

Abstract
Objective
We assessed the role of glycogen synthase kinase-3β(GSK-3β) and kinin B2 receptor in mediating tissue kallikrein’s protective effects against cardiac hypertrophy.
Methods
We investigated the effect and mechanisms of tissue kallikrein using hypertrophic animal models of rats as well as mice deficient in kinin B1 or B2 receptor after aortic constriction (AC).
Results
Intramyocardial delivery of adenovirus containing the human tissue kallikrein gene resulted in expression of recombinant kallikrein in rat myocardium. Kallikrein gene delivery improved cardiac function and reduced heart weight/body weight ratio and cardiomyocyte size without affecting mean arterial pressure 28 days after AC. Icatibant and adenovirus carrying a catalytically inactive GSK-3βmutant (Ad.GSK-3β-KM) abolished kallikrein’s effects. Kallikrein treatment increased cardiac nitric oxide (NO) levels and reduced NAD(P)H oxidase activity and superoxide production. Furthermore, kallikrein reduced the phosphorylation of apoptosis signal-regulating kinase1, mitogen-activated protein kinases (MAPKs), Akt, GSK-3β, and cAMP-response element binding (CREB) protein, and decreased nuclear factor-κB (NF-κB) activation in the myocardium. Ad.GSK-3β-KM abrogated kallikrein’s actions on GSK-3βand CREB phosphorylation and NF-κB activation, whereas icatibant blocked all kallikrein’s effects. The protective role of kinin B2 receptor in cardiac hypertrophy was further confirmed in kinin receptor knockout mice as heart weight/body weight ratio and cardiomyocyte size increased significantly in kinin B2 receptor knockout mice after AC compared to wild type and B1 receptor knockout mice.
Conclusions
These findings indicate that tissue kallikrein, through kinin B2 receptor and GSK-3β signaling, protects against pressure overload-induced cardiomyocyte hypertrophy by increased NO formation and oxidative stress-induced Akt-GSK-3β-mediated signaling events, MAPK and NF-κB activation.
Keywords: Tissue Kallikrein, Kinin B2 receptor, Hypertrophy, Glycogen Synthase Kinase-3β Nuclear Factor-κB
1. Introduction
Cardiac hypertrophy is an adaptive physiological response to pressure or volume overload [1,2]. Although cardiac hypertrophy is a temporary beneficial adaptive response of the heart, it eventually leads to dilated cardiomyopathy and congestive heart failure. Upon increased workload, cardiomyocytes increase in size [3]. Glycogen synthase kinase3β(GSK-3β) has been shown to be a negative regulator of cardiomyocyte hypertrophy, a major risk factor for cardiovascular mortality and morbidity [4]. GSK-3β, which belongs to the serine/threonine kinase family, is active in unstimulated cells. However, during hypertrophic conditions, it is inactivated by phosphorylation of serine 9 [5]. Hypoxia/reoxygenation or hydrogen peroxide treatment was demonstrated to induce hypertrophy in cultured cardiomyocytes via increased GSK-3β in its inactivation [6]. These results indicate that oxidative stress causes cardiomyocyte hypertrophy via inactivation of GSK-3β. In cultured cardiomyocytes, angiotensin II (Ang II) was shown to stimulate reactive oxygen species (ROS) production leading to increased expression or activity of several critical regulators of cardiac hypertrophy, such as mitogen activated protein kinases (MAPKs), transforming growth factor (TGF)-βand nuclear factor (NF)-κB [7]. NF-κB activation has also been shown to play a critical role in ROS-induced cardiac hypertrophy [8, 9]. Moreover, evidence suggests that apoptosis signal-regulating kinase-1 (ASK-1), extracellular signal regulated kinase (ERK), c-jun N-terminal kinase (JNK) and p38MAPK may be responsible for myocyte hypertrophy and gene reprogramming [10, 11]. These findings indicate a role of oxidative stress in induction of Akt-GSK-3β-mediated signaling events, ASK-1, MAPK and NF-κB activation in cardiomyocyte hypertrophy. Furthermore, a regulatory role of cAMP-response element binding protein (CREB), a transcription factor, has been implicated in cardiomyocyte gene expression [12]. For example, a previous report showed that phenylephrine, a hypertrophic agonist, stimulated the transcriptional activity of CREB through the MAPK pathway in cardiac cells [13]. In addition, a critical role of CREB was demonstrated in Ang II-induced hypertrophy through ERK/p38MAPK activation in vascular smooth muscle cells [14]. However, the role of CREB in GSK-3β-mediated cardiac hypertrophy has not yet been elucidated.
Tissue kallikrein is a serine proteinase that cleaves low molecular weight kininogen to release the kinin peptide. Kinin binds to the kinin B2 receptor and activates second messengers such as nitric oxide (NO) and prostacyclin leading to a broad spectrum of biological effects [15, 16]. A previous study showed that transgenic rats overexpressing human tissue kallikrein reduced cardiac hypertrophy induced by isoproterenol [17]. In addition, kallikrein gene delivery attenuated cardiac hypertrophy, in conjunction with increased NO levels and reduced JNK/ERK activation in the heart of spontaneously hypertensive rats [18]. Recently, we demonstrated that a kinin B2 receptor antagonist abolished the benefits of kallikrein in inhibiting cardiomyocyte apoptosis after acute myocardial ischemia/reperfusion in association with reduced oxidative stress and Akt and GSK-3β signaling [19]. Based on these results, we assessed the hypothesis that tissue kallikrein through kinin B2 receptor protects against cardiac hypertrophy by maintaining GSK-3β activity using pressure-overload models of rats and kinin B1 and B2 knockout mice.
2. Methods
2.2. Preparation of replication-deficient adenoviral vectors
Adenoviral vectors harboring the human tissue kallikrein cDNA under the control of cytomegalovirus enhancer/promoter (Ad.TK) and control adenovirus without a reporter gene (Ad.Null) were prepared as previpously described [20]. A replication-defective adenoviral vector expressing catalytically inactive GSK-3β(Ad.GSK-3β-KM) was kindly provided by Dr. Kenneth Walsh, St. Elizabeth’s Medical Center in Boston. Lysine residues at positions 85 and 86 were mutated to methionine and alanine, respectively, to create the inactive GSK-3β. The replication-deficient recombinant adenoviruses were propagated in 293 cells and purified by CsCl density gradient centrifugation.
2.2. Abdominal aortic constriction in rats
Male Wistar rats (200-250 g body weight) were obtained from Harlan Inc. (Indianapolis, IN). All procedures complied with the standards for care and use of animal subjects as stated in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Resources, National Academy of Sciences, Bethesda, MD). Before surgery, the animals were anesthetized intraperitoneally with pentobarbital at a dose of 50 mg/kg body weight and the aorta was exposed through a midline abdominal incision. A blunt 23-gauge needle was then placed adjacent to the abdominal aorta above the renal arteries. A ligature was tightened around the aorta and the needle was removed after ligation [21]. Rats were sacrificed at 3, 7 14 and 28 days after aortic constriction in order to evaluate time-dependent changes of cardiac hypertrophy. The sham control rats underwent the same surgical protocol but without aortic ligation. Rats received Ad.Null or Ad.TK at a dose of 2 × 1010 plaque forming units per rat by direct injection to the left ventricle wall in several different sites immediately after aortic banding. To investigate the roles of kinin B2 receptor and GSK-3βin mediating kallikrein’s effects on cardiac hypertrophy, rats subjected to aortic banding were randomly divided into four groups and received one of the following treatments: (1) Ad.Null injection, (2) Ad.TK injection, (3) Ad.TK injection along with infusion of the kinin B2 receptor antagonist icatibant (1.3 μg/hr) by osmotic minipump implanted subcutaneously, or (4) Ad.TK and Ad.GSK-3β-KM (2 × 1010 plaque forming units) co-injection. Rats were sacrified 28 days after AC and heart tissues were processed for morphological analysis. Expression of recombinant human tissue kallikrein in the heart was measured by immunohistochemistry and enzyme-linked immunosorbent assay (ELISA) [19].
2.3. Abdominal aortic constriction in kinin B1 and B2 receptor deficient mice
To determine the role of kinin B1 and B2 receptor in cardiac hypertrophy, wild type (WT), kinin B1 receptor- and B2 receptor knockout mice were employed in this study. Mice (body weight, 25-30 g), including WT mice (B6129SF2/J) and B2 receptor knockout mice (derived from breeding pairs of C57BL/6 mice on a 129/J genetic background), were purchased from Jackson Laboratories (Bar Harbor, ME). Kinin B1 receptor knockout mice were kindly provided by Professor Miachael Bader, Max-Debruck Center for Molecular Medicine, Berlin-Buch, Germany. Mice were anesthetized by inhaled Isoflurane and underwent the same surgical protocol as outlined above to isolate abdominal aorta. A blunt 30-gauge needle was then placed adjacent to the abdominal aorta close to renal bifurcations. A ligature was tightened around the aorta and the needle was removed after ligation. Mice were sacrified at 28 days after AC.
2.4. In vivo hemodynamic measurements
Hemodynamic parameters and cardiac function were measured 28 days after AC. Rats underwent left ventricular catheterization via left common carotid artery by a 2.5 French micro-manometer (Millar Instrument, Houston, TX) advanced into the LV cavity. Mean arterial pressure (MAP), left ventricular end-diastolic pressure (LVEDP), and ± LV dP/dt max were recorded and analyzed by a polygraph system (BIOPAC, Santa Barbara, CA) [19].
2.5. Histological analyses
At the end of the experiment, all rats were anesthetized and perfused with saline through the left ventricle. Hearts were then removed, fixed with 4% formaldehyde, embedded in paraffin and sectioned at 4 μm thickness. Heart sections were incubated with antibodies against recombinant human tissue kallikrein and the monocyte/macrophage marker ED-1 (Cell Signaling Technology, Beverly, MA) using a Vectastain Universal Elite ABC kit (Vector Laboratories, Burlingame, CA) following the manufacturer’s instructions. For the measurement of cardiomyocyte size, rat heart sections were stained by the Gordon and Sweet silver method. Next, 100 cardiomyocytes were chosen randomly per slide and traced using NIH Image software at 400X magnification. Then, sections from each rat were scanned and analyzed with a digital image analyzer. Heart sections also underwent Sirius red staining for the determination of the extent of fibrosis. The percent area of fibrosis was calculated as previously described [19]. In each group, 70 small (internal diameter <100 μm) and 30 large (>100 μm) arteries were examined for perivascular fibrosis and vascular wall thickening (wall area/vessel area) [22].
Cardiomyocyte size in mice at 4 weeks after AC was examined by staining with Alexa Fluor 488-labeled wheat germ agglutinin (Invitrogen, Carlsbad, CA) and left ventricular cardiac myocyte surfaces were observed by fluorescent microscopy. Cardiocyocyte size was analyzed using NIH Image software. Cross-sectional area of 80 cardiomyocytes from each heart was calculated.
2.6. Western blot analysis
Heart tissues were homogenized in lysis buffer (10 mM Tris, pH 7.4, 100 mM sodium chloride [NaCl], 1 mM ethylenediamine tetraacetic acid [EDTA], 20 mM sodium pyrophosphate [Na4P2O7], 2 mM sodium orthovanadate [Na3VO4], 1% Triton X-100) containing 1:100 protease inhibitor cocktail (Sigma, St. Louis, MO) and centrifuged at 1300 × g at 4°C for 5 minutes. After centrifugation, the supernatant (containing the cytosolic fraction) was removed and stored at -80°C. Nuclear proteins were extracted by suspending the pellet in an equal volume of lysis buffer and adding NaCl to a final concentration of 0.6 M. After incubation on ice for 1 hr, lysates were centrifuged and the supernatant, containing the nuclear proteins, was removed and stored at -80°C. Protein concentrations in tissue homogenates were measured using Bio-Rad DC Protein Assay kit (Bio-Rad Laboratories, Hercules, CA). Western blot analysis was performed using cytosolic fractions for ASK-1, phospho-ASK-1, GSK-3β, phospho-GSK-3β, Akt, phospho-Akt, JNK, phospho-JNK, p38MAPK, phospho-p38MAPK, ERK, phospho-ERK (Cell Signaling), atrial natriuretic peptide (ANP) (Biodesign, Saco, ME), and nuclear extracts for CREB and phospho-CREB (1:1000 dilution) (Cell Signaling). Blots were incubated with secondary antibody conjugated to LumiGLO chemiluminescent reagent. Chemiluminescence was detected using an ECL-Plus kit (Perkin-Elmer Life Sciences, Boston, MA) and exposing the blot to Kodak X-ray film.
2.7. Measurements of nitrate/nitrite (NOx) levels and NADH/NADPH oxidase activity and superoxide formation
Production of NO (in the form of nitrate/nitrite) in heart extracts was measured as previously described [23]. NADH and NADPH oxidase activities were measured by lucigenin-enhanced chemiluminescence [24]. Superoxide levels were measured by a spectrophotometric assay based on rapid reduction of ferricytochrome c to ferrocytochrome c. Non-superoxide-dependent reduction of cytochrome c was corrected for by deducting the activity not inhibited by superoxide dismutase [25].
2.8. Electrophoretic mobility shift assay (EMSA) for NF-κB activation
NF-κB DNA binding site oligonucleotides (AGTTGAGGGGACTTTCCCAGGC, Integrated DNA Technologies, Coralville, IA) were labeled using the Biotin 3’ End DNA Labeling kit (Pierce, Rockford, IL) according to the manufacturer’s protocol. The gel shift assay was performed using the Light Shift Chemiluminescent electrophoretic mobility shift analysis kit (Pierce) following the manufacturer’s instructions. After the membrane was incubated with LightShift Stabilized Streptavidin-Horseradish Peroxidase Conjugate and the Luminol/Enhancer and Stable Peroxide Solution, the NF-κB DNA complexes were detected by exposure of the membrane to X-ray film. The specificity of the identified NF-κB DNA binding activity in the nuclear extracts was confirmed by using 200-fold molar excess of unlabeled NF-κB. The relative nuclear NF-κB-DNA binding activities were quantified by scanning densitometry.
2.9. GSK-3βkinase activity assay
For GSK-3βactivity assay, 10 μg of protein from cardiac tissue extracts were incubated at 37 °C for 30 min with 8 mM MOPS, 0.2 mM EDTA, 10 mM magnesium acetate, 62.5 μM GSK-3β substrate peptide (Upstate Biotechnology, Lake Placid, NY) and 1 μCi/10μl [γ-32P] ATP (Perkin-Elmer Life Sciences). Samples were transferred to P-81 paper and washed three times with 0.75% phosphoric acid followed by a final rinse with acetone. Radioactivity was measured using a scintillation counter [19].
2.10. Statistical analysis
Results are expressed as mean ± SEM. Comparisons among groups were made by ANOVA followed by Fisher’s PLSD (protected least significant difference) or by unpaired student t-test. Differences were considered significant at P<0.05.
3. Results
3.1. Human tissue kallikrein expression in rat hearts
Expression of recombinant human tissue kallikrein expression was localized in rat hearts at 3, 7 and 28 days after local delivery of the kallikrein gene (Figure 1). Immunoreactive human tissue kallikrein levels in the heart measured by ELISA showed that the highest human kallikrein levels were at 3 days (1.55 ± 0.21 ng/mg protein), declined at 7 days (0.35 ± 0.04 ng/mg protein), and were barely detectable at 28 days (0.05 ± 0.01 ng/mg protein) after kallikrein gene delivery. Human tissue kallikrein expression was not detected by immunohistochemical staining or by ELISA in the hearts of rats injected with control adenovirus (Ad.Null).
Figure 1.
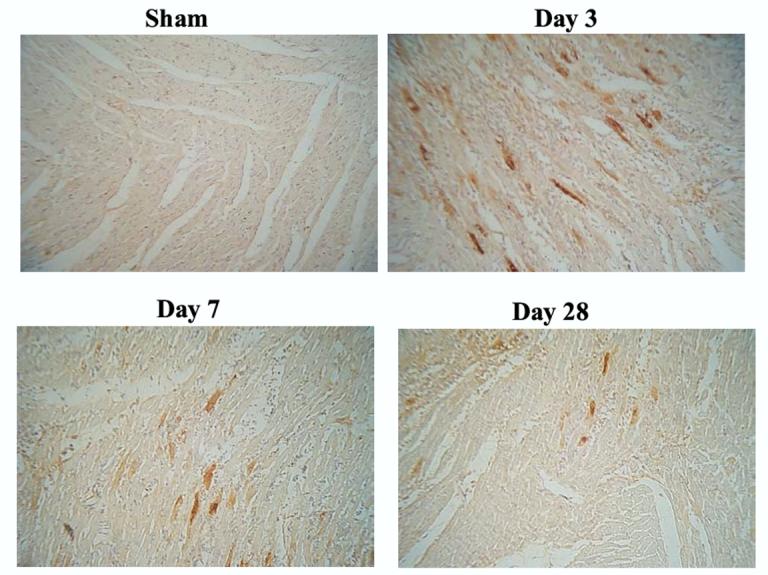
Immunostaining of recombinant human tissue kallikrein in rat heart at 3, 7 and 28 days after kallikrein gene delivery.
3.2. Kallikrein gene delivery improves hemodynamic parameters
Time-dependent increases in cardiomyocyte size and perivascular collagen deposition were identified in rats from 3 to 28 days after aortic banding, while inflammatory cell accumulation around blood vessels peaked at day 3 (data not shown). At 28 days after AC, tissue kallikrein gene delivery improved hemodynamic parameters as shown in Table 1. Kallikrein gene transfer significantly improved cardiac contractility ( ± dP/dt max), and reduced left ventricular end diastolic pressure (LVEDP) and heart weight to body weight ratio (HW/BW), as well as septal thickness, left ventricle free wall thickness and apical thickness compared to aortic-banded rats receiving the control virus. Icatibant infusion or Ad.GSK-3β-KM co-injection, however, abolished kallikrein’s effects on these values. Although aortic banding elevated mean arterial pressure (MAP) compared to sham rats, MAP was similar among groups receiving AC surgery. These results indicate that kallikrein through the kinin B2 receptor protects against AC-induced cardiac dysfunction by maintaining GSK-3βactivation independent of blood pressure.
Table 1.
Effect of Kallikrein Gene Transfer on Hemodynamic Parameters in Pressure-Overloaded Rats 28 days after Aortic Banding
| Parameters | AC |
||||
|---|---|---|---|---|---|
| Sham | Ad.Null | Ad.TK | Ad.TK/Icatibant | Ad.TK/GSK-3β-KM | |
| LVEDP (mmHg) | 4.3 ± 0.8 | 14.2 ± 1.9* | 5.6 ± 0.9 | 14.8 ± 1.9* | 12.5 ± 1.2* |
| LV dP/dt max (mmHg/s) | 3802 ± 146 | 2415 ± 238* | 3201 ±115 | 2364 ± 194* | 2471 ± 156* |
| LV dP/dt min (mmHg/s) | 3117 ± 184 | 1940 ± 148* | 2513 ±108 | 1564 ± 168* | 1759 ± 151* |
| MAP (mmHg) | 122 ±1 | 163 ±3 | 163 ± 3 | 158 ±10 | 160 ± 6 |
| HW/BW (mg/g) | 2.8 ± 0.0 | 4.1 ± 0.3* | 3.4 ± 0.1 | 4.2 ± 0.1* | 4.4 ± 0.2 |
| Septal thickness (mm) | 2.1 ± 0.2 | 3.9 ± 0.1* | 2.2 ± 0.2 | 3.6 ± 0.1* | 3.7 ± 0.2* |
| Free wall thickness (mm) | 3.8 ± 0.1 | 5.4 ± 0.2* | 3.9 ± 0.2 | 5.3 ± 0.2* | 6.1 ± 0.6* |
| Apex thickness (mm) | 1.0 ± 0.1 | 1.8 ± 0.1* | 1.1 ± 0.1 | 2.2 ± 0.3* | 2.2 ± 0.1* |
LVEDP, left ventricular end-diastolic pressure; dP/dtmax, maximum first derivative of pressure; dP/dtmin minimun first derivative of pressure; MAP, mean arterial pressure; HW, heart weight; BW, body weight. Values are expressed as mean ± SEM.
P<0.05 vs Sham & Ad.TK, n= 6
3.3. Kallikrein gene delivery attenuates cardiomyocyte size and vascular remodeling
Kallikrein gene transfer reduced cardiomyocyte size after AC as identified by silver stained heart sections, and icatibant or catalytically inactive GSK-3βabrogated kallikrein’s effect (Figure 2A). Quantitative analysis confirmed these findings. Rats receiving Ad.Null had larger cardiomyocytes than rats in the sham group (558.8 ± 13.2 vs. 355.6 ± 8.6 μm2, n=6, P<0.05), but kallikrein gene delivery significantly reduced cardiomyocyte size compared to the Ad.Null group (440.0 ± 13.7 vs. 558.8 ± 13.2 μm2, n=6, P<0.05). Cardiomyocytes in rats injected with Ad.TK and receiving icatibant infusion (Ad.TK/Icatibant) or GSK-3β-KM co-injection (Ad.TK/GSK-3β-KM) were similar in size as those in the Ad.Null group (Figure 2B).
Figure 2.

Effect of kallikrein gene delivery on cardiac hypertrophy 28 days after aortic constriction. (A) Gordon and Sweet silver staining of heart sections; magnification is 400X. (B) Quantitative analysis of cardiomyocyte size. Data are expressed as mean ± SEM (n=6).
Heart sections were stained with Sirius red for examination of the extent of vascular fibrosis 28 days after AC surgery (Figure 3A). Morphologically, collagen deposition surrounding blood vessels appeared to increase in response to aortic banding, but was attenuated by kallikrein gene delivery. Icatibant infusion and GSK-3β-KM injection, however, appeared to block the effect of kallikrein. These observations were confirmed by quantitative analysis of large blood vessels or small arteries (data not shown) in the heart. Kallikrein gene delivery markedly reduced perivascular fibrosis and vascular wall thickening in both small and large blood vessels compared to the control aortic-banded rats. No significant difference was observed among the Ad.Null, Ad.TK/Icatibant and Ad.TK/GSK-3β-KM groups (Figure 3 B and C).
Figure 3.
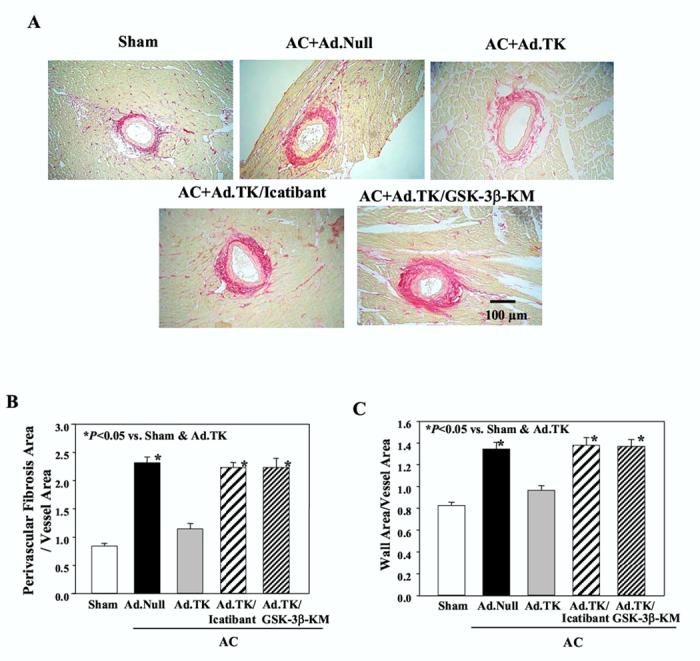
Effect of kallikrein gene delivery on vascular remodeling 28 days after aortic constriction. (A) Representative photographs of Sirius red staining; magnification is 100X. (B) Perivascular fibrosis and (C) wall to vessel area ratio were examined in large (internal diameter >100 μm) arteries. Bar=100 m. Data are expressed as mean ± SEM (n=6).
3.4. Effect of kallikrein on NOx levels, NADH/NADPH oxidase activity and superoxide production
As shown in Figure 4A, aortic constriction (Ad.Null) did not alter nitrate/nitrite (NOx) levels compared with sham rats, although kallikrein gene delivery increased NOx levels above those in the Ad.Null group. Co-injection of GSK-3β-KM along with Ad.TK also increased NOx levels over those in the Ad.Null group. However, the effect of kallikrein on NO production was blocked by icatibant.
Figure 4.
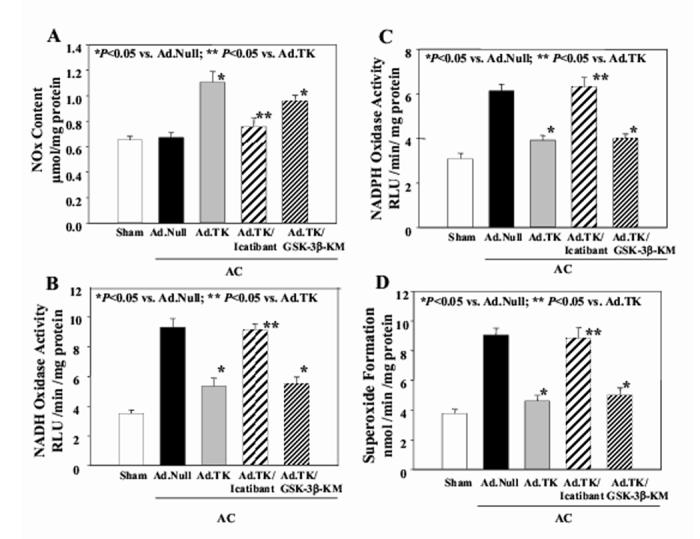
Effect of kallikrein gene delivery on nitric oxide formation and oxidative stress in heart tissues. (A) NO production, (B) NADH oxidase activity, (C) NADPH oxidase activity and (D) superoxide formation in the heart 28 days after aortic banding. Data are expressed as mean ± SEM (n=6).
In order to determine the effect of kallikrein gene transfer on oxidative stress in the heart, NADH/NADPH oxidase activity and superoxide levels were measured in cardiac extracts. As shown in Figure 4B &C, rats in the Ad.Null group had significantly higher NADH and NADPH oxidase activity than both sham rats and rats injected with the kallikrein gene. Rats receiving both Ad.TK and GSK-3β-KM injection also had lower NADH/NADPH oxidase activity than rats in the Ad.Null group. However, icatibant infusion completely reversed kallikrein’s effect on NADH/NADPH oxidase activity. Superoxide formation paralleled NADH/NADPH oxidase activity (Figure 4D). Rats in the Ad.Null group had higher superoxide levels compared to sham rats. Kallikrein gene delivery, with or without GSK-3β-KM injection, significantly lowered superoxide levels compared to rats receiving empty virus. However, kinin B2 receptor antagonism blocked kallikrein’s ability to lower AC-induced superoxide formation. These results indicate that GSK-3βis a downstream target of oxidative stress and that kallikrein suppresses oxidative stress through the kinin B2 receptor.
3.5. Effect of kallikrein on MAPK activation
Western blot analyses were performed in order to determine the effect of kallikrein gene transfer on ASK-1 and MAPK (JNK, p38MAPK, ERK) activation (Figure 5A-D). Aortic constriction caused a significant increase in the phosphorylation of ASK-1, JNK, p38MAPK and ERK. Kallikrein gene delivery markedly reduced the phosphorylation of ASK-1, JNK, p38MAPK and ERK, but icatibant abolished kallikrein’s effects. Catalytically inactive mutant form of GSK-3βreversed kallikrein’s ability to reduce ASK-1 phosphorylation, but had no effect on phosphorylated levels of JNK, p38MAPK or ERK. Quantitative analysis confirmed these results. These data clearly indicate that GSK-3βis a downstream target of MAPK phosphorylation.
Figure 5.
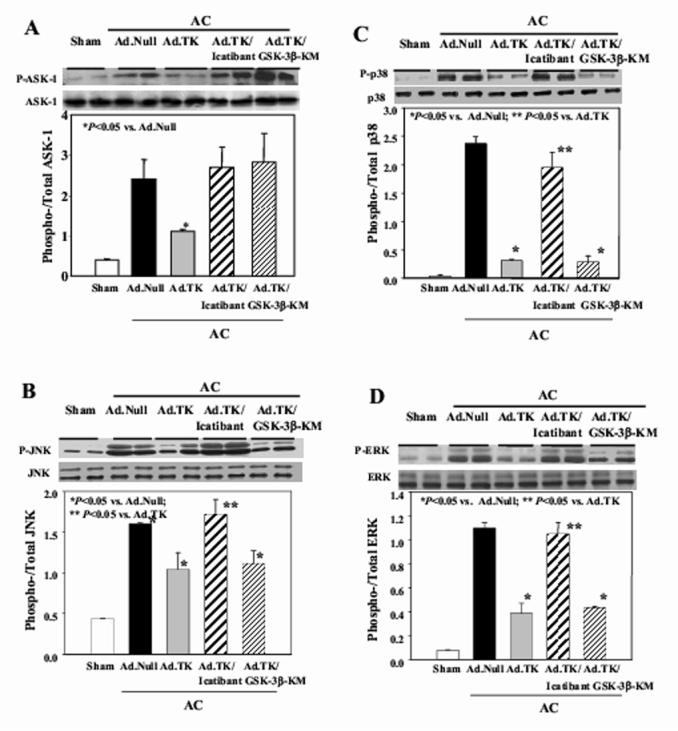
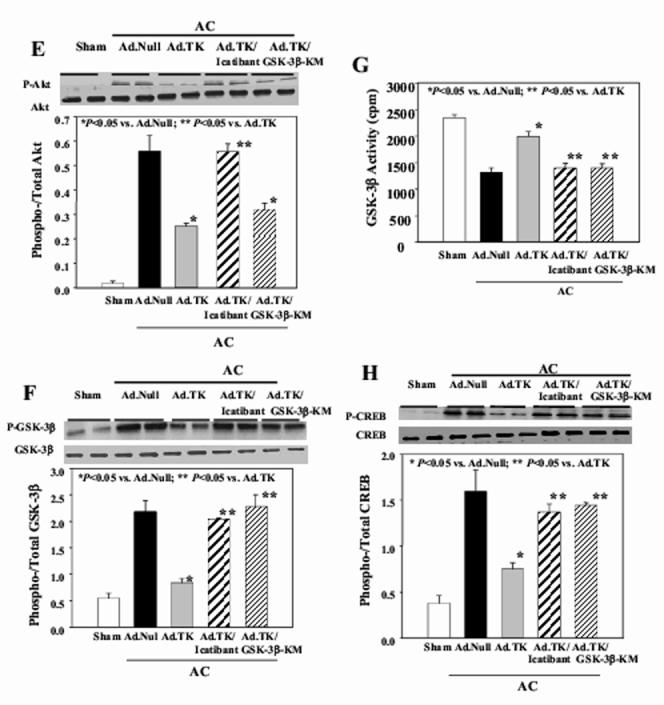
Effect of kallikrein gene delivery on the phosphorylation of (A) ASK-1, (B) JNK, (C) p38MAPK, (D) ERK, (E) Akt, (F) GSK-3β, (G) GSK-3βactivity and (H) CREB phosphorylation in heart tissues. Upper panel: Representative Western blots. Lower Panel: quantitative analysis. Data are expressed as mean ± SEM (n=6).
3.6. Effect of kallikrein on Akt, GSK-3β;, and CREB signaling phosphorylation
Western blot analyses showed that aortic constriction caused a significant increase in Akt, GSK-3βand CREB phosphorylation (Figure 5E,F,H). Kallikrein gene delivery markedly reduced this effect, yet kallikrein’s actions were abolished by icatibant. Injection of both Ad.TK and Ad.GSK-3β-KM caused a significant depression in phosphorylated levels of Akt, but did not affect GSK-3βor CREB phosphorylation. Furthermore, reduced GSK-3βactivity was associated with increased phosphorylation of GSK-3β. Kallikrein gene transfer significantly increased GSK-3β activity compared with Ad.Null (Figure 5G). Furthermore, representative electrophoretic mobility shift assay showed that AC-induced NF-κB binding activity in the heart was blocked by tissue kallikrein, and icatibant and GSK-3βinactivation reversed kallikrein’s effect (Figure 6A). This finding was also supported by quantitative analysis (Figure 6B). Taken together, these results indicate that GSK-3βis a downstream target of Akt and an upstream regulator of CREB and NF-κB nuclear translocation and activation.
Figure 6.
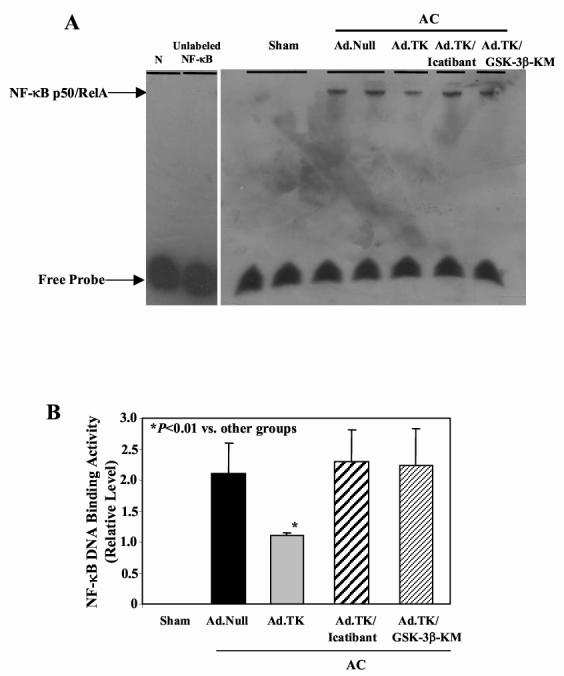
Effect of kallikrein gene delivery on NF-κB-DNA binding activity. (A) EMSA and (B) quantitative analysis. N indicates biotin labeled NF-κB oligonucleotide probe with no protein extract. Data are expressed as mean ± SEM (n=6).
3.7. Cardiac hypertrophy in kinin B1 and B2 receptor knockout mice
Thus far, we have shown that kallikrein, via a kinin B2 receptor-mediated event, reduces cardiac hypertrophy in rats after aortic banding. To further verify the role of kinin B2 receptor in mediating pressure-induced cardiac hypertrophy, we employed WT and B1 and B2 knockout mice subjected to aortic constriction. The ratio of heart weight/body weight of B2-/-mice without pressure overload was significantly higher than the age- and sex-matched WT and B1-/-mice. At 4 weeks after AC, heart weight/body weight ratio of B2-/-mice was also significantly higher than WT and B1-/-mice (Figure 7A). In addition, Western blot analysis was performed to determine ANP levels, a hypertrophic marker, in the hearts of wild-type, B1R-/- and B2R-/-mice. The results showed that cardiac ANP levels were barely detectable in mice not subjected to abdominal aortic ligation, but markedly increased at one month after aortic banding (Figure 7B). Histological staining and quantitative analysis confirmed that cardiomyoyte size in B2-/-mice after AC was significantly larger that in WT and B1-/-mice (215.3 ± 12.9 vs. 178.5 ± 7.2 and 169.3 ± 9.2 μm2, n = 6, P<0.05) (Figure 7C & D). These combined results indicate that the kinin B2 receptor, but not B1 receptor, mediates the protective effect of tissue kallikrein in pressure overload-induced cardiac hypertrophy.
Figure 7.
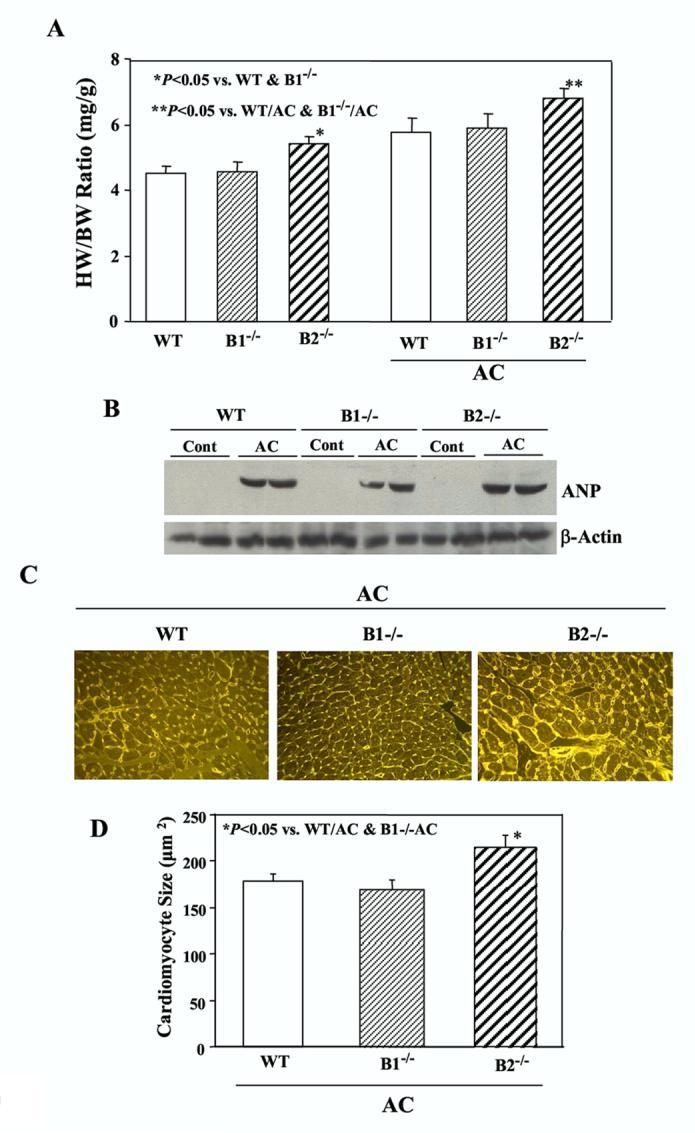
Cardiac hypertrophy is increased in kinin B2 receptor knockout mice at 28 days after aortic constriction. (A) Heart weight/body weight ratio in wild-type (WT), kinin B1-/- and kinin B2-/-mice with and without aortic constriction (n= 9-16). (B) Representative Western blot for ANP. (C) Representative cardiomyocytes stained with Alexa Fluor 488-labeled wheat germ agglutinin; magnification is 400X. (D) Quantitative analysis of cardiomyocyte size. Data are expressed as mean ± SEM (n= 9-16).
4. Discussion
This is the first study to demonstrate that tissue kallikrein through kinin B2 receptor activation protects against pressure overload-induced cardiac hypertrophy by preventing GSK-3βinactivation. Kallikrein gene transfer also improved cardiac function and vascular remodeling in rats after aortic constriction without apparent effect on blood pressure. Icatibant, a kinin B2 receptor antagonist, blocked kallikrein’s effect on cardiac hypertrophy. Using kinin receptor knockout mice, we confirmed that the kinin B2 receptor, but not the B1 receptor, plays a role in the protection of cardiac hypertrophy induced by aortic constriction. Similarly, a previous study showed that kinin B2 receptor is involved in age-related changes of cardiac hypertrophy and microvascular deficit [26]. Left ventricular hypertrophy and heart failure constitute major risks of cardiovascular complications and sudden death among hypertensive patients. These combined findings support an important role of kinin B2 receptor, but not B1 receptor, in the development of cardiac hypertrophy and heart failure.
In the present study, we detected the expression and localization of recombinant human tissue kallikrein in the hypertrophic heart by immunohistochemistry and ELISA at 3, 7 and 28 days after local gene delivery. These results are consistent with our previous finding that the highest expression levels of human tissue kallikrein in rats occurs at 3 to 8 days after adenovirus-mediated gene delivery and declines to a low level at 28 days [18]. Cardiac hypertrophy induced by aortic constriction has been shown to be initiated at the first week and continue to develop in the second and third weeks [21]. Therefore, the expression of recombinant kallikrein in the first two weeks could prevent the initiation and formation of cardiac hypertrophy at the early stage after aortic binding.
Inactivation of GSK-3βby phosphorylation of serine 9 plays a key role in the process of cardiac hypertrophy in mice induced by pressure overload and in cultured cardiomyocytes [27]. In this study, we showed that expression of recombinant tissue kallikrein in the heart prevented AC-induced cardiac hypertrophy as evidenced by reduced heart weight/body weight ratio, ventricular wall thickness and cardiomyocyte size. Administration of GSK-3β-KM or icatibant reversed the anti-hypertrophic actions of kallikrein in the heart. The protective effect of kallikrein on cardiac hypertrophy was accompanied by increased GSK-3βactivity and reduced GSK-3βphosphorylation, indicating that maintaining GSK-3βactivity is crucial in mediating kallikrein’s actions.
Oxidative stress is known to play a critical role in inducing myocardial remodeling and hypertrophy in animal models of chronic pressure overload [9, 28]. Many of the deleterious cellular phenotypes present in hypertrophied and failing hearts have been attributed to increased oxidative stress, characterized by increased superoxide formation [29]. Ang II is a well-known inducer of oxidative stress by stimulating NADH/NADPH oxidase activity, which in turn produces superoxide [28, 30, 31]. It has also been observed that kinin can block Ang II-induced cardiomyocyte hypertrophy through the release of NO from endothelial cells [32]. In this study, we showed that kallikrein gene delivery, through kinin B2 receptor activation, leads to increased NO formation and reduced NADH/NADPH oxidase activity and superoxide anion formation in the hypertrophied heart after aortic banding. Nitric oxide may protect against oxidative stress by inhibiting the assembly of NADH/NADPH oxidase subunits, thereby attenuating the generation of superoxide [33]. ROS has been shown to mediate NF-B tivation ac in TNF-α-induced cardiomyocyte hypertrophy [9]. Moreover, activation of NF-κB is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes [8]. Consistent with these findings, we showed that tissue kallikrein inhibits the development of cardiac hypertrophy in association with reduced NF-κB activation induced by AC and the effect was blocked by icatibant. Together, these findings indicate that kallikrein, through kinin B2 receptor signaling, protects against cardiac hypertrophy via increased NO generation and suppression of oxidative stress-induced NF-B activation.
In cultured cardiomyocytes, it has been shown that hydrogen peroxide induces the phosphorylation of JNK, p38MAPK, ERK and Akt [34]. Similarly, El Jamali et al [6] demonstrated that reoxygenation after severe hypoxia induced cardiomyocyte hypertrophy, possibly through GSK-3β phosphorylation, downstream of Akt. In addition, acute and chronic isoproterenol infusion in rats was found to induce cardiac remodeling by provoking cardiac oxidative stress and activation of MAPKs [35]. It is well-known that Akt is an upstream regulator of GSK-3βand that both MAPKs and Akt-GSK-3βsignaling participate in the development of cardiac hypertrophy [36]. ASK-1, a MAPK kinase, has also been recently observed to play a critical role in the development of cardiac hypertrophy and remodeling [10, 11]. PD98059, an ERK inhibitor, was shown to block Akt phosphorylation, suggesting that ERK is an upstream regulator of Akt [6]. In this study, we showed that kallikrein gene delivery reduced the phosphorylation of MAPKs and Akt, whereas co-injection of dominant-negative GSK-3βmutant with the kallikrein gene abrogated the protective actions of kallikrein in cardiac remodeling without affecting phosphorylation of MAPKs and Akt. Whether MAPKs act as upstream regulators of Akt-GSK-3βhas yet to be elucidated. Moreover, the effect of kallikrein on NO and reactive oxygen species (ROS) formation was reversed by icatibant, but not by GSK-3β-KM, indicating that NO and oxidative stress are upstream regulators of GSK-3β.Furthermore, kallikrein reduced AC-induced phosphorylation of ASK-1, p38MAPK, JNK, ERK as well as Akt, GSK-3βand CREB. Kallikrein’s effects were all blocked by icatibant, indicating a kinin B2 receptor-mediated event. However, catalytically inactive GSK-3βmutant only reversed kallikrein’s effect on MAPK and Akt activation, but not on GSK-3βphosphorylation. These results imply that GSK-3βis a downstream mediator of MAPK and Akt. Moreover, GSK-3β-KM, in conjunction with kallikrein gene delivery, did not affect CREB phosphorylation, indicating that CREB is a downstream mediator of GSK-3βsignaling.
In conclusion, this study presents the novel finding that kallikrein through kinin B2 receptor signaling improves cardiac function and protects against the development of cardiac hypertrophy by preventing GSK-3β inactivation/phosphorylation. The anti-hypertrophic action of kallikrein is mediated by increased NO formation and reduced oxidative stress, leading to suppression of ASK-1, MAPK and Akt-GSK-3β-CREB signaling cascades.
Acknowledgements
This work was supported by National Institutes of Health grants HL29397, DK066350, and C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources.
Footnotes
Huey-Jiun Li and Hang Yin have equal contributions
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Anversa P, Ricci R, Olivetti G. Quantitative structural analysis of the myocardium during physiologic growth and induced cardiac hypertrophy: a review. J Am Coll Cardiol. 1986;7:1140–9. doi: 10.1016/s0735-1097(86)80236-4. [DOI] [PubMed] [Google Scholar]
- [2].Gerdes AM. The use of isolated myocytes to evaluate myocardial remodeling. Trends Cardiovasc Med. 1992;2:152–5. doi: 10.1016/1050-1738(92)90023-L. [DOI] [PubMed] [Google Scholar]
- [3].Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–31. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, et al. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol. 2000;151:117–30. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Badorff C, Ruetten H, Mueller S, Stahmer M, Gehring D, Jung F, et al. Fas receptor signaling inhibits glycogen synthase kinase 3 beta and induces cardiac hypertrophy following pressure overload. J Clin Invest. 2002;109:373–81. doi: 10.1172/JCI13779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].El Jamali A, Freund C, Rechner C, Scheidereit C, Dietz R, Bergmann MW. Reoxygenation after severe hypoxia induces cardiomyocyte hypertrophy in vitro: activation of CREB downstream of GSK-3beta. FASEB J. 2004;18:1096–8. doi: 10.1096/fj.03-1054fje. [DOI] [PubMed] [Google Scholar]
- [7].Wenzel S, Taimor G, Piper HM, Schluter KD. Redox-sensitive intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-beta expression in adult ventricular cardiomyocytes. FASEB J. 2001;15:2291–3. doi: 10.1096/fj.00-0827fje. [DOI] [PubMed] [Google Scholar]
- [8].Purcell NH, Tang G, Yu C, Mercurio F, DiDonato JA, Lin A. Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci U S A. 2001;98:6668–73. doi: 10.1073/pnas.111155798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Higuchi Y, Otsu K, Nishida K, Hirotani S, Nakayama H, Yamaguchi O, et al. Involvement of reactive oxygen species-mediated NF-kappa B activation in TNF-alpha-induced cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2002;34:233–40. doi: 10.1006/jmcc.2001.1505. [DOI] [PubMed] [Google Scholar]
- [10].Izumiya Y, Kim S, Izumi Y, Yoshida K, Yoshiyama M, Matsuzawa A, et al. Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin II-induced cardiac hypertrophy and remodeling. Circ Res. 2003;93:874–83. doi: 10.1161/01.RES.0000100665.67510.F5. [DOI] [PubMed] [Google Scholar]
- [11].Higuchi Y, Otsu K, Nishida K, Hirotani S, Nakayama H, Yamaguchi O, et al. The small GTP-binding protein Rac1 induces cardiac myocyte hypertrophy through the activation of apoptosis signal-regulating kinase 1 and nuclear factor-kappa B. J Biol Chem. 2003;278:20770–7. doi: 10.1074/jbc.M213203200. [DOI] [PubMed] [Google Scholar]
- [12].Markou T, Hadzopoulou-Cladaras M, Lazou A. Phenylephrine induces activation of CREB in adult rat cardiac myocytes through MSK1 and PKA signaling pathways. J Mol Cell Cardiol. 2004;37:1001–11. doi: 10.1016/j.yjmcc.2004.08.002. [DOI] [PubMed] [Google Scholar]
- [13].Gusterson R, Brar B, Faulkes D, Giordano A, Chrivia J, Latchman D. The transcriptional co-activators CBP and p300 are activated via phenylephrine through the p42/p44 MAPK cascade. J Biol Chem. 2002;277:2517–24. doi: 10.1074/jbc.M104626200. [DOI] [PubMed] [Google Scholar]
- [14].Funakoshi Y, Ichiki T, Takeda K, Tokuno T, Iino N, Takeshita A. Critical role of cAMP-response element-binding protein for angiotensin II-induced hypertrophy of vascular smooth muscle cells. J Biol Chem. 2002;277:18710–7. doi: 10.1074/jbc.M110430200. [DOI] [PubMed] [Google Scholar]
- [15].Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- [16].Regoli D, Rhaleb NE, Dion S. Kinin receptor subtypes. J Cardiovasc Pharmacol. 1990;15(Suppl 6):S30–8. [PubMed] [Google Scholar]
- [17].Silva JA, Jr, Araujo RC, Baltatu O, Oliveira SM, Tschope C, Fink E, et al. Reduced cardiac hypertrophy and altered blood pressure control in transgenic rats with the human tissue kallikrein gene. FASEB J. 2000;14:1858–60. doi: 10.1096/fj.99-1010fje. [DOI] [PubMed] [Google Scholar]
- [18].Bledsoe G, Chao L, Chao J. Kallikrein gene delivery attenuates cardiac remodeling and promotes neovascularization in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2003;285:H1479–88. doi: 10.1152/ajpheart.01129.2002. [DOI] [PubMed] [Google Scholar]
- [19].Yin H, Chao L, Chao J. Kallikrein/kinin protects against myocardial apoptosis after ischemia/reperfusion via Akt-glycogen synthase kinase-3 and Akt-Bad.14-3-3 signaling pathways. J Biol Chem. 2005;280:8022–30. doi: 10.1074/jbc.M407179200. [DOI] [PubMed] [Google Scholar]
- [20].Chen LM, Chao L, Chao J. Adenovirus-mediated delivery of human kallistatin gene reduces blood pressure of spontaneously hypertensive rats. Hum Gene Ther. 1997;8:341–7. doi: 10.1089/hum.1997.8.3-341. [DOI] [PubMed] [Google Scholar]
- [21].Zou Y, Hiroi Y, Uozumi H, Takimoto E, Toko H, Zhu W, Kudoh S, Mizukami M, Shimoyama M, Shibasaki F, Nagai R, Yazaki Y, Komuro I. Calcineurin Plays a Critical Role in the Development of Pressure Overload-Induced Cardiac Hypertrophy. Circulation. 2001;104:97–101. doi: 10.1161/01.cir.104.1.97. [DOI] [PubMed] [Google Scholar]
- [22].Takemoto M, Egashira K, Tomita H, Usui M, Okamoto H, Kitabatake A, et al. Chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade: effects on cardiovascular remodeling in rats induced by the long-term blockade of nitric oxide synthesis. Hypertension. 1997;30:1621–7. doi: 10.1161/01.hyp.30.6.1621. [DOI] [PubMed] [Google Scholar]
- [23].Misko TP, Schilling RJ, Salvemini D, Moore WM, Currie MG. A fluorometric assay for the measurement of nitrite in biological samples. Anal Biochem. 1993;214:11–6. doi: 10.1006/abio.1993.1449. [DOI] [PubMed] [Google Scholar]
- [24].Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- [25].Kato K, Yin H, Agata J, Chao L, Chao J. Adrenomedullin gene delivery attenuates myocardial infarction and apoptosis after ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2003;285:H1506–14. doi: 10.1152/ajpheart.00270.2003. [DOI] [PubMed] [Google Scholar]
- [26].Maestri R, Milia AF, Salis MB, Graiani G, Lagrasta C, Monica M, et al. Cardiac hypertrophy and microvascular deficit in kinin B2 receptor knockout mice. Hypertenstion. 2003;41:1151–5. doi: 10.1161/01.HYP.0000064180.55222.DF. [DOI] [PubMed] [Google Scholar]
- [27].Hap S, Michael A, Andreucci M, Bhattacharya K, Dotto P, Walters B, et al. Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc Natl Acad Sci U S A. 2003;100:4610–5. doi: 10.1073/pnas.0835895100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wu R, Laplante MA, De Champlain J. Prevention of angiotensin II-induced hypertension, cardiovascular hypertrophy and oxidative stress by acetylsalicylic acid in rats. J Hypertens. 2004;22:793–801. doi: 10.1097/00004872-200404000-00023. [DOI] [PubMed] [Google Scholar]
- [29].Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol. 2002;34:379–88. doi: 10.1006/jmcc.2002.1526. [DOI] [PubMed] [Google Scholar]
- [30].Varin R, Mulder P, Tamion F, Richard V, Henry JP, Lallemand F, et al. Improvement of endothelial function by chronic angiotensin-converting enzyme inhibition in heart failure: role of nitric oxide, prostanoids, oxidant stress, and bradykinin. Circulation. 2000;102:351–6. doi: 10.1161/01.cir.102.3.351. [DOI] [PubMed] [Google Scholar]
- [31].Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2003;35:851–9. doi: 10.1016/s0022-2828(03)00145-7. [DOI] [PubMed] [Google Scholar]
- [32].Ritchie RH, Schiebinger RJ, LaPointe MC, Marsh JD. Angiotensin II-induced hypertrophy of adult rat cardiomyocytes is blocked by nitric oxide. Am J Physiol. 1998;275(4 Pt 2):H1370–4. doi: 10.1152/ajpheart.1998.275.4.H1370. [DOI] [PubMed] [Google Scholar]
- [33].Fujii H, Ichimori K, Hoshiai K, Nakazawa H. Nitric oxide inactivates NADPH oxidase in pig neutrophils by inhibiting its assembling process. J Biol Chem. 1997;272:32773–8. doi: 10.1074/jbc.272.52.32773. [DOI] [PubMed] [Google Scholar]
- [34].Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H2O2 regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol. 2003;35:615–21. doi: 10.1016/s0022-2828(03)00084-1. [DOI] [PubMed] [Google Scholar]
- [35].Zhang GX, Kimura S, Nishiyama A, Shokoji T, Rahman M, Yao L, et al. Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovasc Res. 2005;65:230–8. doi: 10.1016/j.cardiores.2004.08.013. [DOI] [PubMed] [Google Scholar]
- [36].Seimi SK, Seinosuke K, Tsuyoshi S, Tomomi U, Tetsuaki H, Miki K, et al. Glycogen synthase kinase-3beta is involved in the process of myocardial hypertrophy stimulated by insulin-like growth factor-1. Circ J. 2004;68:247–53. doi: 10.1253/circj.68.247. [DOI] [PubMed] [Google Scholar]