biological Chemistry. 1998;273:4516–4522. doi: 10.1074/jbc.273.8.4516. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, St Clair D, Wermer M, Yen HC, Oberley T, Yang L, Flint Beal M. Manganese superoxide dismutase overexpression attenuates MPTP toxicity. Neurobiology of Disease. 1998;5:253–258. doi: 10.1006/nbdi.1998.0191. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Role of reactive oxygen species in hippocampal long-term potentiation: contributory or inhibitory? Journal of Neuroscience Research. 2002;70:1–7. doi: 10.1002/jnr.10371. [DOI] [PubMed] [Google Scholar]
- Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Brucato FH, Crapo JD. Molecular overexpression of extracellular superoxide dismutase increases the dependency of learning and memory performance on motivational state. Behavior Genetics. 2000;30:95–100. doi: 10.1023/a:1001947003299. [DOI] [PubMed] [Google Scholar]
- Levin ED, Christopher NC, Crapo JD. Memory decline of aging reduced by extracellular superoxide dismutase overexpression. Behavior Genetics. 2005;35:447–453. doi: 10.1007/s10519-004-1510-y. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. Journal of Neurochemistry. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- Maier CM, Chan PH. Role of superoxide dismutases in oxidative damage and neurodegenerative disorders. Neuroscientist. 2002;8:323–334. doi: 10.1177/107385840200800408. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Maragos WF, Jakel R, Chesnut D, Pocernich CB, Butterfield DA, St Clair D, Cass WA. Methamphetamine toxicity is attenuated in mice that overexpress human manganese superoxide dismutase. Brain Research. 2000;878:218–222. doi: 10.1016/s0006-8993(00)02707-4. [DOI] [PubMed] [Google Scholar]
- Marklund SL. Extracellular superoxide dismutase and other superoxide dismutase isoenzymes in tissues from nine mammalian species. Biochemical Journal. 1984;222:649–655. doi: 10.1042/bj2220649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. Journal of Cellular Physiology. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- Melov S, Doctrow SR, Schneider JA, Haberson J, Patel M, Coskun PE, Huffman K, Wallace DC, Malfroy B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. Journal of Neuroscience. 2001;21:8348–8353. doi: 10.1523/JNEUROSCI.21-21-08348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misane I, Tovote P, Meyer M, Spiess J, Ogren SO, Stiedl O. Time-dependent involvement of the dorsal hippocampus in trace fear conditioning in mice. Hippocampus. 2005;15:418–426. doi: 10.1002/hipo.20067. [DOI] [PubMed] [Google Scholar]
- Multhaup G, Ruppert T, Schlicksupp A, Hesse L, Beher D, Masters CL, Beyreuther K. Reactive oxygen species and Alzheimer's disease. Biochemical Pharmacology. 1997;54:533–539. doi: 10.1016/s0006-2952(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. Journal of Neuroscience. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscoli C, Cuzzocrea S, Riley DP, Zweier JL, Thiemermann C, Wang ZQ, Salvemini D. On the selectivity of superoxide dismutase mimetics and its importance in pharmacological studies. British Journal of Pharmacology. 2003;140:445–460. doi: 10.1038/sj.bjp.0705430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberley TD, Coursin DB, Cihla HP, Oberley LW, el-Sayyad N, Ho YS. Immunolocalization of manganese superoxide dismutase in normal and transgenic mice expressing the human enzyme. Histochemical Journal. 1993;25:267–279. doi: 10.1007/BF00159118. [DOI] [PubMed] [Google Scholar]
- Oury TD, Card JP, Klann E. Localization of extracellular superoxide dismutase in adult mouse brain. Brain Research. 1999;850:96–103. doi: 10.1016/s0006-8993(99)02103-4. [DOI] [PubMed] [Google Scholar]
- Petersen SV, Oury TD, Valnickova Z, Thogersen IB, Hojrup P, Crapo JD, Enghild JJ. The dual nature of human extracellular superoxide dismutase: one sequence and two structures. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13875–13880. doi: 10.1073/pnas.2436143100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratico D. Lipid peroxidation and the aging process. Sci Aging Knowledge Environ. 20022002:re5. doi: 10.1126/sageke.2002.50.re5. [DOI] [PubMed] [Google Scholar]
- Quick KL, Dugan LL. Superoxide stress identifies neurons at risk in a model of ataxia-telangiectasia. Annals of Neurology. 2001;49:627–635. [PubMed] [Google Scholar]
- Roberson ED, English JD, Sweatt JD. A biochemist's view of long-term potentiation. Learning and Memory. 1996;3:1–24. doi: 10.1101/lm.3.1.1. [DOI] [PubMed] [Google Scholar]
- Sentman ML, Granstrom M, Jakobson H, Reaume A, Basu S, Marklund SL. Phenotypes of mice lacking extracellular superoxide dismutase and copper- and zinc-containing superoxide dismutase. Journal of Biological Chemistry. 2006;281:6904–6909. doi: 10.1074/jbc.M510764200. [DOI] [PubMed] [Google Scholar]
- Staniek K, Nohl H. Are mitochondria a permanent source of reactive oxygen species? Biochimica et Biophysica Acta. 2000;1460:268–275. doi: 10.1016/s0005-2728(00)00152-3. [DOI] [PubMed] [Google Scholar]
- Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. Journal of Biological Chemistry. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- Sun J, Molitor J, Tower J. Effects of simultaneous over-expression of Cu/ZnSOD and MnSOD on Drosophila melanogaster life span. Mechanisms of Ageing and Development. 2004;125:341–349. doi: 10.1016/j.mad.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Sun J, Folk D, Bradley TJ, Tower J. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics. 2002;161:661–672. doi: 10.1093/genetics/161.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiels E, Urban NN, Gonzalez-Burgos GR, Kanterewicz BI, Barrionuevo G, Chu CT, Oury TD, Klann E. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. Journal of Neuroscience. 2000;20:7631–7639. doi: 10.1523/JNEUROSCI.20-20-07631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Remmen H, Richardson A. Oxidative damage to mitochondria and aging. Experimental Gerontology. 2001;36:957–968. doi: 10.1016/s0531-5565(01)00093-6. [DOI] [PubMed] [Google Scholar]
- Weisiger RA, Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. Journal of Biological Chemistry. 1973;248:4793–4796. [PubMed] [Google Scholar]
- Werner E, Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. Journal of Cell Biology. 2002;158:357–368. doi: 10.1083/jcb.200111028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nature Neuroscience. 2001;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. Journal of Clinical Investigation. 1996;98:1253–1260. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Anderson CM, Chen Y, Stein BA, Fahlman CS, Copin JC, Chan PH, Swanson RA. Differing effects of copper, zinc superoxide dismutase overexpression on neurotoxicity elicited by nitric oxide, reactive oxygen species, and excitotoxins. Journal of Cerebral Blood Flow and Metabolism. 2000;20:359–368. doi: 10.1097/00004647-200002000-00018. [DOI] [PubMed] [Google Scholar]
- Zeevalk GD, Bernard LP, Song C, Gluck M, Ehrhart J. Mitochondrial inhibition and oxidative stress: reciprocating players in neurodegeneration. Antioxid Redox Signal. 2005;7:1117–1139. doi: 10.1089/ars.2005.7.1117. [DOI] [PubMed] [Google Scholar]