

Abstract
ABCG1 promotes cholesterol efflux from cells, but ABCG1-/- bone marrow transplant into ApoE-/- and LDLr-/- mice reduces atherosclerosis. To further investigate the role of ABCG1 in atherosclerosis, ABCG1 transgenic mice were crossed with LDLr-KO mice and placed on a high-fat western diet. Increased expression of ABCG1 mRNA was detected in liver (1.8-fold) and macrophages (2.7-fold) and cholesterol efflux from macrophages to HDL was also increased (1.4-fold) in ABCG1xLDLr-KO vs. LDLr-KO mice. No major differences were observed in total plasma lipids. However, cholesterol in the IDL-LDL size range was increased by approximately 50% in ABCG1xLDLr-KO mice compared to LDLr-KO mice. Atherosclerosis increased by 39% (10.1±0.8 vs 6.1±0.9 % lesion area, p=0.02), as measured by en face analysis, and by 53% (221±98 vs 104±58 ×103 um2, p=0.01), as measured by cross section analysis in ABCG1xLDLr-KO mice. Plasma levels for MCP-1 (1.5-fold) and TNF-alpha (1.2-fold) were also increased in ABCG1xLDLr-KO mice. In summary, these findings suggest that enhanced expression of ABCG1 increases atherosclerosis in LDLr-KO mice, despite its role in promoting cholesterol efflux from cells.
Keywords: ABCG1, cholesterol efflux, atherosclerosis, cytokines
Introduction
ABCG1 is a member of the G sub-family of ATP-binding cassette (ABC) transporters. It is a half-transporter with a single six transmembrane domain and a nucleotide-binding domain at the amino terminus. To be functional ABCG1 has to form either a homodimer or a heterodimer. Several studies demonstrated that ABCG1 can form and function as a homodimer: (a) overexpression of ABCG1 alone can induce cholesterol efflux to HDL[1], (b) cross-linking of ABCG1 produced a complex that had twice the molecular weight of ABCG1[2], and (c) homodimerization of ABCG1 in CHO cells was recently demonstrated[3], using ABCG1 constructs with two different immunoaffinity tags. ABCG1 may also potentially dimerize with other members of the G transporter family, such as ABCG4, although expression of ABCG4 in macrophages is markedly lower compared to ABCG1, even after stimulation with LXR agonists[1]. ABCG1 is highly expressed in macrophages, endothelial cells, kidney and brain, and to a lesser degree in the liver, where expression is higher in Kupfer cells than in hepatocytes[4]. In both hepatocytes and macrophages, ABCG1 expression is upregulated by cholesterol loading[5; 6] and by treatment with LXR agonists[7; 8]. Immunohistochemical studies, using an antibody to ABCG1 or epitope-tagged ABCG1, have revealed that ABCG1 protein is mostly localized in intracellular membranes, and to a lesser extent in the plasma membrane unlike some other ABC transporters, such as ABCA1[9]. Based on its localization, it has been proposed that ABCG1 may be involved in redistribution of cholesterol to cell surface domains prior to efflux to HDL[2].
The role of ABCG1 in regulating macrophage cholesterol homeostasis has been investigated by several groups. Transient overexpression of human or mouse ABCG1 in cultured cells resulted in increased efflux of cholesterol to HDL2 and HDL3, but not to lipid-free ApoAI [2; 10; 11]. Conversely, decreased expression of ABCG1, using siRNA, resulted in decreased cholesterol efflux to HDL[1]. Gelissen et al[3] demonstrated a sequential synergistic role of ABCG1 in promoting cholesterol efflux to phospholipid rich nascent HDL particles first generated by the lipidation of ApoA-I by the ABCA1 transporter. Because of its role in promoting cholesterol redistribution to plasma membrane and the subsequent efflux of excess cholesterol to HDL, it would be predicted that ABCG1 would have an anti-atherogenic effect. Unexpectedly, transplantation of ABCG1-/- bone marrow cells was recently found in several studies to reduce aortic atherosclerotic lesions in both ApoE-/- and LDLr-/- mice [12; 13], although one study found instead a modest increase atherosclerosis in transplanted LDLr-/- mice[14].
To further investigate the role of ABCG1 in atherosclerosis, the effect of increased ABCG1 expression was examined in ABCG1 transgenic mice crossed with LDLr-KO mice on a high-fat diet. Consistent with the previous knock out studies[12; 13] ABCG1 overexpression was found to increase atherosclerosis in LDLr-KO mice on a western diet, despite the observed enhancement of cholesterol efflux from macrophages.
Materials and methods
Animals and Diets
A fully sequenced 140-Kb BAC clone (BAC clone 201 (020), GS Control Number-26458; Incyte Genomics, Palo Alto, CA), encoding the complete mouse ABCG1 protein was used to generate transgenic mice in the C57BL/6 background. To generate LDLr-KO mice overexpressing mouse ABCG1, transgenic mice were crossed with LDLr-KO mice in the C57Bl/6 background. Mice were genotyped by dot-blot hybridization analysis using a specific mouse ABCG1 cDNA (forward primer 5’-CTGAACTGCCCTACCTACCAC-3’, reverse primer 5’-GAAGATGCCCAGGACGAT-3’) and by real-time PCR using TaqMan primers (forward primer 5’-CTTTCATGTCCCGCTGCAT-3’, reverse primer 5’-GTCACGGGACCCACAAATG-3’ and TaqMan MGB Probe 6-FAM-TCACTCCAGGTTGCG) generated with Primer Express 3.0 software (ABI). ABCG1xLDLr-KO transgenic mice were found to contain approximately 30 copies of the ABCG1 gene. Control and transgenic mice (2-months of age) were fed a Western Diet (TD 88137, Harlan Teklad, Madison, WI) containing 0.2% cholesterol and 21.2% fat for 12 weeks. All experiments were performed according to a research protocol approved by the Animal Care and Use Committee of the NHLBI, NIH.
Northern Blot and Real Time RT-PCR Analysis
Total RNA was isolated (TRIzol, Invitrogen) from age and sex-matched mice and further purified, using the RNeasy Mini Kit (Qiagen, Valencia, CA). RNA (10 μg) was subjected to Northern blot analysis, using a 32P-labeled cDNA probes specific for mouse ABCG1 and cyclophilin. mRNA was quantified, using a scanning densitometer (Molecular Dynamics, Sunnyvale, CA). RT-PCR analyses were performed using TaqMan technology on an ABI 7300 real-time PCR system (Applied Biosystems, Foster, CA). cDNA was synthesized from 2μg RNA with TaqMan reverse transcription system and random hexamer primers. Quantitative PCR was performed using probe and primer sets from ABI (mABCG1, Mm00437390; β-Actin, 4352341E), according to the manufacturer’s protocol. Both samples and standards were performed in duplicate.
Western Blot Analysis
5-20ug of protein were separated under reducing conditions on a Novex 3-8% Tris-Acetate gel (Invitrogen, Carlsbad, CA) and transferred to PVDF membranes (Invitrogen). Blots were incubated with rabbit anti-human/mouse ABCG1; anti-human/mouse β-Actin (BioLegend, San Diego, CA) was used to control for equal loading and transfer.
Plasma Lipids Analysis and Fast Protein Liquid Chromatography
Plasma was obtained after a 4-hour fast. Lipid levels and plasma lipoproteins were analyzed as previously described[15].
Cholesterol Efflux Studies
Resident mouse peritoneal macrophages were seeded at a density of 400,000 cells per well in a 24-well plate (Primeria cultureware, BD, Franklin Lakes, NJ), and maintained in DMEM medium (Gibco) containing 10% LPDS and antibiotics (Pen/Strep/L-glutamine, Sigma). Macrophages were loaded with medium containing 1uCi/ml 3H-cholesterol for 4 hours. Efflux of 3H-cholesterol was performed using HDL2, HDL3 (50ug/ml), apoA-I (10ug/ml) or BSA (0.2%) as acceptors[16]. Data were normalized to cell protein and cholesterol efflux to the cell media was expressed as percent of total cell counts.
Analysis of Aortic Lesions
The heart and attached section of ascending aorta were dissected en bloc and prepared as previously described[17]. Three mm sections of the aortic root and ascending aorta were stained with oil-red-O for neutral lipids and hematoxylin for nucleic tissue. Five sections per animal were evaluated to determine the mean cross-sectional area of lesions for each animal. The methods for en face aortic lesion analysis was adapted from Teupser [18] and Jiang [19]. The left ventricle of the heart was perfused first with PBS and then with a fixative solution (4% paraformaldehyde, 5% sucrose, 20 mM EDTA, pH 7.4). The aorta was dissected from the origin of the heart to the ileal bifurcation and was then further fixed overnight with fixative solution, stained with Sudan IV solution for 25 min, destained for 25 min in 80% ethanol, and washed with water. After removal of any remaining adventitial fat, the aortas were cut longitudinally, placed on a glass slide embedded with glycerin, covered by a microscope cover glass and sealed with nail polish. Quantification of plaques from the ileal bifurcation to the origin (not including branching vessels) of each animal was performed in a blind fashion and in triplicate, using the Image-Pro Plus version 4.1 software (Media Cybernetics, Inc., MD). Data are reported as the percentage of the aortic surface covered by lesions (total surface area of the atherosclerotic lesions divided by the total surface area of the aorta).
Measurements of Cytokines and Chemokines
A multiplex, sandwich-type ELISA assay was used for the measurement of cytokines and chemokines in mouse plasma (Pierce Biotechnology) as previously described[20]
Statistical Analysis
All data are expressed as means +/-SEM. Statistically significant differences between control and transgenic mice were assessed by Student’s t-test and defined as a two-tailed probability of less than 0.05. Non-parametric data was analyzed by the Mann-Whitney test (Instat Software, Graphpad, Inc., San Diego, CA).
Results
ABCG1 mRNA and protein expression
Mouse ABCG1 mRNA expression was measured in the liver and macrophages of LDLr-KO and ABCG1xLDLr-KO mice placed on high-fat western diet for 12 weeks. Northern Blot analysis (Fig.1A) showed increased ABCG1 expression in ABCG1xLDLr-KO compared to LDLr-KO mice in both the liver (1.8-fold) and macrophages (2.7-fold). Real-time-PCR (Fig.1B) also confirmed increased expression of ABCG1 in both liver (1.7-fold, p=0.04) and macrophages (1.2-fold, p<0.01). Western blot analysis (Fig.1C) revealed a 1.3-fold and 1.5-fold increased ABCG1 protein in liver and macrophages of ABCG1xLDLr-KO transgenic compared to LDLr-KO control mice. Overall, these results are consistent with a modest increase in the level of ABCG1 expression in the liver and macrophages of ABCG1 transgenic mice.
Fig. 1.
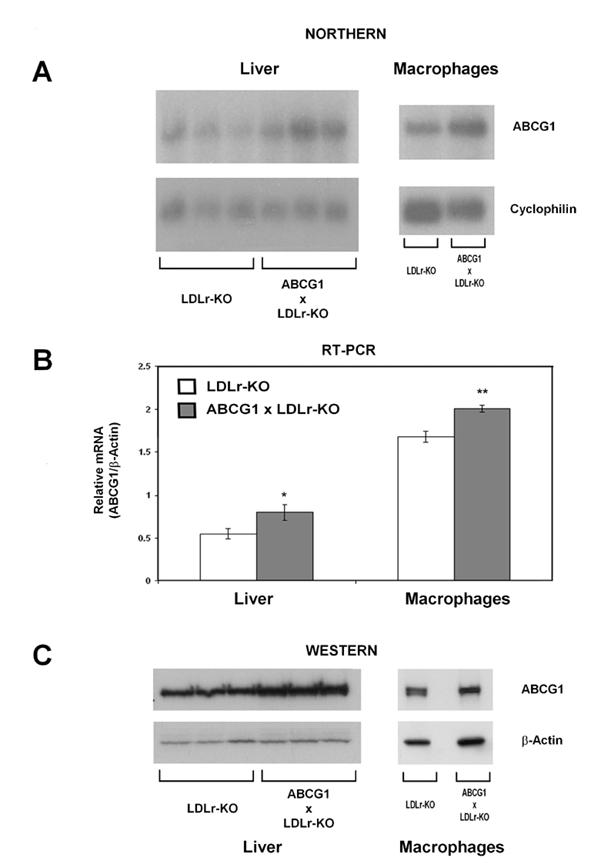
Overexpression of ABCG1 in Liver and Macrophages of LDLr-KO Mice on a Western Diet. A, Northern analysis of ABCG1 expression in LDLr-KO or ABCG1xLDLr-KO mice fed a Western diet for 12 weeks. Total RNA from peritoneal macrophages was extracted from a pool of macrophages (n = 6 male mice per genotype). Radiolabeled mouse ABCG1 and cyclophilin cDNA probes were used. B, Real-time PCR analysis of mouse ABCG1 in liver and macrophages of LDLr-KO (n=3) and ABCG1xLDLr-KO (n=3) mice * p<0.01; ** P=0.03. C, Western blot analysis of hepatic and macrophages ABCG1 expression
Total plasma lipids and FPLC lipoprotein profile
Analysis of plasma lipids (Fig.2A) revealed no major differences in the levels of TC (2072±294 vs 2338±109mg/dL), TG (1162±194 vs 985±101 mg/dL), PL (1124±123 vs 1197±46 mg/dL), FC (1037±309 vs 1113±160 mg/dL), and CE (1211±257 vs 1225±242 mg/dL) between ABCG1xLDLr-KO and LDLr-KO mice on a western diet. FPLC analysis of pool plasma (Fig 2B), however, showed significantly increased cholesterol in the IDL-LDL fraction of ABCG1xLDLr-KO compared to control mice (Fig 2B inset: 36.4±4.5 vs 54.5±5.3 ug/ml, p=0.02). VLDL cholesterol was also increased but did not reach statistically significance (56.8±15.2 vs 75.2±21.3 ug/ml). No differences were observed in the lipid composition or size of HDL particles between the two groups of mice.
Fig. 2.
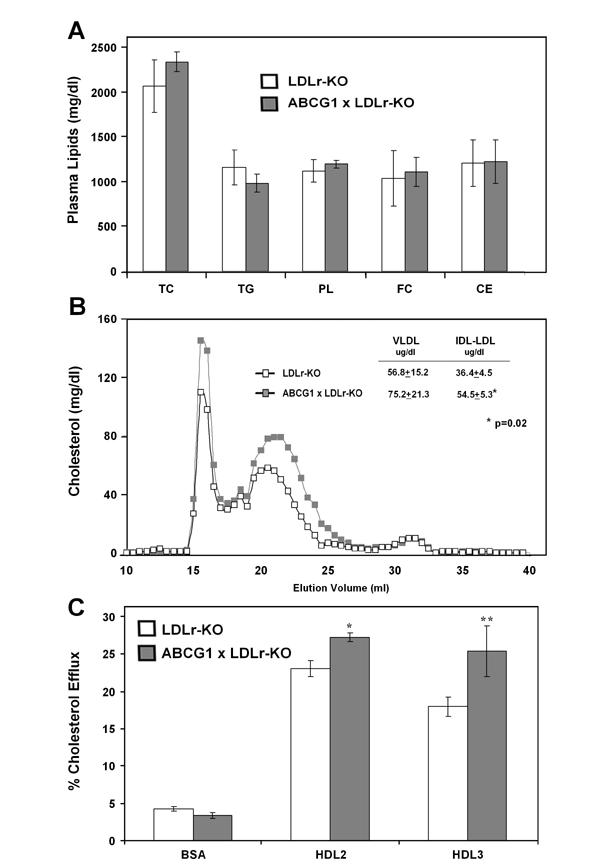
Lipids, Lipoprotein Analysis and Macrophages Cholesterol Efflux in ABCG1xLDLr-KO Mice on a Western Diet. A. Plasma lipids of fasted LDLr-KO (n=7) and ABCG1xLDLr-KO (n=7) mice after 12 weeks of a western diet were quantified. TC, total cholesterol; TG, triglycerides; PL, phospholipids; FC, free cholesterol; CE, cholesterol ester; C, cholesterol. B. Distribution of total cholesterol in plasma lipoproteins after FPLC separation of 30 ul of pooled plasma from fasted mice (n=5 each genotype). FPLC fractions corresponding to VLDL, IDL/LDL, and HDL are shown. C. Macrophages were harvested from 6 mice per each genotype at the 12th week of western diet and plated it for three hours. Cells were loaded overnight with 3H-labeled cholesterol (1uCi/ml), washed with PBS and cholesterol efflux was measured in cell culture media after 4 hours incubation with medium containing either BSA, HDL2 or HDL3 (50ug/ml). *p<0.01, **p=0.03
Increased macrophage cholesterol efflux to HDL
To assess the effect of the increased ABCG1 expression, cholesterol efflux to HDL from peritoneal macrophages isolated from ABCG1xLDLr-KO and LDLr-KO mice was measured (Fig.2C). Macrophages from the ABCG1 transgenic mice showed increased cholesterol efflux compared to the control mice for both HDL2 (27.3±0.6 vs 23.1±1.1%, p<0.01) and HDL3 (25.4±3.4 vs 18.0±1.3%, p=0.03). No differences in the efflux of cholesterol to lipid-free ApoA-I was observed from peritoneal macrophages from either mouse strain (data not shown).
Increased atherosclerosis in ABCG1xLDLr-KO mice
The aortic atherosclerotic lesion area was quantified by two different methods (Fig 3), namely by en face analysis of the entire aortic tree (Panel A) and by cross-section analysis of the proximal aorta (Panel B). After 12 weeks on the western diet, ABCG1 transgenic mice were found to have significantly increased atherosclerosis compared to the control LDLr-KO mice, as evaluated by both by en face analysis (10.1±0.8 vs 6.1±0.9 % lesion area, p=0.02) (Fig.3 A) and by oil-red-O staining of five cross sections of the proximal aorta (221±98 vs 104±58 ×103 um2, p=0.01) (Fig.3 B).
Fig. 3.
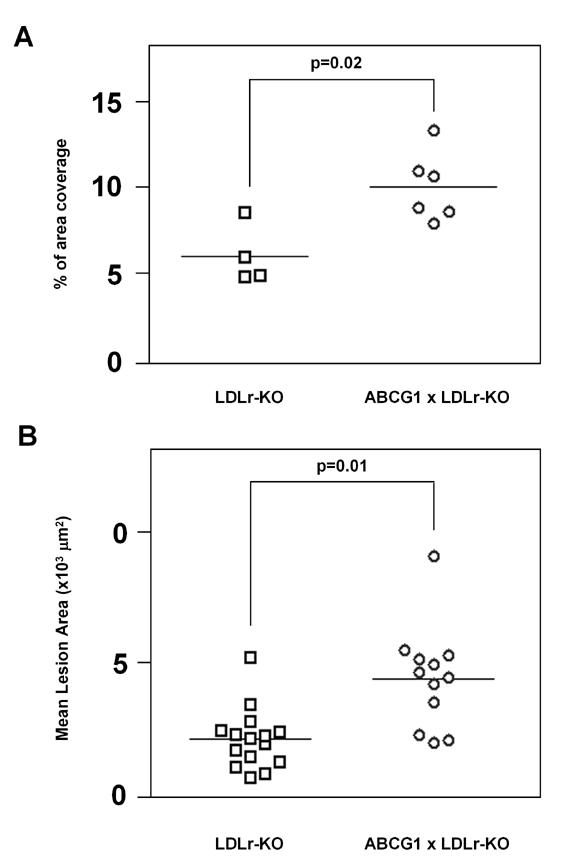
Measurement of Proximal Aortic Lesions in ABCG1xLDLr-KO and LDLr-KO Mice Fed a Western Diet for 12 weeks. Mean aortic lesion area was measured by en face analysis (Panel A) (LDLr-KO n=4, ABCG1xLDLr-KO n=6) and by cross-sectional analysis (Panel B) (LDLr-KO n=15, ABCG1xLDLr-KO n=12) in 5 months old female LDLr-KO (squares) and ABCG1 x LDLr-KO (circles) mice on a Western Diet. Data are expressed as the mean ± SEM.
Increased pro-inflammatory cytokines in ABCG1xLDLr-KO mice
To evaluate the possible role of increased inflammation, the plasma concentration of pro-inflammatory cytokines was measured in the two mouse lines after the 12 weeks of western diet (Fig.4). ABCG1xLDLr-KO transgenic had increased levels of MCP-1 (27.0±1.9 vs 39.6±2.0 pg/ml; p=0.05) and TNF-alpha (8.9±0.2 vs 10.6±0.1 pg/ml; p=0.03) compared to LDLr-KO control mice. IL-6 was also increased and IL-10 was reduced in ABCG1 transgenic mice but did not reach statistical significance (data not shown).
Fig. 4.
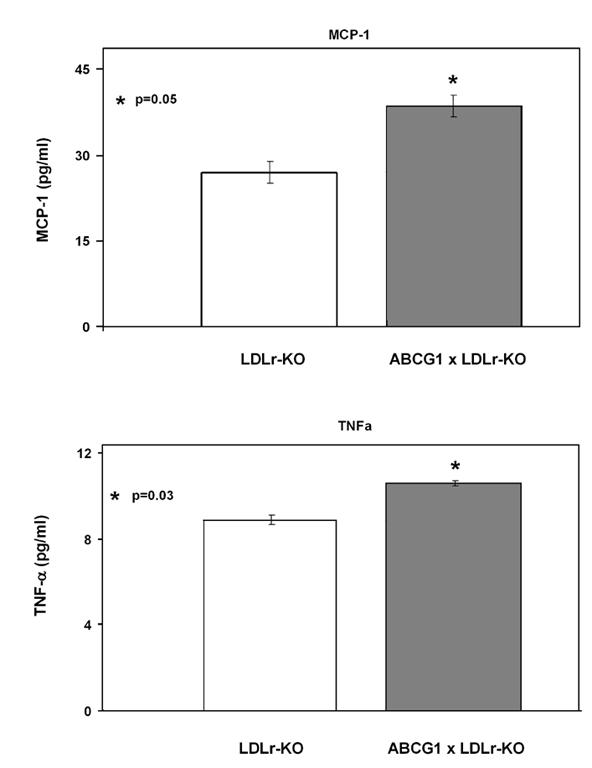
Measurement of Plasma MPC-1 and TNF-αin ABCG1xLDLr-KO and LDLr-KO Mice on a Western Diet. Plasma from 4 mice per each genotype at the 12th week of western diet was used to measure MCP-1 (Panel A) and TNF-α (Panel B)
Discussion
The major finding of this study is that increased expression of ABCG1 was found to increase atherosclerosis in LDLr-KO mice fed a high-fat western diet. This was true, even though as previously shown in other studies[12; 13], ABCG1 was found to increase cholesterol efflux from macrophages. ABCG1 traffics to the plasma membrane, where it alters the distribution of membrane cholesterol, thus making it more accessible for efflux to mature HDL particles in the extracellular fluid[2]. The ability of ABCG1 to promote cholesterol efflux has been proposed as the explanation for the observed decrease in tissue lipid accumulation in ABCG1 transgenic mice, and conversely for the massive deposition of neutral lipids and phospholipids in liver, lungs and macrophages of ABCG1-/- mice. The role of ABCG1 in cholesterol efflux would suggest that ABCG1 overexpression would protect against development of foam cells and atherosclerosis. Recently, however, two studies showed impaired development of atherosclerosis in both LDLr-/- and ApoE-/- mice transplanted with ABCG1-/- bone marrow[12; 13], which is consistent with the main finding of this paper and suggests that in vivo ABCG1 in some way acts as a proatherogenic factor.
One possible explanation for the increased atherosclerosis in the ABCG1xLDLr-KO transgenic mice is that ABCG1 altered the plasma lipoprotein profile. No major changes were observed in total plasma lipids, but FPLC analysis revealed that ABCG1xLDLr-KO transgenic mice had increased cholesterol level in the VLDL and IDL-LDL fractions. This increase in non-HDL cholesterol is known to be proatherogenic, and may have contributed to increase atherosclerosis in the ABCG1xLDLr-KO transgenic mice. Recently, Joyce et al [21] have shown that in ABCA1 transgenic mice the cholesterol effluxed to HDL is readily transferred to apoB-containing lipoproteins, which, in the absence of LDL-r, results in the accumulation of non-HDL proatherogenic lipoproteins. Similarly, in the present study, the increased cholesterol efflux to HDL in ABCG1xLDLr-KO mice may have accelerated the development of atherosclerosis because the effluxed cholesterol was eventually transferred to apoB-containing lipoproteins, which, in the absence of the LDLr, accumulate and become proatherogenic. Interestingly, decreased non-HDL lipoproteins were reported in ABCG1-KO mice, and it was proposed that this was due to increased secretion of apoE , the main ligand for hepatic uptake of these lipoproteins [13]. Furthermore, ABCG1 expression was shown by a post-translational mechanism to reduce the secretion of apoE [13]. Therefore, in addition, to a possible role of ABCG1 in the biogenesis of non-HDL lipoproteins, decreased apoE secretion by ABCG1 could also possibly account for the increase in non-HDL lipoproteins observed in Fig. 2.
In addition to changes in the plasma lipoprotein profile, the enhanced expression of ABCG1 was also found to increase the level of several cytokines, namely MCP-1 and TNF-alpha. Both of these cytokines are known to have important pro-atherogenic roles[22; 23] and, therefore, they could also be contributing to the increased atherosclerosis in ABCG1xLDLr-KO mice. In support of this hypothesis, Ranalletta et al have shown that macrophages from ABCG1-/- mice appear to have a compensatory change in the expression of several genes, particularly those related to cholesterol metabolism[13]. It is not clear, however, whether the increased level of pro-inflammatory cytokines in our study simply reflect the increased presence of atherosclerosis in the ABCG1xLDLr-KO mice or whether they are also playing a role in the initiation of atherosclerosis. Macrophages from ABCG1-/- mice have also been shown to undergo increased apoptosis when loaded with excess cholesterol by treatment with ox-LDL[12]. This suggests that ABCG1, perhaps by enhancing cholesterol efflux, can alter various macrophage functions, which may also include their ability to respond to and secrete inflammatory cytokines. Apoptosis in itself is also known to be an important process in the pathogenesis of atherosclerosis[24; 25], and increased macrophages apoptosis in ABCG1-/- mice was hypothesized to lead to the decreased atherosclerosis in these mice[12]. Conversely, the increased atherosclerosis in ABCG1xLDLr-KO mice observed in this study could possibly be due to decreased macrophage apoptosis from increased ABCG1 expression and enhanced cholesterol efflux. Future detailed studies aimed at the effect of ABCG1 on various macrophage functions will likely be a useful approach for better understanding the role of ABCG1 in the pathogenesis of atherosclerosis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc.Natl.Acad.Sci.U.S.A. 2004;101:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vaughan AM, Oram JF. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J Biol Chem. 2005;280:30150–30157. doi: 10.1074/jbc.M505368200. [DOI] [PubMed] [Google Scholar]
- [3].Gelissen IC, Harris M, Rye KA, Quinn C, Brown AJ, Kockx M, Cartland S, Packianathan M, Kritharides L, Jessup W. ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I Arterioscler. Thromb Vasc Biol. 2006;26:534–540. doi: 10.1161/01.ATV.0000200082.58536.e1. [DOI] [PubMed] [Google Scholar]
- [4].Hoekstra M, Kruijt JK, Van EM, Van Berkel TJ. Specific gene expression of ATP-binding cassette transporters and nuclear hormone receptors in rat liver parenchymal, endothelial, and Kupffer cells. J.Biol.Chem. 2003;278:25448–25453. doi: 10.1074/jbc.M301189200. [DOI] [PubMed] [Google Scholar]
- [5].Klucken J, Buchler C, Orso E, Kaminski WE, Porsch-Ozcurumez M, Liebisch G, Kapinsky M, Diederich W, Drobnik W, Dean M, Allikmets R, Schmitz G. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc.Natl.Acad.Sci.U.S.A. 2000;97:817–822. doi: 10.1073/pnas.97.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lorkowski S, Rust S, Engel T, Jung E, Tegelkamp K, Galinski EA, Assmann G, Cullen P. Genomic sequence and structure of the human ABCG1 (ABC8) gene. Biochem Biophys Res Commun. 2001;280:121–131. doi: 10.1006/bbrc.2000.4089. [DOI] [PubMed] [Google Scholar]
- [7].Sabol SL, Brewer HB, Jr., Santamarina-Fojo S. The human ABCG1 gene: identification of LXR response elements that modulate expression in macrophages and liver. J Lipid Res. 2005;46:2151–2167. doi: 10.1194/jlr.M500080-JLR200. [DOI] [PubMed] [Google Scholar]
- [8].Venkateswaran A, Repa JJ, Lobaccaro JM, Bronson A, Mangelsdorf DJ, Edwards PA. Human white/murine ABC8 mRNA levels are highly induced in lipid-loaded macrophages. A transcriptional role for specific oxysterols. J Biol Chem. 2000;275:14700–14707. doi: 10.1074/jbc.275.19.14700. [DOI] [PubMed] [Google Scholar]
- [9].Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney AM, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie J, Santamarina-Fojo S, Brewer HB., Jr. Cellular localization and trafficking of the human ABCA1 transporter. J.Biol.Chem. 2001;276:27584–27590. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- [10].Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- [11].Wang N, Ranalletta M, Matsuura F, Peng F, Tall AR. LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL Arterioscler. Thromb Vasc Biol. 2006;26:1310–1316. doi: 10.1161/01.ATV.0000218998.75963.02. [DOI] [PubMed] [Google Scholar]
- [12].Baldan A, Pei L, Lee R, Tarr P, Tangirala RK, Weinstein MM, Frank J, Li AC, Tontonoz P, Edwards PA. Impaired Development of Atherosclerosis in Hyperlipidemic Ldlr-/- and ApoE-/-Mice Transplanted With Abcg1-/-Bone Marrow Arterioscler. Thromb.Vasc.Biol. 2006 doi: 10.1161/01.ATV.0000240051.22944.dc. [DOI] [PubMed] [Google Scholar]
- [13].Ranalletta M, Wang N, Han S, Yvan-Charvet L, Welch C, Tall AR. Decreased Atherosclerosis in Low-Density Lipoprotein Receptor Knockout Mice Transplanted With Abcg1-/-Bone Marrow Arterioscler. Thromb.Vasc.Biol. 2006 doi: 10.1161/01.ATV.0000242275.92915.43. [DOI] [PubMed] [Google Scholar]
- [14].Out R, Hoekstra M, Hildebrand RB, Kruit JK, Meurs I, Li Z, Kuipers F, Van Berkel TJ, Van EM. Macrophage ABCG1 Deletion Disrupts Lipid Homeostasis in Alveolar Macrophages and Moderately Influences Atherosclerotic Lesion Development in LDL Receptor-Deficient Mice Arterioscler. Thromb.Vasc.Biol. 2006 doi: 10.1161/01.ATV.0000237629.29842.4c. [DOI] [PubMed] [Google Scholar]
- [15].Basso F, Freeman L, Knapper CL, Remaley A, Stonik J, Neufeld EB, Tansey T, Amar MJ, Fruchart-Najib J, Duverger N, Santamarina-Fojo S, Brewer HB., Jr. Role of the hepatic ABCA1 transporter in modulating intrahepatic cholesterol and plasma HDL cholesterol concentrations. J.Lipid Res. 2003;44:296–302. doi: 10.1194/jlr.M200414-JLR200. [DOI] [PubMed] [Google Scholar]
- [16].Remaley AT, Schumacher UK, Stonik JA, Farsi BD, Nazih H, Brewer HB., Jr. Decreased reverse cholesterol transport from Tangier disease fibroblasts. Acceptor specificity and effect of brefeldin on lipid efflux Arterioscler. Thromb.Vasc.Biol. 1997;17:1813–1821. doi: 10.1161/01.atv.17.9.1813. [DOI] [PubMed] [Google Scholar]
- [17].Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- [18].Teupser D, Persky AD, Breslow JL. Induction of atherosclerosis by low-fat, semisynthetic diets in LDL receptor-deficient C57BL/6J and FVB/NJ mice: comparison of lesions of the aortic root, brachiocephalic artery, and whole aorta (en face measurement) Arterioscler. Thromb.Vasc.Biol. 2003;23:1907–1913. doi: 10.1161/01.ATV.0000090126.34881.B1. [DOI] [PubMed] [Google Scholar]
- [19].Jiang XC, Qin S, Qiao C, Kawano K, Lin M, Skold A, Xiao X, Tall AR. Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency. Nat.Med. 2001;7:847–852. doi: 10.1038/89977. [DOI] [PubMed] [Google Scholar]
- [20].Moody MD, Van Arsdell SW, Murphy KP, Orencole SF, Burns C. Array-based ELISAs for high-throughput analysis of human cytokines. Biotechniques. 2001;31:186–4. doi: 10.2144/01311dd03. [DOI] [PubMed] [Google Scholar]
- [21].Joyce CW, Wagner EM, Basso F, Amar MJ, Freeman LA, Shamburek RD, Knapper CL, Syed J, Wu J, Vaisman BL, Fruchart-Najib J, Billings EM, Paigen B, Remaley AT, Santamarina-Fojo S, Brewer HB., Jr. ABCA1 overexpression in the liver of LDLr-KO mice leads to accumulation of Pro-atherogenic lipoproteins and enhanced atherosclerosis. J.Biol.Chem. 2006 doi: 10.1074/jbc.M604526200. [DOI] [PubMed] [Google Scholar]
- [22].Kowala MC, Recce R, Beyer S, Gu C, Valentine M. Characterization of atherosclerosis in LDL receptor knockout mice: macrophage accumulation correlates with rapid and sustained expression of aortic MCP-1/JE. Atherosclerosis. 2000;149:323–330. doi: 10.1016/s0021-9150(99)00342-1. [DOI] [PubMed] [Google Scholar]
- [23].Nashed B, Yeganeh B, HayGlass KT, Moghadasian MH. Antiatherogenic effects of dietary plant sterols are associated with inhibition of proinflammatory cytokine production in Apo E-KO mice. J.Nutr. 2005;135:2438–2444. doi: 10.1093/jn/135.10.2438. [DOI] [PubMed] [Google Scholar]
- [24].Arai S, Shelton JM, Chen M, Bradley MN, Castrillo A, Bookout AL, Mak PA, Edwards PA, Mangelsdorf DJ, Tontonoz P, Miyazaki T. A role for the apoptosis inhibitory factor AIM/Spalpha/Api6 in atherosclerosis development. Cell Metab. 2005;1:201–213. doi: 10.1016/j.cmet.2005.02.002. [DOI] [PubMed] [Google Scholar]
- [25].Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice Arterioscler. Thromb.Vasc.Biol. 2005;25:174–179. doi: 10.1161/01.ATV.0000148548.47755.22. [DOI] [PMC free article] [PubMed] [Google Scholar]