

Abstract
While cocaine’s interaction with the dopamine (DA) transporter and subsequent increase in DA transmission are usually considered key factors responsible for its locomotor stimulatory and reinforcing properties, many centrally-mediated physiological and psychoemotional effects of cocaine are resistant to DA receptor blockade, suggesting the importance of other, non-DA mechanisms. To explore the role of cocaine’s interaction with Na+ channels, rats were used to compare locomotor stimulatory and temperature (NAcc, temporal muscle and skin) effects of repeated iv injections of cocaine (1 mg/kg) with those induced by procaine (PRO 5 mg/kg), a short-acting local anesthetic with negligible effect on the DA transporter, and cocaine-methiodide (COC-MET 1.31 mg/kg), a quaternary cocaine derivative that is unable to cross the blood-brain barrier. While PRO, unlike cocaine, did not induce locomotor activation, it mimicked cocaine in its ability to increase brain temperature following the initial injection and to induce biphasic, down-up fluctuations following repeated injections. This similarity suggests that both these effects of cocaine may be driven by its action on Na+ channels, a common action of both drugs. While COC-MET also did not affect locomotor activity, it shared with cocaine and PRO their ability to increase brain temperature but failed to induce temperature decreases after repeated injections. These findings point toward activation of peripheral Na+ channels as the primary mechanism of rapid excitatory effects of cocaine and inhibition of centrally-located Na+ channels as a the primary mechanism for transient inhibitory effects of cocaine. DA receptor blockade (SCH23390 + eticlopride) fully eliminated locomotor stimulatory and temperature-increasing effects of cocaine, but its temperature-decreasing effects remained intact. Surprisingly, DA receptor blockade also altered the temperature fluctuations caused by PRO and COC-MET, suggesting that some of the central effects triggered via Na+ channels are in fact DA-dependant. Finally, repeated administration of PRO to animals that had previous cocaine experience led to conditioned locomotion and potentiated temperature-increasing effects of this drug.
It appears, therefore, that, in addition to the central effects of cocaine mediated via interaction with the DA transporter and potentiation of DA uptake, interaction with peripheral and central Na+ channels is important for the initial physiological and, perhaps, affective effects of cocaine, likely contributing to the unique abuse potential of this drug.
Keywords: brain temperature, behavior, dopamine uptake inhibition, procaine, cocaine methiodide, metabolic brain activation and inhibition, rats
1. Introduction
Although cocaine acts on multiple neural substrates in the brain and the periphery, neuropharmacological evidence strongly suggests that cocaine’s inhibition of dopamine (DA) uptake underlies its locomotor stimulatory and reinforcing effects (Ritz et al., 1987; Taylor and Ho, 1978; Wise and Bozarth, 1987). There are, however, two problems when attempting to invoke this drug action as the sole mechanism responsible for changes in DA transmission and reinforcement during cocaine self-administration. First, cocaine is a pure uptake inhibitor, with no effect on DA release in vitro (Heikkila et al., 1975; Rothman et al., 2001) and inhibits impulse-dependent DA release in vivo as suggested by DA cell recordings following intravenous (iv) administration (Einhorn et al., 1988; Pitts and Marwah, 1987). Although cocaine potentiates evoked DA release (Lee et al., 2001; Stamford et al., 1989; Venton et al., 2006), it remains unclear how cocaine per se might increase DA levels via an uptake-inhibiting mechanism if impulse-dependent DA release is inhibited. The drug action that induces this release, and whether this action even exists, remains unclear. Second, cocaine has a high affinity for the DA transporter (IC50=0.3-0.8 μM; Hennings et al., 1999; Rothman et al., 2001). While a single iv cocaine injection results in a rapid increase in brain cocaine levels (∼26 μM as suggested by direct PET measurements [Fowler et al., 1998]), leading to almost complete inhibition of DA re-uptake (Volkow et al., 1996), brain levels of cocaine achieved during repeated self-injections (up to 80-100 μM as calculated from measured peaks and half-lives of radioactive cocaine in the striatum [Fowler et al., 1998]) are enough to totally saturate all DA transporters. Further injections, consequently, may leave cocaine available to act via other mechanisms during self-administration. Therefore, while important for drugseeking behavior and the initiation of drug taking, cocaine’s action on the DA transporter does not sufficiently explain the tight regulation of highly cyclical drug intakes in self-administering animals.
Our previous work showed that nucleus accumbens (NAcc) DA-dependent electrochemical signals increase after the initial self-administration of cocaine, but fluctuate biphasically (down-up) at elevated plateau levels during subsequent injections (Kiyatkin and Stein, 1995). Similar, though less pronounced, changes also occurred during repeated passive cocaine injections (Kiyatkin and Stein, 1996). While the pattern of tonic DA signal changes generally mimics that of extracellular DA levels evaluated by microdialysis (Hemby et al., 1997; Wise et al., 1995), the nature of rapid post-cocaine signal decreases remained unclear. Since brain temperature fluctuates in response to various stimuli and reflects metabolic neural activation (Kiyatkin et al., 2002), we recently used this parameter to clarify this issue. Matching electrochemical findings, brain temperature increased following the initial and fluctuated following repeated cocaine self-injections (Kiyatkin and Brown, 2003). Similar, albeit less pronounced, brain temperature fluctuations also occurred during repeated passive cocaine injections (Kiyatkin and Brown, 2004). While the inhibiting action of iv cocaine on monoamine uptake might be responsible for its locomotor stimulatory and brain temperature-increasing effects, a direct inhibition of central Na+ channels may underlie rapid, transient decreases in brain temperature after each repeated drug administration. Although cocaine’s affinity for Na+ channels is lower than that for monoamine transporters (IC50=14-17 μM; Gifford and Johnson, 1992; Ritz et al., 1987), rapid and strong increases in brain cocaine levels after each repeated injection are high enough for full manifestation of this action. When locally applied by iontophoresis, cocaine potently inhibits striatal neurons (Kiyatkin and Rebec, 2000). These inhibitions are especially strong during glutamate-induced neuronal excitation, totally resistant to systemic DA receptor blockade, and mimicked by iontophoretic procaine (PRO), a short-acting blocker of Na+ channels with minimal effects on DA uptake (Ritz et al., 1987). In contrast to the relatively slow and prolonged action on DA uptake (Heron et al., 1994; Kiyatkin et al., 2000; Pogun et al., 1991), cocaine’s action on Na+ channels is rapid and transient, tightly following rapid fluctuations in brain cocaine levels. Therefore, it may be hypothesized that both these functionally antagonistic central actions, acting in concert, are sufficient to drive and regulate cocaine self-administration behavior.
In support of this hypothesis, we recently showed that systemic DA receptor blockade completely eliminates locomotor-stimulatory and temperature-increasing effects of iv cocaine in the brain, but fails to affect its temperature-decreasing effects in both the brain and skin (Kiyatkin and Brown, 2005). Instead of rapid increases and biphasic, down-up fluctuations following repeated iv injections seen in control animals, during DA receptor blockade cocaine only resulted in phasic decreases in brain temperature after each injection. Although these findings support a role for DA in locomotor stimulatory and temperature-increasing effects of cocaine and suggest that DA-independent drug actions might be responsible for cocaine-induced decreases in brain and skin temperature, the exact role of cocaine’s interaction with Na+ channels in mediating its behavioral and physiological effects remain unclear. This study was designed to clarify these issues by comparing the behavioral and temperature effects of iv cocaine, PRO and cocaine methiodide (COC-MET). While PRO can be used to assess the role of cocaine’s action on central and peripheral Na+ channels, COC-MET, a quaternary derivative of cocaine that is unable to cross the brain-blood barrier (Shriver and Long, 1971), is important for the examination of peripheral mechanisms that may mediate cocaine’s central effects.
Similar to our previous studies (Kiyatkin and Brown, 2004, 2005), cocaine was given to awake, unrestrained rats at the optimal self-administration dose (1 mg/kg) as a series of 10 repeated passive iv injections delivered in a pattern that mimicked the timing of self-administration behavior (8-min inter-injection interval). PRO was delivered at an equipotential anesthetic dose (5 mg/kg) and COC-MET at an equimolar dose (1.31 mg/kg) according to the same experimental protocol. Since DA receptor blockade dramatically alters the behavioral and temperature effects of cocaine, we also assessed how DA receptor blockade, induced by a mixture of D-1 and D-2 selective DA antagonists (SCH23390 and eticlopride) affects the behavioral and temperature effects of PRO and COC-MET. Finally, to understand the potential role cocaine’s interaction with Na+ channels may play in a conditioned response to the drug, we studied the behavioral and temperature effects of PRO in rats previously treated with cocaine.
2. Materials and Methods
2.1. Subjects and Surgery
Nineteen Long-Evans male rats (Taconic, Germantown, NY), weighing 420-480 g and housed in a 12 h light cycle (lights on at 0700) with ad libitum food and water, were used. Protocols were performed in compliance with the Guide for the Care and Use of Laboratory Animals (NIH, Publications 865-23) and were approved by the Animal Care and Use Committee, NIDA-IRP.
Each rat underwent the same procedure of thermocouple electrode and jugular iv catheter implantation. Thermocouples were prepared as previously described (Kiyatkin and Brown, 2003). Animals were anesthetized with Equithesin (3.3 ml/kg i.p.) and mounted in a stereotaxic apparatus. Holes were drilled through the skull over NAcc shell (1.2 mm anterior to the bregma, 0.9 mm lateral to the bregma) according to the coordinates of Paxinos and Watson (1998). The dura matter was retracted and a thermocouple probe was slowly lowered to the desired target depth (7.4 mm). A second thermocouple probe was implanted subcutaneously along the nasal ridge with the tip approximately 15 mm from bregma. A third thermocouple probe was implanted in deep temporal muscle (musculus temporalis). The probes were secured with dental cement to three stainless steel screws threaded into the skull. For jugular catheter implantation, a 10 mm incision was made in the neck to expose the jugular vein. A catheter was then inserted into, and secured to, the vein, and the catheter was run subcutaneously to the head mount and secured with dental cement. Rats were allowed three days recovery and one day of habituation (6 h session) to the testing environment before the start of testing.
2.2. Experimental Protocol
All tests occurred inside a Plexiglas chamber (32×32×32 cm), equipped with four infrared motion detectors (Med Associates, Burlington, VT), placed inside of a sound attenuation chamber with an environmental temperature maintained at 23±0.5 °C. Rats were brought to the testing chamber at 9:00 and attached via a flexible cord and electrical commutator to thermal recording hardware (Thermes 16, Physitemp, Clifton, NJ). A catheter extension was also attached to the internal catheter, thereby allowing remote iv injections. Temperatures were recorded with a time resolution of 10 s and movement was recorded as the number of infrared beam breaks per 1 min.
All animals were divided into three equal groups according to the drug being tested (cocaine, PRO and COC-MET; n=7, 6 and 6, respectively). Animals of each group were exposed to three similar recording sessions; one additional session was given in the cocaine group. After a habituation period (at least 3 hrs), rats were given a sc injection of either saline (Session 1 and 3) or a mixture of DA antagonists (Session 2) and 30 min later they were exposed to a series of ten repeated iv injections of one of three drugs [(-) cocaine HCl, procaine HCl or PRO, (-) cocaine methiodide or COC-MET]. Intervals between injections were 8 min, corresponding to the modal value of inter-injection intervals during cocaine self-administration (Kiyatkin and Brown, 2003). Each drug was dissolved in saline and delivered as a 0.15 ml injection over 16 s. The cocaine dose was 1 mg/kg, which is the optimal dose for maintaining self-administration, COC-MET was used at an equimolar dose (1. 31 mg/kg), and PRO was used at a dose (5mg/kg) that is equipotential to cocaine’s local anesthetic action. At this dose, procaine has virtually no effects on DA uptake because its affinity for the DA transporter is more than 100 times less than that for cocaine (153:1; Wilcox et al., 2001; 260:1; Ritz et al., 1987). Recording was continued for at least 120 min after the last iv injection. To eliminate the influence of day-to-day differences in basal temperatures (Kiyatkin et al., 2002) and increase data sample, animals of each group were exposed to two identical saline-pretreatment sessions (Day 1 and 3) and one DA antagonists-pretreatment session (Day 2). One free day was allowed after the session with DA antagonists to eliminate any possible consequences of this treatment. In the cocaine group, one additional session was conducted after two free days. During this session, 30-min after a saline injection, rats were exposed to a series of repeated PRO injections (5 mg/kg with 8-min inter-injection intervals). During free days before this session, the catheter was flushed twice with saline to remove any remaining drug.
To induce DA receptor blockade, we used a mixture of D1-like selective (SCH23390 0.2 mg/kg) and D2-like selective (eticlopride or ETI 0.2 mg/kg) antagonists. As previously reported, a SCH+ETI mixture at these doses effectively attenuated striatal neuronal responses to iontophoretic DA (Kiyatkin and Rebec, 1999), strongly decreased spontaneous movement activity and fully blocked locomotor activation induced by iv cocaine (Kiyatkin and Brown, 2005). These drugs were freshly dissolved in saline before each treatment session.
2.3. Histology and Data Analysis
When recording was completed, all rats were anesthetized, decapitated, and had their brains removed for sectioning and confirmation of probe placement. Brains were cut on a cryostat into 50μ slices and placed on glass slides. All brain probes were located within NAcc shell, as described in Paxinos and Watson (1998).
Temperature and movement data were analyzed with 1-min time bins and presented as both absolute and relative changes. We analyzed both tonic changes during a drug session (vs. pre-injection baseline) and phasic changes associated with either the first or repeated (2-10) drug injections of each tested drug. ANOVA with repeated measures, followed by post-hoc Fisher tests, was used for statistical evaluation of drug-induced changes in temperature and movement. Student’s t-test was used for comparisons of between-site, between-drug and between-treatment (saline vs. DA antagonists) differences in temperature and locomotion.
3. Results
3.1. Behavioral and temperature effects of cocaine, procaine and cocaine methiodide
3.1.1. Tonic changes
As shown in Fig. 1A, NAcc temperature significantly increased after the first injection of each tested drug and remained elevated during subsequent repeated injections. This tonic increase was weakest for cocaine (∼0.3°C) and larger for PRO (∼0.6°C) and COC-MET (∼0.5°C). Skin temperature rapidly decreased after the first injection of each drug and then either fluctuated biphasically at levels below baseline (cocaine) or near baseline (PRO and COC-MET). Temperature changes in muscle for the most part were similar to those in the NAcc. With each drug NAcc-muscle differentials rapidly increased after the first injection of each drug and remained at higher levels during subsequent repeated injections (Fig. 1B). In contrast, skin-muscle differentials robustly decreased after the first injection and either became cyclical (cocaine) or slowly returned toward pre-injection baseline (PRO and COC-MET) following subsequent injections. Locomotor activity was greatly increased and phasic during repeated cocaine administration but was altered little by repeated PRO and COC-MET (Fig. 1C).
Fig. 1.
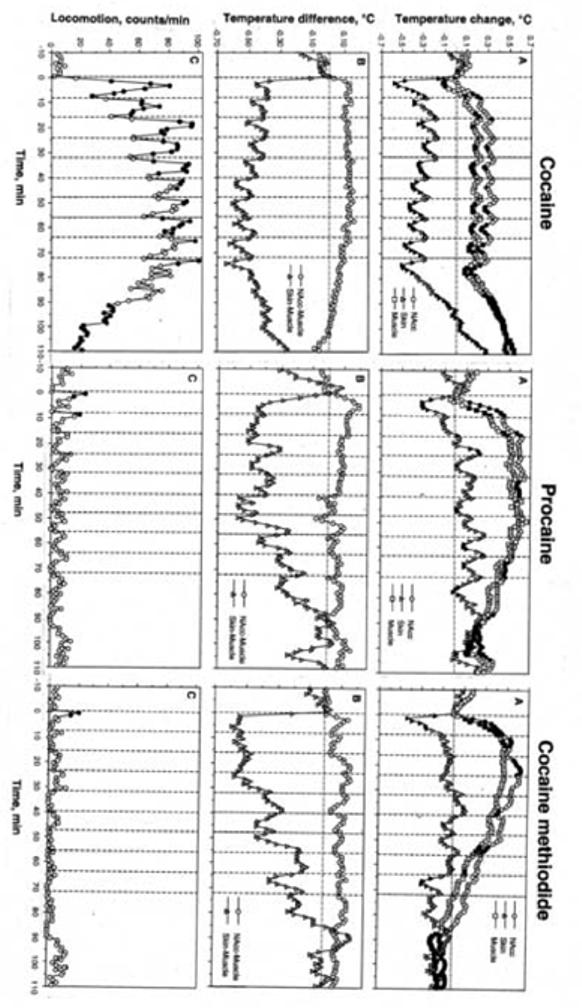
Mean changes in temperature (A, B) and locomotion (C) following a series of ten iv injections of cocaine, procaine and cocaine methiodide in awake, unrestrained rats in drug-naive conditions. A= relative temperature change with respect to the last pre-injection value. B= NAcc-Muscle and Skin-Muscle temperature differentials. Filled symbols show values significantly different (p<0.05) from the last-pre-injection value. The effect of time on both temperature and movement was statistically evaluated for each drug with one-way ANOVA with repeated measures. F values (NAcc, skin, muscle, and movement, respectively) were for cocaine: F20,650=11.40, 14.29, 24.43, and 15.98; procaine: F20,650=11.40, 14.29, 24.43, and 15.98; and for cocaine-methiodide: F20,650=11.40, 14.29, 24.43, and 15.98. All values indicate significant effect, with p<0.01. Vertical hatched lines show the moments of drug injection and horizontal hatched line show basal values preceding the first drug injection.
3.1.2. Phasic changes
As can be seen in Fig. 2A, for each drug brain and muscle temperatures significantly increased and skin temperature significantly decreased after the first injection of a series. The NAcc temperature increase was larger and more rapid with PRO and COC-MET than with cocaine. In contrast, skin temperature decrease was equally rapid with each drug and maximal in amplitude (-0.6°C) after cocaine. Muscle temperature generally followed brain temperature, but the increase was weaker and delayed with each drug. With cocaine, but not with PRO and COC-MET, muscle temperature decreased significantly immediately after injection.
Fig. 2.
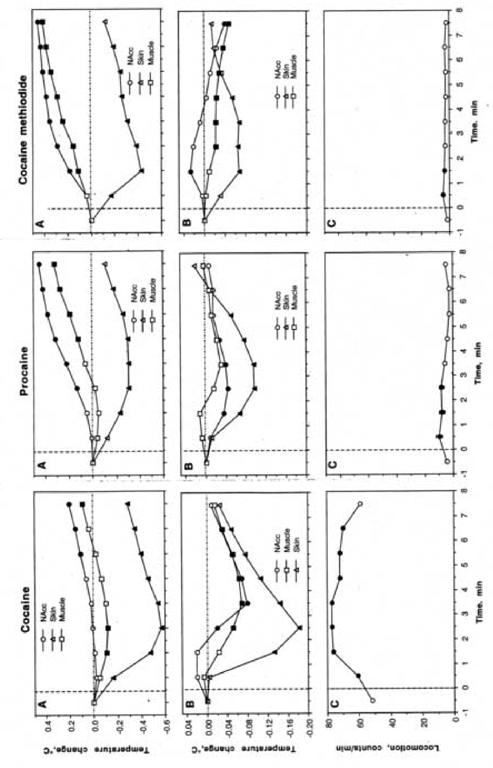
Mean changes in temperature following the initial (A) and repeated (B) injections of cocaine, procaine and cocaine methiodide in drug-naive conditions. C=changes in locomotion following repeated injections of each drug. Filled symbols show values significantly different from the last pre-injection value (p<0.05; Scheffe post-hoc test based on significant time effects evaluated by ANOVA with repeated measures). F values of the effect of the initial injections were presented in abbreviations to Fig. 1. F values (NAcc, skin, muscle, and movement, respectively) for repeated injections were for cocaine: F125,1133=19.61, 88.39, 19.34, and 33.15 (each p<0.001), for procaine: F53,485=2.94, 6.18, 1.68, and 4.54 (each, expect muscle, is significant with p<0.01); for cocaine-methiodide: F125,1133=6.03, 2.45, 4.61, and 2.12 (each is significant with p<0.05).
When repeated injections (2-10) are averaged, it is apparent that cocaine induced significant biphasic temperature fluctuations in each location, with maximal change in the skin (Fig. 2B). Repeated cocaine injections also induced biphasic fluctuations in locomotion, though it was the inverted, mirror image of temperature fluctuations (Fig. 2C). While PRO had little effect on locomotion, it induced similar, although weaker, biphasic temperature fluctuations in the NAcc and skin, with no changes in the muscle. Similar to cocaine, post-PRO temperature decrease was maximal in skin. In contrast to cocaine and PRO, COC-MET showed a weak, transient increase in NAcc temperature with a gradual decrease in skin and muscle temperatures. Changes in skin temperature followed the same pattern as with cocaine and PRO, but the change was smaller.
3.1.3. Between-drug comparisons
NAcc temperature increase following the first injection for both PRO and COC-MET was significantly more rapid and stronger than that for cocaine (Fig. 3). Although each drug induced a similar decrease in skin temperature, it was strongest for cocaine and weakest for PRO. In contrast to the first injection, NAcc temperature transiently decreased during repeated injections of both cocaine and PRO; the decrease was more rapid but weaker for PRO than for cocaine. The COC-MET-induced change was significantly different from that of cocaine, showing a delayed biphasic fluctuation. Similar to the first injection, each drug transiently decreased skin temperature after repeated injections, but the effect of COC-MET was minimal, significantly weaker than that of cocaine.
Fig. 3.
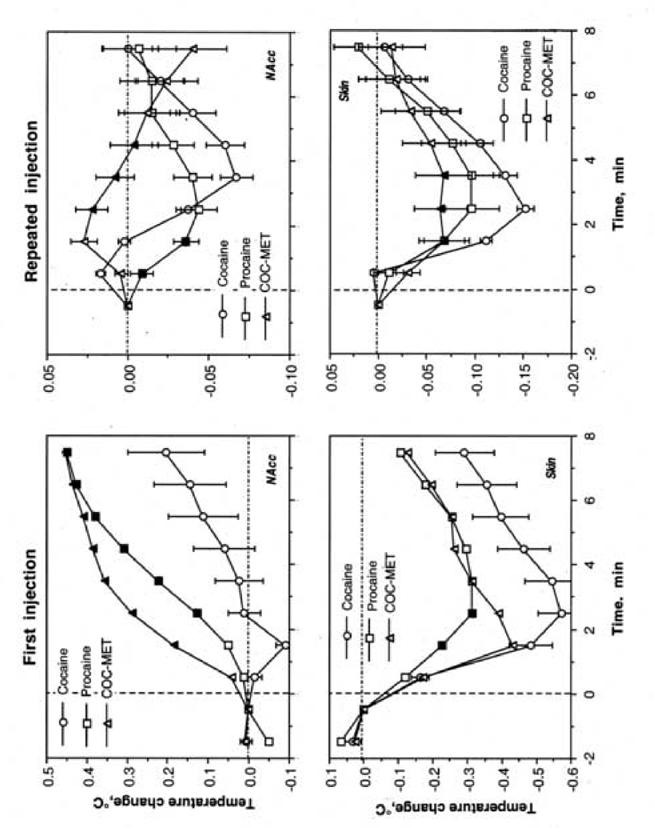
Between-drug differences in temperature changes in the NAcc (top) and skin (bottom) following the initial injection of cocaine, procaine and cocaine methiodide. Filled symbols show values significantly different from those of cocaine.
3.2. The effects of DA receptor blockade on drug-induced changes in temperature and locomotion
3.2.1. Tonic changes
DA receptor blockade dramatically altered temperature changes induced by all drugs (Fig. 4A,B) and completely eliminated drug-induced locomotor responses (Fig. 4C). In contrast to moderate increases in brain and muscle temperature induced by cocaine in control conditions, these parameters decreased under DA receptor blockade. However, the temperature-decreasing effects of cocaine remained intact, resulting in a negative temperature trend. In contrast to the stable increase in brain-muscle differential seen in control conditions, this parameter decreased below the pre-injection baseline. Similar to control, during DA receptor blockade skin-muscle differential also decreased during repeated cocaine injections, though this decrease was greater in control.
Fig. 4.
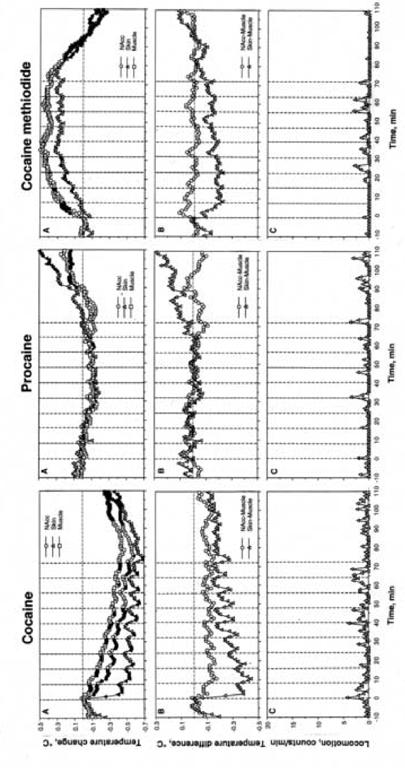
Mean changes in temperature (A,B) and locomotion (C) following a series of ten iv injections of cocaine, procaine and cocaine methiodide in awake, unrestrained rats during DA receptor blockade. A= relative temperature change with respect to the last pre-injection value. B= NAcc-Muscle and Skin-Muscle temperature differentials. Filled symbols show values significantly different (p<0.05) from the last-pre-injection value. The effect of time on both temperature and movement was statistically evaluated for each drug with one-way ANOVA with repeated measures. F values (NAcc, skin, muscle, and movement, respectively) were for cocaine: F20,650=11.40, 14.29, 24.43, and 15.98; procaine: F20,650=11.40, 14.29, 24.43,15.98; for cocaine-methiodide: F20,650=11.40, 14.29, 24.43, and 15.98. All values indicate significant effect, with p<0.01. Vertical hatched lines show the moments of drug injection and horizontal hatched line show basal values preceding the first drug injection.
Pre-treatment with DA antagonists completely eliminated the gradual increases in brain and muscle temperature and decreases in skin temperature induced by repeated PRO injections seen in control conditions (Fig. 4). Similarly, during this treatment PRO-induced changes in brain-muscle and skin-muscle differential were completely eliminated.
In contrast, DA receptor blockade had minimal effect on brain and muscle temperature changes induced by COC-MET (Fig. 4A). The increase, however, was less sharp, but more tonic than in control. Skin temperature decrease was blocked and this parameter instead followed brain and muscle temperatures. Changes in Nacc- and Skin-muscle differential were weaker. The effect was especially evident for Skin.
3.2.2. Phasic changes
DA receptor blockade totally eliminated brain and muscle temperature increase following the first injection of cocaine, decreasing temperature in both sites (Fig. 5A). Cocaine’s effect on skin temperature was similar to the control condition without the return toward baseline seen before the next injection. DA receptor blockade also eliminated all temperature change following the first PRO injection; NAcc, muscle and skin temperatures all remained at baseline levels. COC-MET had almost the same effect as PRO under DA receptor blockade: skin temperature did not change, and brain and muscle temperature increased, though less than they did under control conditions.
Fig.5.
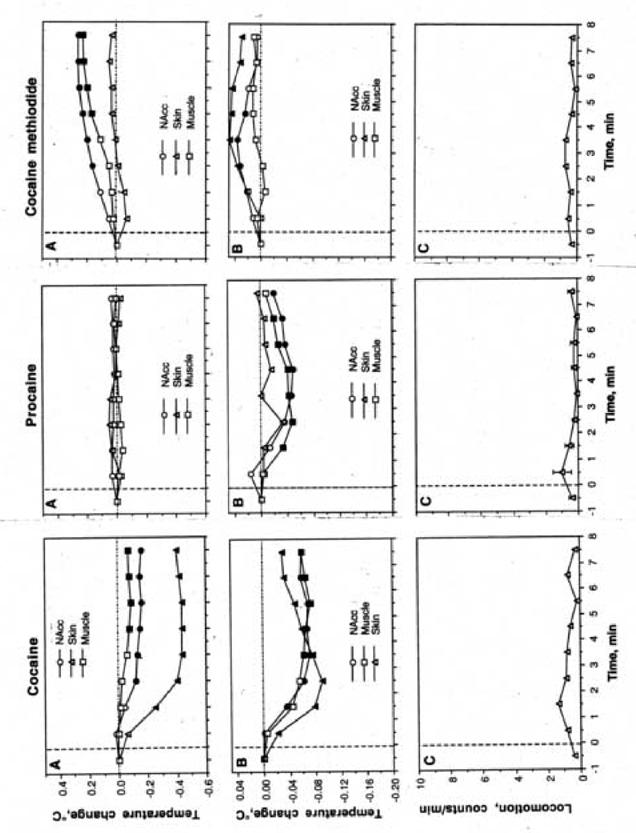
Mean changes in temperature following the initial (A) and repeated (B) injections of cocaine, procaine and cocaine methiodide during DA receptor blockade. C=changes in locomotion following repeated injections of each drug. Filled symbols show values significantly different from the last pre-injection value (p<0.05; Scheffe post-hoc test based on significant time effects evaluated by ANOVA with repeated measures). F values of the effect of the initial injections were presented in abbreviations to Fig. 4. F values (NAcc, skin, muscle, and movement, respectively) for repeated injections were for cocaine: F44,404=15.81, 8.28, 7.81, and 0.66 (each, expect movement, is significant with p<0.001); for procaine: F53,485=12.05, 0.62, 9.05, and 0.71 (all, except muscle and movement, are significant with p<0.001); and for cocaine-methiodide: F53,485=2.45, 3.51, 0.85, and 0.87 (each, expect muscle and movement, is significant with p<0.05).
Repeated injections were also altered by DA receptor blockade (Fig. 5B,C). Under this condition, cocaine still caused a decrease in temperature in all three sites, but skin temperature did not decrease as greatly and all three temperatures did not return to baseline as occurred under control conditions. Temperatures following repeated PRO administration did fluctuate but were more muted under DA receptor blockade. Finally, repeated COC-MET administration led to a slight increase in skin and NAcc temperature and no change in muscle temperature after pre-treatment with DA antagonists. Additionally, with all three drugs locomotor activity was nearly non-existent during DA receptor blockade.
3.2.3. Between-condition comparisons
Figure 6 compares differences in temperature effects of the initial injection of cocaine, PRO and COC-MET in control conditions and during DA receptor blockade separately for NAcc (top) and skin (bottom). DA antagonists significantly altered cocaine-induced change in NAcc temperature. Rather than increasing, NAcc temperature slightly decreased. Skin temperature was not significantly changed. DA antagonists also significantly altered PRO-induced changes in NAcc and skin temperatures. In both sites, PRO had no effect on temperature, which remained near baseline levels. Although the increase in NAcc temperature induced by initial COC-MET injection during DA receptor blockade was weaker than in control, this difference was not significant. In contrast, DA receptor blockade almost completely eliminated the decrease in skin temperature induced by the initial administration of this drug.
Fig. 6.
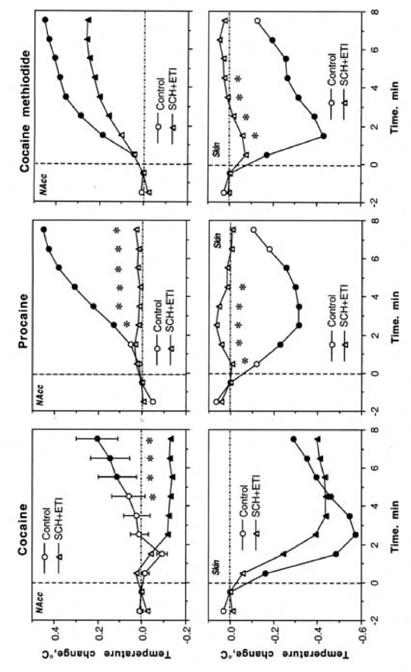
Differences in temperature changes induced by the initial injection of cocaine, procaine and cocaine methiodide in drug-naive conditions (control) and during DA receptor blockade (SCH+ETI). Filled symbols show values significantly different from the last pre-injection values and asterisks show between-group differences (p<0.05; Student’s t-test).
The same, within-drug comparison for repeated drug injections is shown in Figure 7. During repeated cocaine injections following DA antagonists, NAcc temperature decreased more quickly and remained at a lower level than it did under control, but skin temperature did not change as much as it did in control. Following repeated PRO, NAcc temperature showed a delayed biphasic response, and skin temperature decrease was completely blocked by DA antagonists. Repeated COC-MET under DA receptor blockade did not result in altered NAcc temperatures. However, skin temperatures increased following an administration and did not fully return to baseline before the next administration.
Fig. 7.
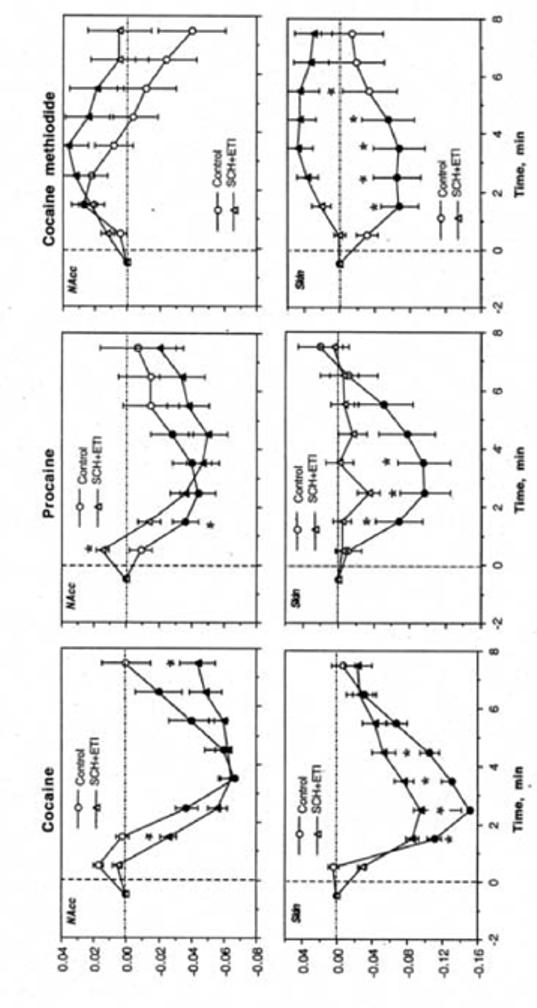
Differences in temperature changes induced by repeated injections of cocaine, procaine and cocaine-methiodide in drug naive conditions (control) and during DA receptor blockade (SCH+ETI). Filled symbols show values significantly different from the last pre-injection values and asterisks show between-group differences (p<0.05; Student’s t-test).
3.3. Locomotor and temperature effects of procaine in cocaine-experienced rats
Figure 8 shows changes in temperature (A, B) and locomotion (C) during a series of repeated PRO injections in cocaine-experienced rats. These rats were exposed to three days of cocaine treatment (Day 1-3; two with saline- and one with SCH+ETI-pretreatment) and tested with repeated PRO on Day 5. With respect to the effects of PRO in cocaine-naive conditions (see Fig. 1), the most important change in cocaine-experienced conditions was an increase in locomotion seen after the first and each subsequent PRO injection. This increase was clearly phasic and significantly higher than that induced by PRO in drug-naive conditions (Fig. 9). However, the pattern of PRO-induced hyperlocomotion was clearly different from that induced by cocaine in control conditions, with which there were phasic increases superimposed on a tonic increase in locomotor activity. Although locomotor responses to each PRO injection in cocaine-experienced animals were larger than in drug-naive controls (Fig. 10), the difference was minimal for the initial injection of a series, grew with subsequent injections (2-6) and decreased for the final injections of a series.
Fig. 8.

Changes in temperature (A, B) and locomotion (C) induced by procaine injections in drug-naive conditions and after cocaine experience. A shows relative temperature changes in cocaine-experienced conditions, B shows between-group differences in NAcc-Muscle differential, and C shows between-group differences in locomotion.
Fig. 9.
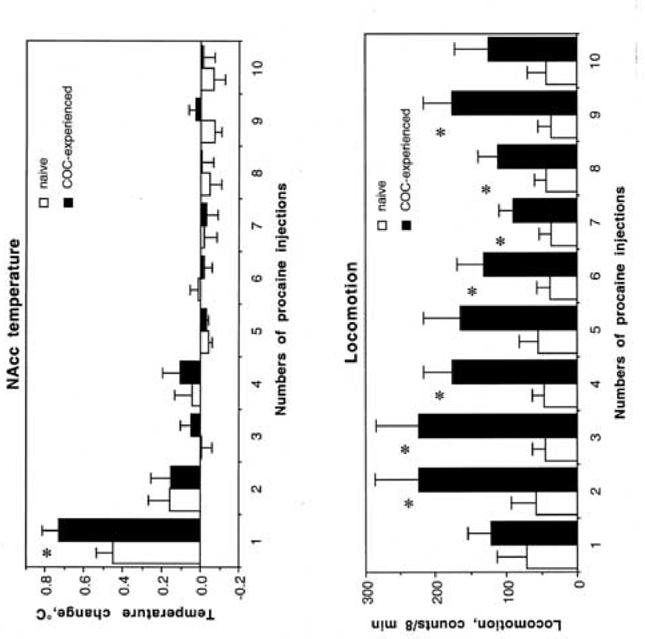
Changes in NAcc temperature (top) and locomotion (bottom) induced by each of ten procaine injections in drug-naïve (open symbols) and cocaine-experienced (black symbols) conditions. Asterisks show between-group differences (p<0.05; Student’s t-test).
Fig. 10.
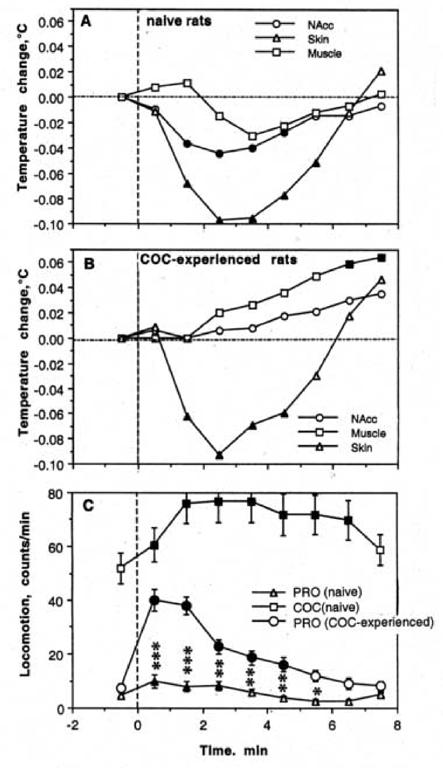
Mean changes in temperature (A, B) and locomotion (C) during repeated procaine injections in drug-naive conditions and after cocaine experience. Filled symbols show values significantly different from the pre-injection baseline. Asterisks show between-group differences (procaine in drug-naive vs. cocaine-experienced conditions) in locomotion (*, p<0.05; **, p<0.01, and *** p<0.001; Student’s t-test).
Although PRO given to cocaine-experienced rats resulted in a similar elevation in temperature as in cocaine-naive rats (compare Fig. 1 and 8), increases in brain and muscle temperature were significantly larger and brain-muscle differentials increased significantly stronger (Fig. 8B). As shown in Fig. 10, these differences in tonic NAcc temperatures resulted from a significantly stronger increase after the first PRO injection. Following subsequent injections, temperatures maintained at the same plateau, but in contrast to control conditions, fluctuations in brain and muscle temperature disappeared and both temperatures had a tendency to tonically increase. In contrast to strong effects on brain and muscle temperature, the effects of PRO on skin temperature in cocaine-experienced conditions were similar to drug-naive conditions. In both conditions, skin temperature strongly and phasically decreased after the first procaine injection, and showed significant down-up fluctuations with no changes in tonic baseline following subsequent injections.
4. Discussion
Although it is generally believed that brain temperature is a stable homeostatic parameter, it shows relatively rapid and strong fluctuations in response to salient somato-sensory stimuli and different drugs as well as during various types of motivated behavior (see Kiyatkin, 2005 for review). Our previous work revealed that cocaine at a typical self-administering dose (1 mg/kg, iv) has different effects on brain temperature depending upon the behavioral context and pattern of administration. While a single injection induces a monophasic temperature increase (Brown and Kiyatkin, 2005), repeated self-administrations result in biphasic, down-up fluctuations maintained at elevated, relatively stable levels (Kiyatkin and Brown, 2003). Because the time course of transient brain temperature decreases induced by repeated cocaine injections matches the time-course of its appearance in the brain upon iv administration (Fowler et al., 1998), we hypothesized that the direct action of cocaine on central Na+ channels might be responsible for this effect. Our finding that DA receptor blockade eliminates the locomotor and brain temperature-increasing effects of repeated cocaine but not its temperature-decreasing effects (Kiyatkin and Brown, 2005) indirectly supports such a mechanism. The present study was designed to further explore the possible role of Na+ channels in mediating the locomotor stimulatory and temperature effects of iv cocaine.
4.1. Thermorecording as a tool for studying metabolic brain activation and central effects of drugs
While the behavioral effects of addictive drugs can be evaluated by locomotion, alterations in brain, skin and muscle temperatures provide information to characterize their physiological effects. Since increased brain metabolism is accompanied by heat production (Laughlin et al., 1998; Ritchie, 1973; Siesjo, 1978), phasic increases in brain temperature induced by salient environmental challenges and drugs appear to reflect metabolic brain activation (see Kiyatkin, 2005 for review). Although such activation also enhances body metabolism and increases systemic and cerebral circulation, metabolic heat generated in the body cannot be delivered to the brain because arterial blood under physiological conditions is always cooler than brain tissue (Delgado and Hanai, 1966; Hayward and Baker, 1968; Kiyatkin et al., 2002; McElligott and Melzack, 1967; Nybo et al., 2002). While alterations in cerebral blood flow provide another indirect measure of metabolic brain activity (Mintun et al., 2001; Raichle, 2003; Trubel et al., 2006), they also affect heat dissipation from the brain, thus modulating brain temperature.
While brain temperature fluctuations depend primarily on brain metabolic activity, alterations in skin temperature are determined by the tone of peripheral vessels as well as changes in arterial blood temperature. Brain hyperthermia induced by salient environmental stimuli is typically accompanied by rapid, transient skin hypothermia (Baker et al., 1976; Kiyatkin, 2005), a result of limiting heat dissipation through vasoconstriction. Therefore, drug-induced changes in skin temperature may reflect not only local actions of drugs on skin vessels, but also a peripheral manifestation of metabolic brain activation and centrally mediated sympathoexcitation.
Finally, temperature changes in temporal muscle are important to determine both the source of initial heat production and direction of heat flow between the brain and the rest of the body. Since this head muscle is supplied with blood from the carotid artery just as the brain is, and as a non-locomotor muscle does not produce heat during movement, its temperature depends primarily on heat delivery by incoming blood. Therefore, the change in brain-muscle temperature differentials is a good indicator of metabolic activity. Hyperthermia induced by salient environmental challenges is always delayed in the muscle relative to the brain, implicating metabolic brain activation rather than blood as the source of brain hyperthermia (see Kiyatkin, 2005). Conversely, pentobarbital-induced hypothermia is stronger and more rapid in the brain than in the muscle, suggesting metabolic brain inhibition rather than decreased arterial blood temperature as its primary cause (Kiyatkin and Brown, 2005).
4.2. Cocaine’s interaction with peripheral Na+ channels as a trigger of its brain temperature-increasing and other central excitatory effects
While inducing negligible locomotor activation, both PRO and COC-MET generally mimicked the temperature-increasing effects of cocaine in the brain and induced similar skin hypothermia. As previously mentioned, rapid skin hypothermia reflects peripheral vasoconstriction, a known effect of iv cocaine and a component of its sympathoexitatory effect (Knuepfer and Branch, 1992; Crandall et al., 2002). Although cocaine is known to release peripheral amines in blood plasma (Robinson et al., 2000), it is well established that these effects are centrally mediated (Knuepfer and Branch, 1992; Szabo et al., 1995; Tella and Goldberg, 1998) and resistant to DA receptor blockade (Kiritsy-Roy et al., 1990; Poon and van den Buuse, 1992). Therefore, although peripheral vasoconstriction diminishes heat dissipation, metabolic brain activation (Breiter et al., 1997; Howell et al., 2002; Robinson et al., 2000; Thomas et al., 1995), appears to be the primary contributor to cocaine-induced brain hyperthermia and peripheral vasoconstriction. Intra-brain heat accumulation is most likely not due to cerebral vasoconstriction since cerebral blood flow increases rather than decreases following iv cocaine in awake animals (Dohi et al., 1990; Robinson et al., 2000; Stein and Fuller, 1993).
Despite ineffectiveness as a monoamine uptake inhibitor, PRO also caused rapid and strong brain temperature elevation and skin hypothermia, suggesting metabolic brain activation. While this similarity with cocaine may be surprising, iv PRO is known to mimic several of the same physiological effects as cocaine: an acute increase in arterial blood pressure (Pitts et al., 1987), rapid and strong EEG activation (Adamec and Stark-Adamec, 1987; Parekh et al., 1995), and powerful increases in cerebral blood flow (Servan-Shreiber et al., 1998). Iv PRO also induces strong subjective effects in healthy individuals (Servan-Shreiber et al., 1998). While these effects differ from those of cocaine, they are equally rapid, combining euphoria, anxiety, and fear as well as powerful sensory and somatic sensations. This centrally mediated sympathoexcitatory action appears to be the cause of skin hypothermia induced by iv PRO, which is not a vasoconstrictor but a vasodilator in vitro (Willatts and Reynolds, 1985). Despite an inability to cross the blood-brain barrier, iv COC-MET also increased brain and decreased skin temperatures as well as fully mimiced the cardiovascular effects of iv cocaine (Dickerson et al., 1999). Since PRO, like cocaine, interacts with Na+ channels (Ritz et al., 1987) and COC-MET acts only in the periphery (Shriver and Long, 1971), it appears that the brain hyperthermia and vasoconstriction that follows iv cocaine results primarily from the drug’s interaction with peripheral Na+ channels.
Although this study did not clarify where these peripheral Na+ channels are located, the afferent terminals of sensory nerves may be a possible substrate. These afferents are dense in venous walls (Goder et al., 1993; Michaelis et al., 1994) and equipped with polymodal receptors that are sensitive to shifting levels of various chemical substances in blood (Lee et al., 2005). Several ionic channels, including Na+ channels, are known to be involved in activating these afferents (see Lee et al., 2005 for review). While the exact mechanisms by which these receptors are activated are unclear, and special work is necessary to verify the role of Na+ channels in their stimulation, this receptor activation produces afferent drive to the CNS, involves thalamo-cortical pathways, and results in rapid and generalized brain activation, which is similar to that induced via activation of cutaneous sensory receptors (Ploner et al., 2002). This action, however, is different from a classic local anesthetic action, which is related to inhibition of transmembranal Na+ transport and block of neuronal activity, and it more resembles the action of somato-sensory stimuli, which induce metabolic brain activation by interacting with peripheral receptors and rapidly transmitting to the CNS via sensory pathways. Temperature responses induced by somato-sensory stimuli and cocaine also shared important similarities: more rapid and stronger temperature increase in brain than muscle, acute, transient skin hypothermia, and dependence of brain temperature increase upon basal brain temperatures (activity state).
Consistent with the key role DA plays in central organization of activational processes (LeMoal and Simon, 1991), we found that DA antagonists completely blocked cocaine-induced locomotor activation and eliminated its temperature-increasing effects, replicating our previous work (Kiyatkin and Brown, 2005). Unexpectedly, we found that DA receptor blockade also completely eliminates temperature-increasing effects of PRO in the brain and muscle, blocks its temperature-decreasing effects in the skin, but does not eliminate its temperature-decreasing effects in the brain and muscle. Therefore, it appears that the temperature-increasing effects of PRO, which are triggered via a DA-independent mechanism, are in fact DA-mediated, despite its inability (at this dose) to affect directly monoamine uptake. If iv PRO induces DA activation, one may assume that iv cocaine also induces rapid DA activation independently from its later action on DA uptake. Via this mechanism cocaine would provide phasic DA release, to be later potentiated by uptake inhibition, resulting in a rise of DA concentration. Such an action may explain the unusually rapid appearance (20-30 s or less) of DA in the NAcc of awake rats following iv cocaine as measured by fast-cyclic voltammetry (Heien et al., 2005). As shown in this study, DA is detected electrochemically at the same time as or even before cocaine reached the brain (∼20 sec) and peaks in arterial blood in striatum (∼120 s; Fowler et al., 1998) and well before it is able to cross the blood-brain barrier, diffuse to the DA transporter and inhibit pre-synaptic uptake. Although direct cocaine interaction with the DA transporter has been postulated as a possible cause of these rapid increases in extrasynaptic DA, this extremely fast effect is inconsistent with the more delayed and slower time-course of DA uptake inhibition (Heron et al., 1994; Kiyatkin et al., 2000; Pogun et al., 1991).
By using rapid neural transmission and inducing generalized brain activation, cocaine action on peripheral Na+ channels may be important in drug sensing and a number of its rapid subjective and physiological effects. Some of these rapid affective and physiological effects, moreover, occur more rapidly that expected based on the dynamics of cocaine concentration in the brain. Cocaine is perceived by humans within seconds after iv injection (Fischman and Schuster, 1983; Zernig et al., 2002) and several of its central effects (i.e., EEG, evoked potential, DA release) in both humans and animals occur too quickly to reflect inhibition of DA uptake or even a direct interaction of the drug with brain substrates (Heien et al., 2005; Mateo et al., 2004; Lukas et al., 1990; Matsuzaki et al., 1978). It is well known that acute subjective effects of iv cocaine are extremely rapid and generally resistant to DA receptor blockade (Gavin, 1986; Sherer et al., 1987). As shown in these studies, DA antagonists affect some of the more prolonged subjective effects, but strong feelings of crash and rush, major components of an euphoric experience, remain intact. This rapid neural action could provide an essential “sensory” drug cue and thus contribute to learning. Our finding that PRO given to cocaine-experienced animals is able to induce conditioned locomotion and significantly stronger brain temperature effects supports this role (see below).
4.3. Cocaine’s interaction with central Na+ channels as a primary mechanism determining its temperature-decreasing and other central inhibitory effects
We previously hypothesized that direct action on central Na+ channels is responsible for transient decreases in brain temperature during repeated cocaine injections. In the present study, repeatedly injected PRO consistently induced brain temperature decreases similar to those seen with cocaine. In contrast, peripherally acting COC-MET failed to induce this effect. The temperature-decreasing effects of both cocaine and PRO, moreover, remained intact during DA receptor blockade. Therefore, these data appear to confirm that cocaine’s action on central Na+ channels is essential for its temperature-decreasing effects. These temperature decreases were rapid and transient, inversely matching the increases in brain cocaine levels (Fowler et al., 1998). While a measured, 26 μM cocaine peak in the striatum following a single iv injection at 1 mg/kg dose is well above the levels for substantial binding to neuronal Na+ channels (Gifford and Johnson, 1992; Reith et al., 1986; Ritz et al., 1987), simple pharmacodynamic calculations suggest that brain cocaine levels during self-administration behavior, similarly fluctuating with each injection, would peak at about 80-100 μM. These levels are about 100 times larger than the ED50 for monoamine transporters (Hennings et al., 1999; Ritz et al., 1987), suggesting their saturation and thus functional inactivation. On the other hand, a direct action on Na+ channels is strongly concentration-dependent and its contribution should exponentially grow with repeated cocaine self-injections.
Work with iontophoresis suggests that locally applied cocaine inhibits impulse activity of central neurons by blocking trans-membrane Na+ transport. Both cocaine and PRO were found to be equally potent at inhibiting spontaneously active striatal neurons in awake, unrestrained rats (Kiyatkin and Rebec, 2000). As shown in this study, this effect was stronger during tonic neuronal excitation induced by continuous, low-current glutamate application and totally resistant to DA receptor blockade (SCH+ETI), which sometimes resulted in even stronger effects perhaps because of increased discharge rate and stronger neuronal responsiveness to glutamate induced by DA antagonists.
While it is clear that cocaine may inhibit central neurons via direct interaction with Na+ channels, it remains unclear which cells are inhibited and what the consequences of this inhibition might be. It is known, however, that the sensitivity of central neurons to local anesthetic action depends upon the size of their somata and terminals, presence or absence of the myelin sheath on their axons, the type of their intrinsic organization (autoactive vs. driven by excitatory inputs), as well as their ongoing activity state (Gasser and Erlanger, 1929; Gifford and Johnson, 1992; Kiyatkin and Rebec, 2000; Ritchie et al., 1965). While direct data are absent, monoamine-containing CNS neurons, which have relatively small somata and highly arborized, thin, nonmyelinated axons, should be more sensitive to local anesthetic action than other cells. It is known that these cells are strongly inhibited by iv cocaine in anesthetized animals (Einhorn et al., 1988; Pitts and Marwah, 1987); under this condition rapid excitatory action appears to be blocked. Although a rise in synaptic monoamine levels because of cocaine action on uptake is believed to be a primary factor determining this response, the time-course of neuronal responses was very rapid and generally similar for each type of monoamine-containing neurons. For example, DA cell inhibition was prominent at ∼30 s after iv injection, peaked at 1-2 min and slowly disappeared over the next 10-12 min. This time-course is consistent with the dynamics of local cocaine levels, but inconsistent with the relatively slow effect of this drug on DA uptake (see above).
4.4. Unique effects of cocaine and their possible mediation
While the similarity in the temperature effects of cocaine, PRO and COC-MET may clarify the role of central and peripheral Na+ channels in their mediation, the unique effects of cocaine, which are absent in PRO and COC-MET, may be related to its specific pharmacological action - a potent inhibition of monoamine uptake in the brain.
In contrast to other drugs, cocaine induced strong and rapid locomotor activation following the initial injection and phasic locomotor responses during each repeated injection. Since these effects were completely blocked by DA antagonists, it is logical to assume that they are related to the DA uptake-inhibiting properties of cocaine. However, cocaine-induced locomotor activation appeared surprisingly fast. Although the activity increased reliably during the first min following the initial cocaine injection, other indices of behavioral activation in most animals were evident at the end of the injection or within seconds after. Locomotor activation, moreover, grew following several cocaine injections and reached a tonic plateau with only phasic fluctuations of progressively decreasing amplitude. Finally, reduced but significant locomotor activation occurred after the initial injection of both PRO and COC-MET; these effects rapidly habituated following subsequent injections. Nevertheless, the increasing effect of PRO on locomotor activity was significant as an average for all repeated injections in the series. Therefore, it may be suggested that although the locomotor stimulatory action of cocaine is obviously related to DA uptake inhibition, it tightly interacts with other cocaine actions mediated via Na+ channels.
Cocaine also differed from PRO in several important aspects of temperature dynamics. While both drugs had temperature-increasing and -decreasing properties, their ratio was different. With repeated injections, cocaine induced stronger temperature decreases in each recording location than PRO, although their time-course was similar. Also, DA antagonists generally blocked most temperature effects of PRO, but made inhibitory effects of cocaine stronger, suggesting that other mechanisms, possibly uptake inhibition of other monoamines, are involved. Brain-muscle differentials following repeated cocaine treatment significantly decreased during DA receptor blockade, suggesting that under this condition this drug acts as a metabolic inhibitor. While it is difficult to speculate on the mechanisms determining these differences, cocaine under this self-administration-like treatment regiment should almost fully saturate all monoamine transporters, making them “functionally inactive” at regulating neurotransmission levels. Under these conditions, local anesthetic mechanisms, which are dose-dependent, continue to act with increasing potency. Therefore, it is possible to speculate that under conditions of monoamine uptake blockade, cocaine’s actions via peripheral and central Na+ channels serve as the primary means for regulating monoamine transmission.
Co-existence of these mechanisms with DA uptake inhibition may explain the dynamics of DA transmission during cocaine self-administration previously suggested by our electrochemical studies (Kiyatkin and Stein, 1995). While NAcc DA-dependent electrochemical signals gradually increase both before (because of drug-independent DA cell activity) and after the initial few cocaine self-injections (because of drug-induced DA cell activation and drug-induced uptake inhibition), they stabilize with consistent, phasic down-up fluctuations tightly related to individual self-injections. Therefore, it is logical to assume that transient post-cocaine decreases in DA suggested by our previous data may result from phasic inhibitions on DA cells because of direct inhibitory action of cocaine via its interaction with Na+ channels.
The contribution of DA-independent, Na+ channel-mediated actions of cocaine may also explain the paradoxical findings that DA transporter knockout animals are able not only to learn cocaine self-administration, but to maintain cyclical drug taking at levels comparable to wild-type animals (Rocha et al., 1998). Although the DA uptake mechanism is permanently inactivated in these animals and basal DA levels are extremely high (Jones et al., 1998; Benoit-Marand et al., 2000), cocaine still acts via Na+ channels and this action appears to be sufficient for the development of drug-taking behavior and its regulation in these hyper-DA animals.
4.5. Conclusions: Possible role of cocaine’s interaction with peripheral and central Na+ channels in the development and regulation of cocaine self-administration
The results of this study suggest that in addition to inhibiting monoamine uptake iv cocaine has two functionally opposite actions triggered via interaction with Na+ channels. The first action, which is presumably mediated via peripheral Na+ channels located on terminals of sensory nerves, is excitatory in nature and results in generalized metabolic brain activation. This action provides a sensory signal of the drug and is reflected in various rapid psychoemotional and physiological effects. Although this action is triggered independently of the DA system, strong attenuation of brain temperature increases resulting from DA receptor blockade, suggests a key role for DA mechanisms in its central organization and mediation. Analogous to other salient environmental stimuli, which phasically activate DA neurons (Kiyatkin, 1988; Strecker and Jacobs, 1985; Steinfels et al., 1983), this rapid drug action may provide phasic DA release, creating the proper setting for the more delayed action of cocaine as a DA reuptake inhibitor. By providing a rapid sensory signal of drug, this action appears to be important for learning and reinforcement, thus contributing to cocaine-induced behavioral sensitization, the development of cocaine self-administration, and its reinstatement in drug-experienced individuals. The appearance of conditioned locomotion and an increased brain temperature response to PRO after previous cocaine experience strongly support this role. Along with this action on peripheral Na+ channels, cocaine’s interaction with central Na+ channels appears to play an important role in regulating drug-taking behavior. By providing rapid neuronal inhibition via direct blockade of Na+ channels, neural activation that precedes drug intake is transiently blocked. This mechanism may play a key role in operant reinforcement and regulation of drug intakes, contributing to drug-induced euphoria and relaxation. Therefore, although cocaine’s interaction with the DA transporter appears to be essential in mediating the primary reinforcing effects of cocaine (Wise and Bozarth, 1987; Ritz et al, 1987), other mechanisms, particularly involving drug’s interaction with peripheral and central Na+ channels, appear to be important in making cocaine a unique reinforcer and cocaine self-administration a highly compulsive motivated behavior.
Acknowledgments
We thank Dr. Barry Hoffer for valuable comments regarding this manuscript. This research was supported by the Intramural Research Program of the NIH, NIDA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamec RE, Stark-Adamec C. The effects of procaine HCl on population cellular and evoked response activity within the limbic system of the cat. Evidence for differential excitatory action of procaine in a variety of limbic circuits. Prog. Neuropsychoparmacol. Biol. Psychiatry. 1987;11:345–364. doi: 10.1016/0278-5846(87)90012-1. [DOI] [PubMed] [Google Scholar]
- Baker M, Cronin M, Mountjoy D. Variability of skin temperature in the waking monkey. Am. J. Physiol. 1976;230:449–455. doi: 10.1152/ajplegacy.1976.230.2.449. [DOI] [PubMed] [Google Scholar]
- Benoit-Marand M, Jaber M, Gonon F. Release and elimination of dopamine in vivo in mice lacking the dopamine transporter: functional consequences. Eur. J. Neurosci. 2000;12:2985–2992. doi: 10.1046/j.1460-9568.2000.00155.x. [DOI] [PubMed] [Google Scholar]
- Breiter HC, Gollub RL, Weisskoff RM, Kennedy DN, Makris N, Berke JD, Goodman JM, Kantor HL, Gastfriend DR, Riorden JP, Mathew RT, Rosen BR, Hyman SE. Acute effects of cocaine on human brain activity and emotion. Neuron. 1997;19:591–611. doi: 10.1016/s0896-6273(00)80374-8. [DOI] [PubMed] [Google Scholar]
- Crandall CG, Vongpatanasin W, Victor RG. Mechanism of cocaine-induced hyperthermia in humans. Ann. Intern. Med. 2002;136:785–791. doi: 10.7326/0003-4819-136-11-200206040-00006. [DOI] [PubMed] [Google Scholar]
- Delgado J, Hanai T. Intracerebral temperatures in free-moving cats. Am. J. Physiol. 1966;211:755–769. doi: 10.1152/ajplegacy.1966.211.3.755. [DOI] [PubMed] [Google Scholar]
- Dickerson LW, Rodak DJ, Kuhn FE, Wahlstrom SK, Tessel RE, Visner MS, Schaer GL, Gillis RA. Cocaine-induced cardiovascular effects: Lack of evidence for a central nervous system site of action based on hemodynamic studies with cocaine methiodide. J. Cardiovasc. Pharmacol. 1999;33:36–42. doi: 10.1097/00005344-199901000-00006. [DOI] [PubMed] [Google Scholar]
- Dohi S, Jones MD, Hudak ML, Traystman RJ. Effects of cocaine on pial arterioles in cats. Stroke. 1990;21:1710–1714. doi: 10.1161/01.str.21.12.1710. [DOI] [PubMed] [Google Scholar]
- Einhorn LC, Johansen PA, White FJ. Electrophysiological effects of cocaine in the mesoaccumbens dopamine system: studies in the ventral tegmental area. J. Neurosci. 1988;8:100–112. doi: 10.1523/JNEUROSCI.08-01-00100.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischman MW, Schuster CR. A comparison of the subjective and cardiovascular effects of cocaine and procaine in humans. Pharmacol. Biochem. Behav. 1983;18:711–716. doi: 10.1016/0091-3057(83)90011-4. [DOI] [PubMed] [Google Scholar]
- Fowler JS, Volkow ND, Logan J, Gatley SJ, Pappas N, King P, Ding Y-S, Wang G-J. Measuring dopamine transporter occupancy by cocaine in vitro: radiotracer considerations. Synapse. 1998;28:111–116. doi: 10.1002/(SICI)1098-2396(199802)28:2<111::AID-SYN1>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Gasser HS, Erlanger J. The role of the fiber size in the establishment of a nerve block by pressure or cocaine. Am. J. Physiol. 1929;88:581–591. [Google Scholar]
- Gawin F. Neuroleptic reduction of cocaine-induced paranoia but not euphoria? Psychopharmacology. 1986;90:142–143. doi: 10.1007/BF00172886. [DOI] [PubMed] [Google Scholar]
- Gifford AN, Johnson KM. Comparison of the role of local anesthetic properties with dopamine uptake blockade in the inhibition of striatal and nucleus accumbens [3H]acetylcholine release by cocaine. J. Pharmacol. Exp. Ther. 1992;263:757–761. [PubMed] [Google Scholar]
- Goder R, Habler HJ, Janig W, Michaelis M. Receptor properties of afferent nerve fibers associated with the rat saphenous vein. Neurosci. Lett. 1993;164:175–178. doi: 10.1016/0304-3940(93)90885-o. [DOI] [PubMed] [Google Scholar]
- Hayward JN, Baker MA. Role of cerebral arterial blood in the regulation of brain temperature in the monkey. Am. J. Physiol. 1968;215:389–403. doi: 10.1152/ajplegacy.1968.215.2.389. [DOI] [PubMed] [Google Scholar]
- Heien ML, Khan AS, Ariansen JL, Cheer JF, Phillips PEM, Wassum KM, Weightman RM. Real-time measurements of dopamine fluctuations after cocaine in the brain of behaving rats. Proc. Natl. Acad. Sci. 2005;102:10023–10028. doi: 10.1073/pnas.0504657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkila RE, Orlanski H, Cohen G. Studies on the distinction between uptake inhibition and release of [3H] -dopamine in rat brain tissue slices. Biochem. Pharmacol. 1975;24:847–852. doi: 10.1016/0006-2952(75)90152-5. [DOI] [PubMed] [Google Scholar]
- Hennings EC, Kiss JP, De Oliveira K, Toth PT, Vizi ES. Nicotinic acetylcholine receptor antagonistic activity of monoamine uptake blockers in rat hippocampal slices. J. Neurochem. 1999;73:1043–50. doi: 10.1046/j.1471-4159.1999.0731043.x. [DOI] [PubMed] [Google Scholar]
- Hemby SE, Co C, Koves TR, Smith JE, Dworkin SI. Differences in extracellular dopamine concentrations in the nucleus accumbens during response-dependent and response-independent cocaine administration in the rat. Psychopharmacology. 1997;133:7–16. doi: 10.1007/s002130050365. [DOI] [PubMed] [Google Scholar]
- Heron C, Costentin J, Bonnet J-J. Evidence that pure uptake inhibitors including cocaine interact slowly with the dopamine neuronal carrier. Eur. J. Pharmacol. 1994;264:391–398. doi: 10.1016/0014-2999(94)00502-8. [DOI] [PubMed] [Google Scholar]
- Howell LL, Hoffman JN, Votaw JR, Landrum AM, Wilcox KM, Lindsey KP. Cocaine-induced brain activation determined by positron emission tomography neuroimaging in conscious rhesus monkeys. Psychopharmacology. 2002;159:154–160. doi: 10.1007/s002130100911. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J. Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritsy-Roy JA, Halter JB, Gordon SM, Smith MJ, Terry LC. Role of the central nervous system in hemodynamic and sympathoadrenal responses to cocaine in rats. J. Pharmacol. Exp. Ther. 1990;256:154–160. [PubMed] [Google Scholar]
- Kiyatkin EA. Functional properties of presumed dopamine-containing and other ventral tegmental area neurons in conscious rats. Int. J. Neurosci. 1988;42:21–43. doi: 10.3109/00207458808985756. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA. Brain hyperthermia as physiological and pathological phenomena. Brain Res. Rev. 2005;50:27–56. doi: 10.1016/j.brainresrev.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Brown PL, Wise R. Brain temperature fluctuation: a reflection of functional neural activation. Eur. J. Neurosci. 2002;16:164–168. doi: 10.1046/j.1460-9568.2002.02066.x. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Brown PL. Fluctuations in neural activity during cocaine self-administration: clues provided by brain thermorecording. Neuroscience. 2003;116:525–538. doi: 10.1016/s0306-4522(02)00711-x. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Brown PL. Brain temperature fluctuations during repeated passive vs. active cocaine administration: Clues for understanding the pharmacological determination of drug-taking behavior. Brain Res. 2004;1005:101–116. doi: 10.1016/j.brainres.2004.01.038. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Brown PL. Dopamine-dependent and dopamine-independent actions of cocaine as revealed by brain thermorecording in freely moving rats. Eur. J. Neurosci. 2005;22:9430–938. doi: 10.1111/j.1460-9568.2005.04269.x. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Brown PL. Brain and body temperature homeostasis during sodium pentobarbital anesthesia with and without body warming in rats. Physiol. Behav. 2005;84:563–570. doi: 10.1016/j.physbeh.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Kiyatkin DE, Rebec GV. Phasic inhibition of dopamine uptake in nucleus accumbens induced by intravenous cocaine in freely behaving rats. Neuroscience. 2000;98:729–741. doi: 10.1016/s0306-4522(00)00168-8. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Rebec GV. Striatal neuronal activity and responsiveness of striatal neurons to dopamine and glutamate after selective blockade of D1 and D2 dopamine receptors in awake rats. J. Neurosci. 1999;19:3594–3609. doi: 10.1523/JNEUROSCI.19-09-03594.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyatkin EA, Rebec GV. Dopamine-independent action of cocaine on striatal and accumbal neurons. Eur. J. Neurosci. 2000;12:1–13. doi: 10.1046/j.1460-9568.2000.00066.x. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Stein EA. Fluctuations in nucleus accumbens dopamine during cocaine self-administration behavior: an in vivo electrochemical study. Neuroscience. 1995;64:599–617. doi: 10.1016/0306-4522(94)00436-9. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Stein EA. Conditioned changes in nucleus accumbens dopamine signal established by intravenous cocaine in rats. Neurosci. Lett. 1996;211:73–76. doi: 10.1016/0304-3940(96)12731-2. [DOI] [PubMed] [Google Scholar]
- Knuepfer MM, Branch CA. Cardiovascular responses to cocaine are initially mediated by the central nervous system in rats. J. Pharmacol. Exp. Ther. 1992;263:734–741. [PubMed] [Google Scholar]
- Laughlin SB, de Ruyter van Steveninck RR, Anderson JC. The metabolic cost of neural information. Nature Neurosci. 1998;1:36–41. doi: 10.1038/236. [DOI] [PubMed] [Google Scholar]
- Lee TH, Balu R, Davidson C, Ellinwood EH. Differential time-course profiles of dopamine release and uptake changes induced by three dopamine uptake inhibitors. Synapse. 2001;41:301–310. doi: 10.1002/syn.1087. [DOI] [PubMed] [Google Scholar]
- Lee Y, Lee C-H, Oh U. Painful channels in sensory neurons. Mol. Cells. 2005;20:315–324. [PubMed] [Google Scholar]
- Le Moal M, Simon H. Mesocortical dopaminergic network: functional and regulatory roles. Physiol Rev. 1991;71:155–234. doi: 10.1152/physrev.1991.71.1.155. [DOI] [PubMed] [Google Scholar]
- Lindoft HH. Investigation of the vascular effect of newer local anesthetics and vasoconstrictors. Oral. Surg. Oral. Med. Oral. Pathol. 1979;48:2192–297. doi: 10.1016/0030-4220(79)90026-4. [DOI] [PubMed] [Google Scholar]
- Lukas SE, Mendelson JH, Amass L, Benedikt R. Behavioral and EEG studies of acute cocaine administration: comparisons with morphine, amphetamine, pentobarbital, nicotine, ethanol and marijuana. NIDA Res. Monogr. 1990;95:146–152. [PubMed] [Google Scholar]
- Mateo Y, Budygin EA, Morgan D, Roberts DCS, Jones SR. Fast onset of dopamine uptake inhibition by intravenous cocaine. Eur. J. Neurosci. 2004;20:2838–2842. doi: 10.1111/j.1460-9568.2004.03736.x. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Spingler PJ, Whitlock EG, Misra AL, Mule SJ. Comparative effects of cocaine and pseudococaine on EEG activities, cardiorespiratory functions, and self-administration behavior in the rhesus monkey. Psychopharmacology (Berl) 1978;57:12–20. doi: 10.1007/BF00426951. [DOI] [PubMed] [Google Scholar]
- McElligott JC, Melzack R. Localized thermal changes evoked in the brain by visual and auditory stimulation. Exp. Neurol. 1967;17:293–312. doi: 10.1016/0014-4886(67)90108-2. [DOI] [PubMed] [Google Scholar]
- Michaelis M, Goder R, Habler HJ, Janig W. Properties of afferent nerve fibers supplying the saphenous vein in the cat. J. Physiol. (Lond) 1994;474:233–243. doi: 10.1113/jphysiol.1994.sp020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintun MA, Lundstrom BN, Snyder AZ, Vlasenko AG, Shulman GL, Raichle ME. Blood flow and oxygen delivery to human brain during functional activity: Theoretical modeling and experimental data. Proc. Natl. Acad. Sci. 2001;98:6859–6864. doi: 10.1073/pnas.111164398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nybo L, Secher NH, Nielson B. Inadequate heat release from the human brain during prolonged exercise with hyperthermia. J. Physiol. 2002;545:697–704. doi: 10.1113/jphysiol.2002.030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh PI, Spencer JW, George MS, Gill DS, Ketter TA, Andreason P, Herscovitch P, Post RM. Procaine-induced increases in limbic rCBF correlate positively with increases in occipital and temporal EEG fast activity. Brain Topogr. 1995;7:209–218. doi: 10.1007/BF01202380. [DOI] [PubMed] [Google Scholar]
- Paxinos J, Watson C. The Rat Brain in Stereotaxic Coordinates. Fourth Edition Academic Press; San Diego: 1998. [Google Scholar]
- Pitts DK, Udom CE, Marwah J. Cardiovascular effects of cocaine in anesthetized and conscious rats. Life Sci. 1987;40:1099–1111. doi: 10.1016/0024-3205(87)90573-x. [DOI] [PubMed] [Google Scholar]
- Pitts DK, Marwah J. Cocaine modulation of central monoaminergic neurotransmission. Pharmacol. Biochem. Behav. 1987;26:453–461. doi: 10.1016/0091-3057(87)90147-x. [DOI] [PubMed] [Google Scholar]
- Ploner M, Holthusen H, Noetges P, Schnitzler A. Cortical representation of venous niciception in humans. J. Neurophysiol. 2002;88:300–305. doi: 10.1152/jn.2002.88.1.300. [DOI] [PubMed] [Google Scholar]
- Pogun S, Scheffel U, Kuhar MJ. Cocaine displaces [3H]WIN 35,428 binding to dopamine uptake sites in vivo more rapidly than mazindol or GBR 12909. Eur. J. Pharmacol. 1991;198:203–205. doi: 10.1016/0014-2999(91)90622-w. [DOI] [PubMed] [Google Scholar]
- Poon J, van den Buuse M. Autonomic mechanisms in the acute cardiovascular effects of cocaine in conscious rats. Eur. J. Pharmacol. 1986;363:147–152. doi: 10.1016/s0014-2999(98)00804-8. [DOI] [PubMed] [Google Scholar]
- Reith MEA, Kim SS, Lajtha A. Structural requirements for cocaine congeners to interact with [3H]batrachotoxin A20-α-benzoate binding sites on sodium channels in mouse brain synaptosomes. J. Biol. Chem. 1986;261:7300–7305. [PubMed] [Google Scholar]
- Raichle ME. Functional brain imaging and human brain functions. J. Neurosci. 2003;23:3959–3962. doi: 10.1523/JNEUROSCI.23-10-03959.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie JM. Energetic aspects of nerve conduction: the relationships between heat production, electrical activity, and metabolism. Prog. Biophys. Mol. Biol. 1973;26:147–187. doi: 10.1016/0079-6107(73)90019-9. [DOI] [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors of dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- Robinson R, Iida H, O’Brien TP, Pane MA, Traystman RJ, Gleason CA. Comparison of cerebrovascular effects of intravenous cocaine injection in fetal, newborn, and adult sheep. Am. J. Physiol. Heart. Circ. Physiol. 2000;279:H1–H6. doi: 10.1152/ajpheart.2000.279.1.H1. [DOI] [PubMed] [Google Scholar]
- Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B, Miller GW, Caron MG. Cocaine self-administration in dopamine-transporter knockout mice. Nat. Neurosci. 1998;1:132–137. doi: 10.1038/381. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Bauman MH, Dersch CM, Romero DV, Rice KC, Carrol FI, Partilla HS. Amphetamine-type central neurvous system stimulants release norephinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Servan-Shreiber D, Perlstein WM, Cohen JD, Mintun M. Selective pharmacological activation of limbic structures in human volunteers: A positron emission tomorgaphy study. J. Neuropsychiatry. 1998;10:148–159. doi: 10.1176/jnp.10.2.148. [DOI] [PubMed] [Google Scholar]
- Siesjo B. Brain Energy Metabolism. Wiley; New York: 1978. [Google Scholar]
- Sherer MA, Kumor KM, Jaffe JH. Effects of intravenous cocaine are partially attenuated by haloperidol. Psychiatry Res. 1989;27:117–125. doi: 10.1016/0165-1781(89)90127-3. [DOI] [PubMed] [Google Scholar]
- Shriver DA, Long IP. A pharmacological comparison of some quaternary derivatives of cocaine. Arch. Int. Pharmacodyn. 1971;189:198–208. [PubMed] [Google Scholar]
- Stamford JA, Kruk ZL, Millar J. Dissociation of the actions of uptake blockers upon dopamine overflow and uptake in the rat nucleus accumbens: in vivo voltammetric data. Neuropharmacology. 1989;28:1383–1388. doi: 10.1016/0028-3908(89)90014-2. [DOI] [PubMed] [Google Scholar]
- Stein EA, Fuller SA. Cocaine’s time action profile on regional cerebral blood flow in the rat. Brain Res. 1993;626:117–126. doi: 10.1016/0006-8993(93)90570-d. [DOI] [PubMed] [Google Scholar]
- Steinfels GF, Heym L, Strecker RE, Jacobs BL. Behavioral correlates of dopaminergic unit activity in freely moving cat. Brain Res. 1983;258:217–228. doi: 10.1016/0006-8993(83)91145-9. [DOI] [PubMed] [Google Scholar]
- Strecker RE, Jacobs BL. Substantia nigra dopaminergic unit activity in behaving cats: effect of arousal on spontaneous discharge and sensory evoked activity. Brain Res. 1985;361:339–350. doi: 10.1016/0006-8993(85)91304-6. [DOI] [PubMed] [Google Scholar]
- Szabo B, Obergfell A, Starke K. Involvement of monoamine uptake inhibition and local anesthesia in the cardiovascular response to cocaine in conscious rabbits. J. Pharmacol. Exp. Ther. 1995;274:128–137. [PubMed] [Google Scholar]
- Taylor D, Ho BT. Comparison of inhibition of monoamine uptake by cocaine, methylphenidate and amphetamine. Res. Comm. Chem. Pathol. Pharmacol. 1978;21:67–75. [PubMed] [Google Scholar]
- Tella SR, Goldberg SR. Monoamine transporter and sodium channel mechanisms in the rapid pressor response to cocaine. Pharmacol. Biochem. Behav. 1998;59:305–312. doi: 10.1016/s0091-3057(97)00448-6. [DOI] [PubMed] [Google Scholar]
- Thomas WL, Cooke ES, Hammer RP. Pretreatment with a dopamine D1-receptor antagonist prevents metabolic activation by cocaine. Neurosci. Lett. 1995;196:161–164. doi: 10.1016/0304-3940(95)11865-t. [DOI] [PubMed] [Google Scholar]
- Trubel HKF, Sacolick LI, Hyder F. Regional temperature changes in the brain during somatosensory stimulation. J. Cerebral Flow & Metabolism. 2006;26:68–78. doi: 10.1038/sj.jcbfm.9600164. [DOI] [PubMed] [Google Scholar]
- Venton BJ, Seipel AT, Phillips PE, Wetsel WC, Gitler D, Greengard P, Augustine GJ, Wightman RM. Cocaine increases dopamine release by mobilization of a synapsin-dependent reserve pool. J. Neurosci. 2006;26:3206–3209. doi: 10.1523/JNEUROSCI.4901-04.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Gatley SJ, Fowler JS, Logan J, Fischman M, Gifford AN, Pappas N, King P, Vitkun S, Ding YS, Wang C-J. Cocaine doses equivalent to those abused by humans occupy most of the dopamine transporters. Synapse. 1996;24:399–402. doi: 10.1002/(SICI)1098-2396(199612)24:4<399::AID-SYN7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Wilcox KM, Paul IA, Ordway GA, Woolverton WL. Role of the dopamine transporter and the sodium channel on the cocaine-like discriminative stimulus effects of local anesthetics in rats. Psychopharmacology. 2001;157:260–268. doi: 10.1007/s002130100796. [DOI] [PubMed] [Google Scholar]
- Willats DG, Reynolds F. Comparison of the vasoactivity of amide and ester local anesthetics. An intradermal study. Br. J. Anaesth. 1985;57:1006–11. doi: 10.1093/bja/57.10.1006. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol. Rev. 1987;94:469–492. [PubMed] [Google Scholar]
- Wise RA, Newton P, Leeb K, Burnette B, Pocock D, Justice JB. Fluctuations in nucleus accumbens dopamine concentration during intravenous cocaine self-administration in rats. Psychopharmacology. 1995;120:10–20. doi: 10.1007/BF02246140. [DOI] [PubMed] [Google Scholar]
- Zernig G, Giacomuzzi S, Riemer Y, Wakonigg G, Sturm K, Saria A. Intravenous drug injection habits: Drug users’ self-reports versus researchers’ perception. Pharmacology. 2003;68:49–56. doi: 10.1159/000068731. [DOI] [PubMed] [Google Scholar]