

Abstract
Amyloid β-peptide (Aβ) is postulated to play a central role in the pathogenesis of Alzheimer’s disease. We recently proposed a pathway of Aβ-induced toxicity that is APP dependent and involves the facilitation of APP complex formation by Aβ. The APP-dependent component requires cleavage of APP at position 664 in the cytoplasmic domain, presumably by caspases or caspase-like proteases, with release of a potentially cytotoxic C31 peptide. In this study we show that Aβ interacted directly and specifically with membrane-bound APP to facilitate APP homo-oligomerization. Using chimeric APP molecules, this interaction was shown to take place between Aβ and its homologous sequence on APP. Consistent with this finding, we demonstrated that Aβ also facilitated the oligomerization of β-secretase cleaved APP C-terminal fragment (C99). We found that the YENPTY domain in the APP cytoplasmic tail and contained within C31 is critical for this cell death pathway. Deletion or alanine- scanning mutagenesis through this domain significantly attenuated cell death apparently without affecting either APP dimerization or cleavage at position 664. This indicated that sequences within C31 are required after its release from APP. As the YENPTY domain has been shown to interact with a number of cytosolic adaptor molecules, it is possible that the interaction of APP, especially dimeric forms of APP, with these molecules contribute to cell death.
Keywords: C31, YENPTY region, APP homo-oligomerization
The accumulation and deposition of amyloid β-protein (Aβ) within senile plaques in brain is one of the histopathological hallmarks of Alzheimer’s disease (AD). Aβ is derived by sequential proteolysis from the amyloid precursor protein (APP). Substantial data suggest that Aβ plays a critical role in initiating the cascade of events that results in AD (reviewed in ref 1). In addition to senile plaques and neurofibrillary tangles, synapse loss and neuronal death are consistently observed, and these latter changes have been hypothesized to be due to the increased levels of Aβ in brain (2). Although the traditional view suggests that Aβ assembled into insoluble and fibrillar forms is cytotoxic, increasing evidence indicates that soluble prefibrillar oligomeric Aβ species are equally, if not more, detrimental to neuronal function in vitro and in vivo (2–6). Many varied mechanisms have been proposed for Aβ-induced toxicity, but no consensus has emerged to account for its deleterious effects (7).
APP is a type I membrane protein whose function has not been clearly defined. It belongs to a gene family that includes its mammalian paralogs APLP1 and APLP2 (amyloid precursor-like proteins 1, 2) (8–10). APLP1 and APLP2 are also type I membrane proteins and share sequence similarity with APP in the N and C termini, but do not contain the Aβ domain.
APP may function as a cell surface receptor and mediate the transduction of extracellular signals into the cell, although a definitive extracellular ligand that activates this proposed pathway has yet to be described. Some biological properties have been attributed to APP, including adhesion, neuronal development, synaptogenesis, neurite outgrowth, neuroprotection, and stimulation of proliferation of neuronal progenitor cells (11). APP was recently shown to be cleaved in the cytoplasmic domain by caspase or caspase-like proteases, suggesting that proteolysis of APP accompanies activation of caspases or the apoptotic program (12–15). This cleavage takes place after the aspartate residue at amino acid position 664 (APP695 numbering) such that mutation of this residue to alanine abrogates this cleavage event (12–14). Further, the intracytoplasmic cleavage of APP at the caspase site is increased in brains of AD individuals and appears to be an early event in AD pathogenesis (16), suggesting that APP proteolysis may play a role in neurodegeneration. This concept would be consistent with the finding that after cleavage, a potentially cytotoxic C-terminal peptide, called C31, is released (12).
To integrate the observations concerning APP cleavage and Aβ-associated cytotoxicity, we recently demonstrated that there is a component of Aβ toxicity that can be attributed to the APP cytoplasmic domain, putatively related to the caspase-mediated cleavage of APP (12). We presented a model of Aβ toxicity wherein Aβ accelerates the multimerization of APP in a manner similar to other known physiological pathways, such as Fas ligand (FasL) -induced cell death and cytokine receptor signaling.
This mechanism is consistent with the recent demonstration that a minor fraction of APP exists as non-covalent dimers and higher order tetramers (17). We further hypothesized that APP complex formation is associated with activation of caspases as well as recruitment of other molecules that increase the susceptibility of cells to injury and death. The proposed mechanism also has parallel to other neurodegenerative disorders. For example, oligomeric species of PrP may facilitate the conversion of PrPC to PrPSc during scrapie infection (18).
What is unclear is how Aβ associates with APP and the mechanism(s) by which APP multimerization increases the susceptibility of neurons to cell death. In this study we show that Aβ interacts with the cognate Aβ domain in APP. In this way, Aβ may act as a ligand to its own precursor to enhance APP complex formation. Subsequent to APP multimerization, we have proposed that there is recruitment of molecules to the complex, some involved in cleavage of APP, but others presumably transduce a cell death-related signal. Our studies show that the -YENPTY- motif in the cytoplasmic domain is required for enhancing Aβ toxicity. This region is not required for cleavage of APP, indicating that C31 may transmit its cytotoxicity via interaction with other molecules via this domain.
MATERIALS AND METHODS
Antibodies
Polyclonal antibodies include the following: CT15 recognizes the C-terminal 15 amino acids of APP (19), APP-α664 recognizes the neoepitope at position 664 after cleavage (20), 863 is directed at the extracellular domain of APP (21), and D2II (Calbiochem, San Diego, CA, USA), a polyclonal antisera raised against bacteria expressed full-length mouse APLP2. Monoclonal antibodies include 26D6, recognizing the Aβ peptide sequence of amino acids 1 to 12 (12), Ab40.1 is an end-specific Aβ Ab recognizing the C terminus of Aβ 40 peptide (43), Ab9 is a pan Aβ Ab recognizing the NH2 terminus of Aβ and the C terminus of APPs (43), anti-GFP (Stressgen, Victoria, Canada), HA tag (Roche, Indianapolis, IN, USA), and FLAG epitope (M2, Sigma-Aldrich).
Plasmid construction and mutagenesis
The APP695, APP-D664A, C99, C99-D664A, and C31 cDNA constructs have been described (12). APP695-KK was engineered by adding the di-lysine retention motif (KKQN) to the C terminus (22). Two chimeric APP-APLP2 constructs were generated by polymerase chain reaction (PCR). In one, APP-APLP2, the region encompassing the Aβ domain from positions 1–28 (DAEF… GSNK from residues 597–624) were substituted by a 44 amino acid region from mouse APLP2 immediately outside the predicted transmembrane domain (residues 616–659). In C99-APLP2, the same Aβ domain was replaced by the identical 44 amino acids from mAPLP2 (see Fig. 2). Additional APP constructs generated by PCR include: APP with HA or FLAG tags appended to the C terminus, APP ΔNPTY where the seven amino motif –GYENPTY- (residues 681–687) was deleted, and APP-APLP2 chimera where the N-terminal Aβ binding domain (residues 18–119) was deleted.
Figure 2.

Schematic of APP-APLP2 chimeric constructs. 44 amino acids from mouse APLP2 were substituted for the 28 juxtamembrane amino acid residues.
Alanine point mutations were accomplished using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). C31-enhanced GFP stabilized constructs were engineered by subcloning into pEGFP-N3 (Clontech, Mountain View, CA, USA).
Cell culture and coimmunoprecipitation
Mouse N2a neuroblastoma cells and rat B103 neuroblastoma cells were cultured under standard conditions. Plasmid constructs were transiently transfected into the various cell lines by Fugene-6 (Roche). Coimmunoprecipitation and Western blot analysis were performed as described previously (12).
Assessment of Aβ-induced cell death
For Aβ toxicity study, cells expressing various constructs were exposed to 10 μm Aβ1–42 (kind gift from Dr. C. Glabe). Aβ42 peptide was prepared by dissolving the lyophilized peptide in DMSO at 1 mM and adding immediately to cell culture medium to give final 10μM concentration. This freshly solubilized Aβ preparation yielded primarily monomeric to oligomeric Aβ species. For aggregated Aβ42, the peptide was incubated in water at 37°C for 72 h. Cell death studies were carried out in transiently transfected cells. A GFP-expressing plasmid was cotransfected with the respective constructs (at 1:1 M ratio) and death positive cells were scored as a percentage of total GFP-positive cells. Apoptosis was assessed by calcein AM/ethidium homodimer stain (live/dead assay, Molecular probes) and bis-benzamide staining (Hoechst assay, Sigma-Aldrich) 24 h after Aβ treatment. For each treatment group, the percentage of dead cells was evaluated from the total population for both Aβ-treated and untreated control cells. Aβ-induced cell death was further calculated as the ratio between these two values. To average across multiple experiments, the ratios were normalized to either full-length APP or C99 transfected cells. The use of this value, Aβ-induced cell death, allows us to control for basal cell death that may vary through different assays. Averages of all experiments ± SD are presented.
Aβ binding assay
Direct Aβ binding assay was conducted through a modified ligand dot blot procedure (23, 34) as follows. In vitro translated products from the various constructs were labeled with 35S using the TnT Reticulocyte Lysate kit (Promega, Madison, WI, USA) and purified through NAP5 sephadex column (Amersham Biosciences, Uppsala, Sweden). Equivalent amounts of in vitro translated products measured by CT15 reactivity on dot blot assay were then incubated with freshly prepared Aβ42 peptide (4 μg) dot blotted onto nitrocellulose membrane. Scrambled Aβ42 (Anaspec, San Jose, CA, USA) was also spotted to determine background, nonspecific binding values for each probe. Radioactivity was quantified in a scintillation counter. The amount of specific binding was calculated by subtracting the radioactivity of the scrambled Aβ42 spot from the Aβ42 spot for each probe. Results were averaged to be expressed as specific binding values (adjusted by relative number of methionine residues). These were normalized to APP = 1 or C99 = 1, respectively.
Cell surface biotinylation
N2a cells were surface biotinylated by incubating with Sulfo-NHS-biotin (Pierce, Rockford, IL, USA) at 2 mg/ml in ice-cold PBS. After 30 min, cells were washed and quenched with PBS containing 25 mM lysine monohydrochloride. Cells were then lysed in NP-40 1% buffer and incubated with streptavidin-agarose beads overnight (Sigma Chemical Company, St. Louis, MO, USA). After washing in NP-40 buffer, the beads were boiled in sample buffer, fractionated in SDS-PAGE and immunoblotted with the indicated antibodies.
RESULTS
AβInduces cell death in cells expressing APP on the cell surface
We previously demonstrated that a component of Aβ toxicity is APP dependent and this pathway requires an intact APP cytoplasmic domain, specifically the aspartate residue at position 664. What is unclear is whether this is due to Aβ interacting directly with APP, presumably at the cell surface, or via an indirect pathway. Accordingly, in this first experiment, we tested whether APP enhancement of Aβ toxicity requires trafficking of APP to the cell surface as would be required if Aβ interacted directly with APP. N2a cells were transiently transfected with either APP wild-type or APP-KK, the latter containing an ER retention signal (-KKQN-) appended to the C terminus of APP. Addition of such a signal prevents APP from being transported beyond the cis-Golgi compartment. Hence, this APP-KK is retained in the ER (22) and not sorted to the cell surface. Accordingly, primarily immature APP species were seen when this mutant was transiently transfected into N2a neuroblastoma cells (Fig. 1A). After surface biotinylation with a cell impermeant cross-linker, APP-KK was essentially excluded from the cell surface, as expected (Fig. 1A). Having demonstrated that APP-KK is retained intracellularly, N2a cells transfected with either APP-KK or APP wild-type control were treated with Aβ to determine the degree of cell death. Only APP wild-type cells showed the expected cytotoxicity (Fig. 1B). There was no cell death induction in the APP-KK transfected cells, indicating that the APP-dependent component of Aβ cytotoxicity requires trafficking of APP to the cell surface, where presumably Aβ interaction takes place.
Figure 1.
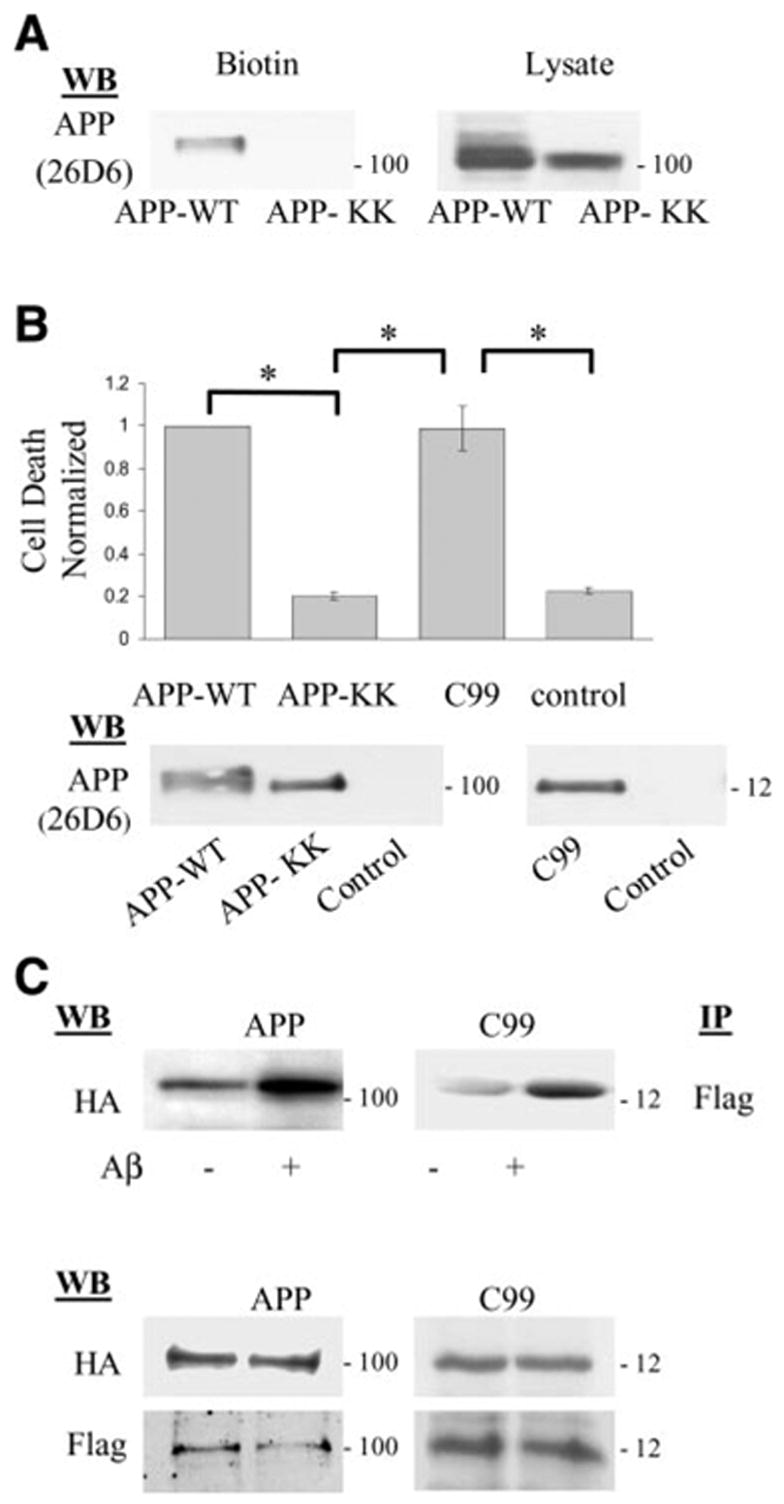
Aβ-induced cell death after expression of various APP constructs. A) Cell surface biotinylation showed that APP mutant with the ER retention signal (APP-KK) when transiently expressed in N2a cells was not sorted to the cell surface compared with wild-type APP (APP wild-type). Western blot of whole cell lysates (right panel) showed less maturation for the APP-KK mutant. B) N2a cells expressing APP wild-type, C99, or APP-KK constructs were treated with Aβ and assayed for cell death as described. Cell death induced by Aβ treatment was normalized to that obtained from APP wild-type construct. Expression of C99 or APP wild-type similarly showed significant cell death, while APP-KK comparable to control cells (mock transfection) showed diminished cell death (*P<0.001, 1-way ANOVA; post hoc Tukey). Error bars indicate SD. C) N2a cells were transiently transfected with HA and Flag tagged APP constructs and APP complexes detected by immunoprecipitation with Flag Ab followed by Western blot with anti-HA Ab. After Aβ treatment, APP complexes increased by ~ 2-fold [1.98±0.59 (n=5) for APP695, and 1.93±0.43 (n=5) for C99]. Lower panels showed that expression levels for HA and Flag tagged APP constructs were comparable in all conditions. Indicated on the right of each Western blot panel is the approximate molecular weight.
The Aβ effect on cell death is mediated through interaction with the cognate Aβ region, APP 597–624
Aβ has been reported previously to interact with APP (25), and one site of interaction was mapped to the NH2 terminus of APP (26). These observations are consistent with the findings described above that APP must traffic to the cell surface to enhance Aβ toxicity. However, we have previously shown that toxicity of the C-terminal fragments of APP such as C99 (also known as “C100”) depends in part on the aspartate residue at position 664 (12). This indicated that the large N-terminal region of APP might be unnecessary for mediating Aβ toxicity. Therefore, we next asked whether N-terminally truncated APP C-terminal fragment (C99), similar to full-length APP, could enhance Aβ toxicity. Accordingly, Aβ was added to N2a cells transiently transfected with APP C99 or full-length APP. Indeed, expression of C99 significantly increased (~5 times) Aβ-induced cell death to a degree comparable to full-length APP695 (Fig. 1B).
Next we asked whether Aβ can amplify C99 dimerization, as has been shown with full-length APP and which (in the case of full-length APP) correlated with cell death. We have previously shown that these APP complexes induced by Aβ were not generated after cell lysis (27). Similar to full-length APP, HA-tagged C99 species can be coimmunoprecipitated with Flag-tagged C99 species (Fig. 1C) and vice versa (not shown). Moreover, like full-length APP, C99 homomeric complexes can be enhanced ~ 2-fold by the addition of Aβ (Fig. 1C), suggesting that much of the extracellular domain of APP is not required for enhancing Aβ cytotoxicity. Conversely, these finding suggest that the 28 amino acid juxtamembrane residues that are present on the cell surface contained within both C99 and APP695 might be the critical sequence for mediating this effect.
To test the hypothesis that the 28 amino acid residues of the Aβ domain in APP are required for mediating Aβ-induced toxicity, two APP chimeric molecules were engineered in which the 28 amino acid region within APP was replaced by a comparable domain from mouse APLP2 (Fig. 2). This region was chosen because it is the most divergent between APP and APLP2 as there is no Aβ domain in the latter. However, both APP and APLP2 undergo similar processing to release the large soluble N-terminal fragment after α-secretory processing, as well as γ-secretase cleavage to generate Aβ and Aβ-like peptide, respectively (28–31).
The resulting two chimeric constructs, APP-APLP2 and C99-APLP2, based on APP695 and C99, respectively, were then transfected into N2a cells, and cell death was assessed after Aβ treatment. Unlike the parental APP or C99 molecules, neither chimeric construct was able to augment Aβ toxicity (Fig. 3A). The lack of cell death in the APP-APLP2 and C99-APLP2 chimeric molecules was not due to altered α or γ-secretase processing of these proteins. This was manifested by the presence of APPs and Aβ (or Aβ-like peptides) in the media of cells expressing the chimeras (Fig. 3B). Moreover, when coimmunoprecipitations of HA and Flag-tagged chimeras were carried out after Aβ treatment, there was no evidence of an increase in APP-APLP2 or C99-APLP2 complexes, unlike what had been observed with APP or C99 (Fig. 4A). Accordingly, no caspase cleavage at position 664 was documented in these constructs by the cleavage specific Ab (Fig. 4B). These results therefore point to the region encompassing Aβ positions 1–28, within APP or C99, as the critical domain for Aβ-induced oligomerization, caspase cleavage, and cell death.
Figure 3.
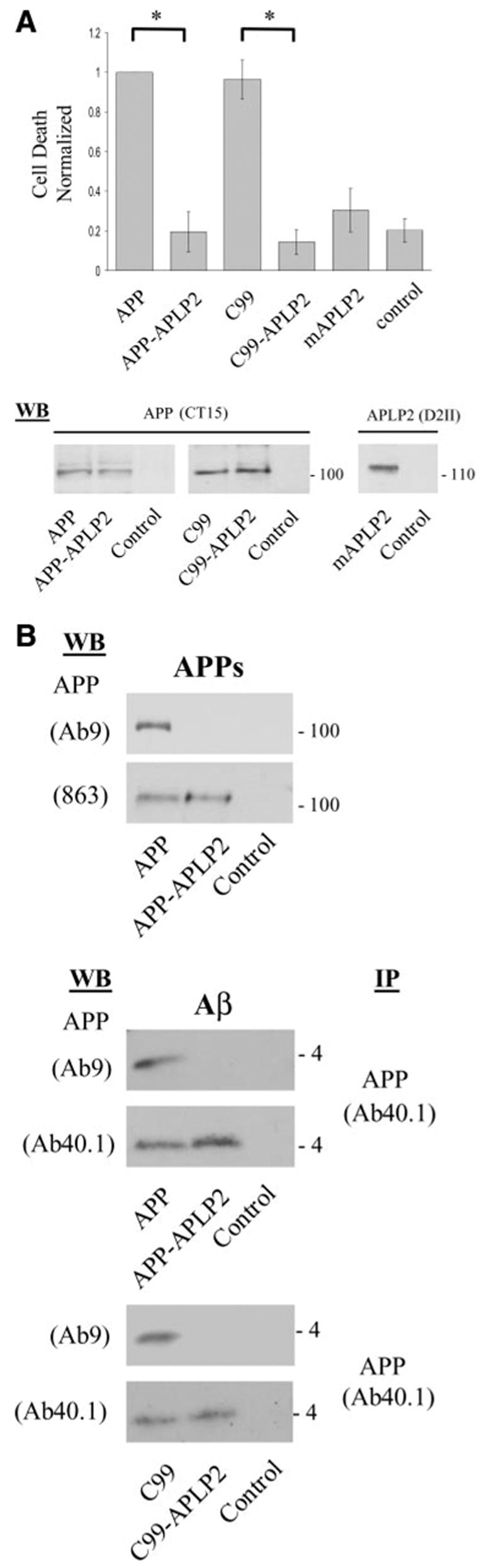
APP 597–624 domain mediates the APP dependent Aβ-induced cell death. A) Cell death enhanced by Aβ treatment was significantly reduced in N2a cells expressing the chimeric constructs (APP-APLP2 and C99-APLP2), down to a concentration comparable to control conditions or mAPLP2 (*P<0.001, 1-way ANOVA; post hoc Tukey). Lower panel shows that expression levels of all the APP-based constructs are comparable. Full-length mAPLP2 expression was also detectable by Western blot. B) The APP-APLP2 chimeras show α- and γ-secretase processing similar to native APP. The top panel shows secretion of the N-terminal APPs fragment. An Ab to the midregion of APP (863) detects the soluble APPs fragment from both APP and APP-APLP2 but not with an Ab to the C terminus of APP (Ab9) within the Aβ domain as this region is absent in the APP-APLP2 chimera. In the middle panel, Aβ and Aβ-like peptides are detected in the medium of cells expressing APP and APP-APLP2 chimera, respectively. These peptides were immunoprecipitated with Ab40.1 MAb recognizing the C terminus of Aβ40 peptide, then Western blotted with either Ab9 or Ab40.1 Ab. As with APPs, Ab9 is unreactive to the Aβ-like peptides as the NH2 terminus has been substituted by sequences from APLP2 but is reactive to the authentic NH2 terminus of Aβ released from APP trans-fected cells. Similar peptides are released from C99 and C99-APLP2 transfected cells and detected with the same Ab combinations as performed above (bottom panel).
Figure 4.
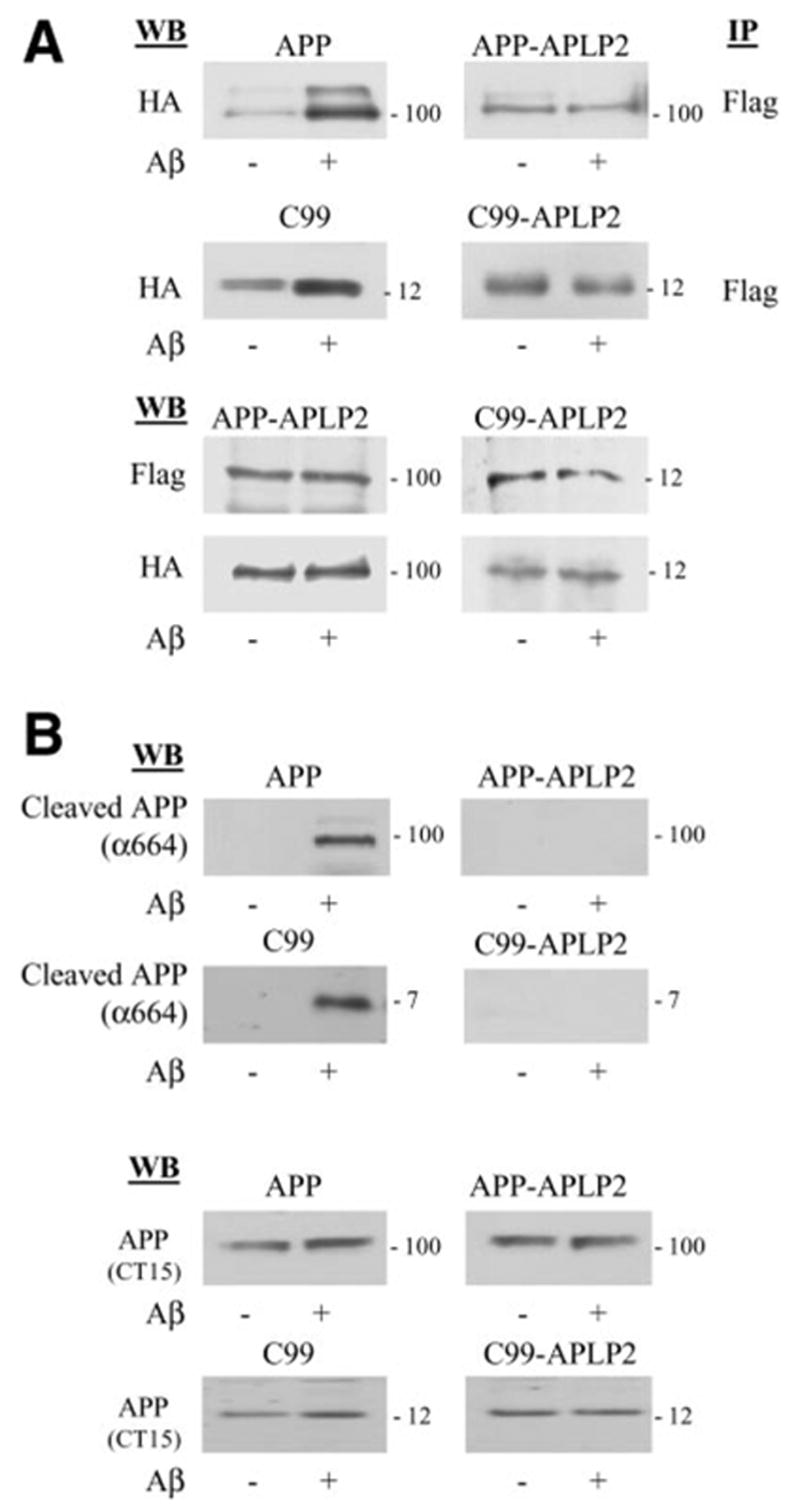
APP 597–624 domain mediates the Aβ-induced APP complex formation and caspase cleavage. A) The APP-APLP2 and C99-APLP2 constructs that failed to show APP-dependent, Aβ-induced cell death also failed to show enhanced complex formation after Aβ treatment. Immunoprecipitation and Western blot was identical to that in Fig. 1. Lower panels show comparable protein expression of the various APLP2 chimeric constructs by Western blot of total cell lysates. B) APP-based chimeras do not undergo caspase cleavage after Aβ treatment. B103 cells expressing wild-type APP, C99, and the respective chimeras were incubated in the presence or absence of Aβ treatment. Cell lysates were blotted with APP Ab (bottom panel) or APP-α664 (upper panel), directed against the neoepitope at position 664 after cleavage.
Finally, although the preceding data point to the importance of the juxtamembrane region of APP as crucial for Aβ interaction, it is unclear whether this is a direct or an indirect interaction requiring an adapter molecule(s). To address this question, 35S-methionine labeled in vitro translated APP constructs corresponding to those transfected into N2a cells were assayed for their ability to interact with Aβ spotted onto nitrocellulose membranes (Fig. 5). In this modified ligand blotting assay, the C99-APLP2 and APP-APLP2 chimeras showed marked reduction in Aβ binding compared with the full-length APP695 and APP C-terminal C99 constructs, respectively. Specifically, C99-APLP2 showed virtually no binding to immobilized Aβ compared with C99 (*P<0.001, t test). With APP-APLP2 construct, there was ~50% reduction in binding compared with full-length APP-695 (*P<0.001, 1-way ANOVA; post hoc Tukey).
Figure 5.
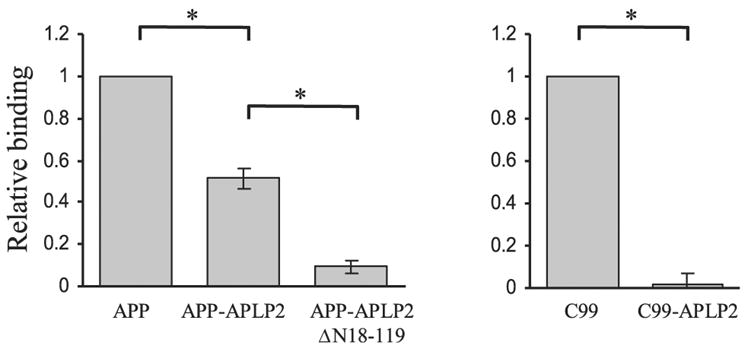
Aβ binds directly to APP at residues 597–624. 35S-labeled APP, C99, and the respective chimeras were expressed in vitro and purified. Ligand binding experiment was conducted by incubating 35S-labeled in vitro translated APP constructs with Aβ peptides immobilized onto nitrocellulose membrane. Bound probe was quantitated by measuring the radioactivity in a scintillation counter. Specific binding to Aβ42 was calculated by subtracting the radioactivity obtained from immobilized scrambled Aβ42 peptide used as a nonspecific binding control. Values were normalized relatively to C99 or APP, respectively. For both chimeras binding was significantly lower than native constructs (*P<0.001 by unpaired t test).
We attribute the residual interaction of APP-APLP2 with Aβ to the presence of an additional binding site within the NH2 terminus of APP that is retained in the APP-APLP2 chimera as reported (26).
To test this notion, a mutant APP-APLP chimera was engineered where the N- terminal 18–119 region was deleted (APP-APLP2ΔN18–119). Consistent with a previous report that binding of APP to Aβ was mediated by this domain within the APP NH2 terminus (26), the interaction between APP-APLP2 ΔN18–119 and Aβ was significantly reduced compared with APP-APLP2 chimeric protein (*P<0.001, 1-way ANOVA; post hoc Tukey) (Fig. 5). Taken together, these results suggest that the 28 amino acid residues immediately outside the transmembrane region, or the Aβ domain, are essential for facilitating APP complex formation, presumably by a homophilic interaction whereby Aβ interacts with its cognate sequences within APP to bring adjacent APP molecules together.
The GYENPTY region is essential for Aβ-induced cell death
In our current working model whereby Aβ interacts directly with APP to increase APP complex formation, the hypothesis is that the APP complex recruits molecules, such as caspase-8, that cleave APP in the C terminus and, in so doing, transduces a potentially cytotoxic signal, one of which may be the release of C31 peptide. What is unclear is how APP or C31 may be cytotoxic because a death domain (as present, for example, in some TNF receptor family members) has not been described in APP. To address this issue, we first asked which domain(s) within APP is critical for this cell death pathway. A number of cytosolic molecules have been shown to interact with APP, including Fe65/Fe65L, x11 (or Mint-1)/X11L, JIP-1B (JNK-interacting protein 1B), and mammalian disabled (mDab1). Most of these molecules interact with the GYENPTY motif within APP (positions 681 to 687), which overlaps with the signal for internalization. Therefore, APP695 or APP751 of the GYENPTY deletion mutants were transfected into N2a cells and tested for their ability to amplify Aβ toxicity. Surprisingly, unlike the full-length APP constructs, both mutants failed to enhance Aβ-induced cell death when expressed at comparable levels as wild-type APP constructs (Fig. 6A). On the other hand, when immunoprecipitations were carried out with epitope-tagged constructs, we found that these APP deletion constructs maintained similar levels of Aβ-induced oligomerization to those of the wild-type proteins (Fig. 6B). This latter observation is not surprising because, as seen in the preceding results, APP oligomerization is amplified by the binding of Aβ to its cognate sequences within the extracellular region of APP, and the cytoplasmic region is presumably dispensable. However, this finding indicated that loss of cytotoxicity is not secondary to the absence of APP homomeric complexes.
Figure 6.
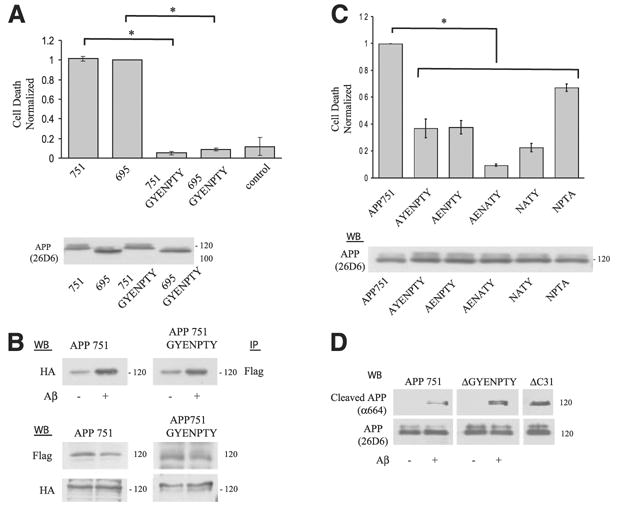
The GYENPTY region in APP is essential for Aβ-induced cell death yet dispensable for APP oligomerization and cleavage. A) Expression of either APP751 or APP695 mutants deleted of the GYENPTY domain reduced cell death to control levels. Lower panel showed that expression of the APP mutant constructs was comparable to the wild-type APP constructs (unpaired t test between both wild-type APP species and the respective deletion mutants). B) APP complex formation was unchanged in the deletion mutant construct. APP751 and APP751 ΔGYENPTY constructs were transiently expressed in N2a cells and the lysates immunoprecipitated with Flag Ab followed by Western blot with anti-HA Ab. APP complex formation in the APP-GYENPTY mutant was increased ~ 2-fold (1.8±0.26) (n=5) after Aβ treatment, comparable to control APP (1.6±0.21) (n=5). Protein expression control is shown in the lower panel. C) Expression of single Alanine point mutants through the –GYENPTY– region in N2a showed reduction in Aβ-induced cell death (*P<0.001, 1-way ANOVA; post hoc Tukey) similar to the deletion mutant shown in panel A. Control for protein expression of the various APP constructs shown in the lower panel. D) APP is cleaved after the D664 residue after Aβ treatment in the APP deletion mutant construct. Cleavage was assayed in B103 cells following transient transfection with APP, APP ΔGYENPTY, and APP ΔC31 control and blotted with APP Ab (bottom panel) or APP-α664 (upper panel) against the neo epitope at position 664 after cleavage. As shown, both APP and APP deletion mutant showed cleavage after Aβ treatment. The C-terminal truncated APP cleavage fragment comigrates with the control APPΔC31 construct, which is deleted of the last 31 amino acids.
We next performed alanine-scanning mutagenesis through the GYENPTY domain to further define the critical sequences. All point mutants (AYENPTY [G681], AENPTY [Y682], AENATY [G681/P685], NATY [P685], and NPTA [Y687]) showed a significant decrease in Aβ-induced cell death compared with wild-type APP (Fig. 6C). Though cell death was significantly attenuated, the magnitude of reduction was not equal, with the NPTA mutant showing the smallest reduction (~40%).
To examine whether APP is cleaved, we next assessed caspase cleavage of these deletion constructs after Aβ treatment by using an end-specific Ab. The APP constructs were transfected into B103 neuroblastoma cells that lack endogenous APP in order to lower the background signal. To our surprise, there was essentially no difference in the concentration of cleaved APP species that reacted to the cleavage-specific Ab, α664, between wild-type APP and the GYENPTY deletion mutant. This observation indicated that loss of cytotoxicity in the GYENPTY mutant was not related to the absence of cleavage at position 664 (Fig. 6D), as a C31-like peptide lacking the GYENPTY domain should have been released from the APP deletion mutant. To confirm this finding, C31 constructs with and without the GYENPTY region were expressed in N2a cells to assess the concentration of cell death. In this experiment, two different starting C31 constructs were examined: C31 alone or C31-enhanced GFP chimera, the latter to stabilize the C31 peptide. Consistent with our notion, deletion of the GYENPTY region decreased the C31 and C31-enhanced GFP mediated toxicity back to control level (Fig. 7).
Figure 7.
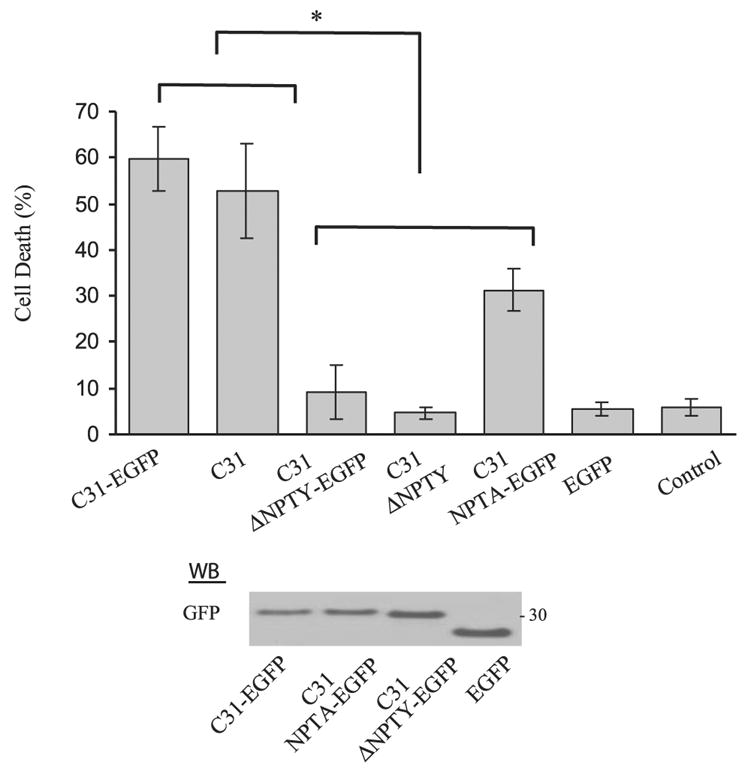
C31 toxicity requires an intact GYENPTY region. Transient expression of C31 in N2a cells caused increased cell death significantly above control. Expression of the C31 and C31-enhanced GFP NPTY deletion mutant showed no enhanced cell death compared with control (*P<0.001, 1-way ANOVA, post hoc Tukey). C31-enhanced GFP NPTA chimera showed an intermediate level of cell death. Fusion of enhanced GFP to the C terminus of C31 stabilized the peptide and these chimeric proteins are readily detected by an Ab to GFP (bottom panel).
DISCUSSION
It has been shown by a number of laboratories that APP can be cleaved by caspases at multiple sites (13, 14, 32, 33). In particular, cleavage in the cytoplasmic domain at position 664 is associated with cell death, putatively mediated through the release of a cytotoxic peptide termed C31 (12, 20). Given the central role of Aβ in the amyloid hypothesis of AD pathogenesis, we recently demonstrated that caspase cleavage of APP is associated with Aβ toxicity. Specifically, we demonstrated that a component of Aβ toxicity is dependent on APP and that Aβ-induced oligomerization of APP increases susceptibility to cell death, presumably involving the caspase cleavage of APP (34).
Although it was reported earlier that Aβ interacts with cell surface APP (25, 26, 35), it is unclear which region within APP interacts with Aβ to initiate this potentially cytotoxic signal. In this study, our results indicate that this interaction takes place when Aβ directly binds to its own sequence on the extracellular domain of APP, analogous to APP acting as a receptor for Aβ, which in turn acts as a pseudo-ligand. In so doing, Aβ can directly facilitate APP complex formation, an event we propose initiates the cell death signal through a pathway that requires the YENPTY motif within the APP cytoplasmic tail.
We previously showed that the Aβ-induced APP complex formation did not require the cytoplasmic domain of APP. This was established by expressing a chimeric APP molecule in which the entire cytoplasmic domain was replaced by the CD3ξ subunit of the T cell receptor (34). In this study, we showed through multiple experiments that Aβ appears to interact directly with APP to enhance APP homo-oligomerization by binding to its homologous sequences, possibly taking place at the cell surface. First, when an ER retention signal (-KKQN-) was appended to APP, this APP-KK construct was largely excluded from the cell surface; but more important, Aβ toxicity was largely attenuated compared with expression of wild-type APP. This suggested that APP must be on the cell surface to facilitate Aβ toxicity. Second, C99, the APP C-terminal fragment generated after β-secretase cleavage, was not only able to form Aβ-induced complexes, but also potentiated Aβ toxicity indistinguishable from that of full-length APP. This indicated that the critical domain in APP mediating Aβ cytotoxicity was within the Aβ sequence and the transmembrane domain, since neither the large extracellular region up to the β-secretase site nor the cytoplasmic domain was necessary. Third, we demonstrated that the 28 amino acid juxtamembrane residues (residues 597–624) present on the cell surface contained within C99 and APP695 were necessary and sufficient to enhance Aβ toxicity. This was accomplished by expressing several APP chimeric molecules in which this region was exchanged with sequences from APLP2, where there was no primary sequence homology. Neither the chimeric C99 nor chimeric APP695 was able to augment Aβ toxicity. Further, neither chimeric construct was able to form complexes by coimmunoprecipitation on Aβ addition. Fourth, to show that Aβ interacted with its own cognate sequences in APP, we performed a ligand blotting experiment in which Aβ was immobilized to nitrocellulose membrane and incubated with various radiolabeled APP constructs. That Aβ can aggregate into oligomeric and aggregated forms is well established. However, it has not been shown before that Aβ can directly interact with APP by binding to its own sequences. Consequently, Aβ can associate with APP through two regions: in the NH2 terminus at residues 18–119 (26, 36) and with its own cognate domain at residues 597–624. Thus, we suggest that the latter domain is essential for APP complex formation, presumably through a homophilic interaction between free Aβ and the Aβ domain within APP.
In mapping the site where Aβ binds to APP, we noticed that APP dimerization can take place through two apparently independent steps. From the coimmunoprecipitation studies, both APP and C99 demonstrated a basal level of oligomerization in the absence of Aβ, consistent with a previous study with full-length APP (17). After the addition of exogenous Aβ, APP complex formation consistently increased by 2- to 3-fold. Although both APP-APLP2 and C99-APLP2 chimera failed to show Aβ-induced oligomerization or cell death, coimmunoprecipitable complexes were detected basally and did not require the Aβ sequence, because these residues had been exchanged with APLP2 (Fig. 4a). This indicated that there are two domains mediating APP dimerization: one induced by Aβ and one that is independent of the Aβ sequence. Identifying the latter domain was not a focus of this study although a recent X-ray crystallographic study indicated the “E2” region of APP, a highly conserved domain containing the N-glycosylation sites, has a high propensity to dimerize (37). On the other hand, this finding would not explain why C99, which does not contain the E2 region, also forms complexes. It is possible that the transmembrane sequence or cytosolic adaptor molecules are able to facilitate APP complex formations as well.
We previously suggested that a component of Aβ toxicity is APP dependent, and this requires an intact caspase cleavage site in the cytoplasmic domain at position 664 of APP (12, 20, 38). Further, we showed that forced dimerization of APP led to increased susceptibility to cell death (27). To understand the mechanism whereby APP dimerization can initiate a cell death signal, we performed deletion and alanine mutagenesis through the –GYENPTY- motif in the APP cytoplasmic tail (residues 681–687). The rationale for focusing on this region is because this is a site contained within C31 and one that interacts with cytoplasmic proteins, including Fe65, X11/Mint1, Jip-1b, and mDab1. Indeed, the APP-dependent component of Aβ toxicity was essentially abrogated when this domain was deleted from APP. Alanine-scanning mutants further confirmed the importance of this motif. Conversely, cell toxicity due to overexpression of C31 was also lost in a C31 construct deleted of this domain. APP complex formation was not perturbed in the APP deletion mutant. Further, cleavage at position 664 was also unaltered after Aβ treatment. This indicated that, according to our proposed scheme, the requirement of this domain occurs downstream of both APP dimerization and cleavage. Why this domain is important for cytotoxicity is unclear. On the one hand, it has been reported that expression of an artificial AICD construct encompassing the C-terminal 58 residues of APP resulted in apoptosis in H4 glioma cells (39). Therefore, it is possible that C31, which is contained entirely within C58, induces cell death simply through a C58-mediated pathway. The latter was shown to require Tip60, possibly through a mechanism involving AICD-Tip60 complex formation and translocation into the nucleus. On the other hand, this pathway did not require either Fe65 or the YENPTY domain because mutagenesis of either tyrosine residues had no effect on cell death. In this regard, our results are more consistent with a proposed pathway in which AICD (C59) or C31 forms a ternary complex with Fe65 and CP2/LSF/LBP1 to induce expression of GSK-3β, which in turn was correlated with apoptosis (40). This is because APP binds to Fe65 through the NPTY domain, and this interaction will be lost when this motif is deleted, as was seen in our studies. However, both C58 and C59 are likely nonphysiologic or at least represent very minor species because ε-cleavage of APP generates AICD, which is 49 to 50 residues in length (41). Therefore, findings of cell death derived from the overexpression of C58 and C59 may be misleading.
The model we have presented here suggests that cleavage of APP at position 664 with release of C31 is associated with cell death. C31 has been difficult to detect, likely because of its short half-life or that it rapidly interacts with other cellular proteins. However, the C-terminal truncated APP species is very stable and can be recognized with cleavage specific antibodies (Figs. 4B, 6D; (12, 14, 20, 27)). It is noteworthy that caspase-cleaved APP N-terminal fragment has been detected in brains of AD individuals and indeed, this fragment colocalizes with senile plaques and neuronal cytoplasmic inclusions known as granulovacuolar degeneration (14, 42). Thus, it has been proposed that this cleavage may represent an early event in AD pathogenesis (16). If the latter speculation is true, then it will be important to understand what regulates the caspase cleavage of APP and mediates the toxicity of the cleaved fragments, whether through a pathway that involves C31 or protein binding partners that interact with the GYENPTY domain of APP.
Acknowledgments
We thank Dr. C. Glabe of the University of California, Irvine, for generous gift of Aβ peptide and Dr. Todd Golde for gift of Ab9 and Ab40.1 antibodies. This work was supported by a grant from the National Institutes of Health AG05131 (EHK and DEB) and the Alzheimer’s Association (EHK).
References
- 1.Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. Medicine—the amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Klein WL. ADDLs & protofibrils—the missing links? Neurobiol Aging. 2002;23:231–235. doi: 10.1016/s0197-4580(01)00312-8. [DOI] [PubMed] [Google Scholar]
- 4.Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol Aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong YS, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid beta-protein fibrillogenesis—structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 8.Sprecher CA, Grant FJ, Grimm G, Ohara PJ, Norris F, Norris K, Foster DC. Molecular-cloning of the cDNA for a human amyloid precursor protein homolog—evidence for a multigene family. Biochemistry. 1993;32:4481–4486. doi: 10.1021/bi00068a002. [DOI] [PubMed] [Google Scholar]
- 9.Wasco W, Gurubhagavatula S, Dparadis M, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE. Isolation and characterization of Aplp2 encoding a homolog of the Alzheimers associated amyloid beta-protein precursor. Nature Genet. 1993;5:95–100. doi: 10.1038/ng0993-95. [DOI] [PubMed] [Google Scholar]
- 10.Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F. identification of a mouse-brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid-beta-protein precursor. Proc Natl Acad Sci USA. 1992;89:10758–10762. doi: 10.1073/pnas.89.22.10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 12.Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nature Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 13.Weidemann A, Paliga K, Durrwang U, Reinhard FBM, Schuckert O, Evin G, Masters CL. Proteolytic processing of the Alzheimer’s disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J Biol Chem. 1999;274:5823–5829. doi: 10.1074/jbc.274.9.5823. [DOI] [PubMed] [Google Scholar]
- 14.Gervais FG, Xu DG, Robertson GS, Vaillancourt JP, Zhu YX, Huang JQ, LeBlanc A, Smith D, Rigby M, Shearman MS, et al. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 15.Pellegrini L, Passer BJ, Tabaton M, Ganjei JK, D’Adamio L. Alternative, non-secretase processing of Alzheimer’s beta-amyloid precursor protein during apoptosis by caspase-6 and-8. J Biol Chem. 1999;274:21011–21016. doi: 10.1074/jbc.274.30.21011. [DOI] [PubMed] [Google Scholar]
- 16.Zhao M, Su J, Head E, Cotman CW. Accumulation of caspase cleaved amyloid precursor protein represents an early neurodegenerative event in aging and in Alzheimer’s disease. Neurobiol Dis. 2003;14:391–403. doi: 10.1016/j.nbd.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J Biol Chem. 2001;276:33923–33929. doi: 10.1074/jbc.M105410200. [DOI] [PubMed] [Google Scholar]
- 18.Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem. 1998;67:793–819. doi: 10.1146/annurev.biochem.67.1.793. [DOI] [PubMed] [Google Scholar]
- 19.Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci. 1993;13:3136–3142. doi: 10.1523/JNEUROSCI.13-07-03136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galvan V, Chen S, Lu D, Logvinova A, Goldsmith P, Koo EH, Bredesen DE. Caspase cleavage of members of the amyloid precursor family of proteins. J Neurochem. 2002;82:283–294. doi: 10.1046/j.1471-4159.2002.00970.x. [DOI] [PubMed] [Google Scholar]
- 21.Marquez-Sterling NR, Lo ACY, Sisodia SS, Koo EH. Trafficking of cell-surface beta-amyloid precursor protein: evidence that a sorting intermediate participates in synaptic vesicle recycling. J Neurosci. 1997;17:140–151. doi: 10.1523/JNEUROSCI.17-01-00140.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maltese WA, Wilson S, Tan Y, Suomensaari S, Sinha S, Barbour R, McConlogue L. Retention of the Alzheimer’s amyloid precursor fragment C99 in the endoplasmic reticulum prevents formation of amyloid beta-peptide. J Biol Chem. 2001;276:20267–20279. doi: 10.1074/jbc.M007238200. [DOI] [PubMed] [Google Scholar]
- 23.Guichet A, Copeland JWR, Erdelyi M, Hlousek D, Zavorszky P, Ho J, Brown S, Percival-Smith A, Krause HM, Ephrussi A. The nuclear receptor homologue Ftz-F1 and the homeodomain protein Ftz are mutually dependent cofactors. Nature. 1997;385:548–552. doi: 10.1038/385548a0. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz CJ, Sampson HM, Hlousek D, Percival-Smith A, Copeland JW, Simmonds AJ, Krause HM. FTZ-factor1 and Fushi tarazu interact via conserved nuclear receptor and coactivator motifs. EMBO J. 2001;20:510–519. doi: 10.1093/emboj/20.3.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lorenzo A, Yuan ML, Zhang ZH, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Sol Vigo F, Sommer B, Yankner BA. Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer’s disease. Nature Neurosci. 2000;3:460–464. doi: 10.1038/74833. [DOI] [PubMed] [Google Scholar]
- 26.Van Nostrand WE, Melchor JP, Keane DM, Saporito-Irwin SM, Romanov G, Davis J, Xu F. Localization of a fibrillar amyloid beta-protein binding domain on its precursor. J Biol Chem. 2002;277:36392–36398. doi: 10.1074/jbc.M204676200. [DOI] [PubMed] [Google Scholar]
- 27.Lu DC, Shaked GM, Masliah E, Bredesen DE, Koo EH. Amyloid beta protein toxicity mediated by the formation of amyloid-beta protein precursor complexes. Ann Neurol. 2003;54:781–789. doi: 10.1002/ana.10761. [DOI] [PubMed] [Google Scholar]
- 28.Eggert S, Paliga K, Soba P, Evin G, Masters CL, Weide-mann A, Beyreuther K. The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves alpha-, beta-, gamma-, and epsilon-like cleavages—modulation of APLP-1 processing by N-glycosylation. J Biol Chem. 2004;279:18146–18156. doi: 10.1074/jbc.M311601200. [DOI] [PubMed] [Google Scholar]
- 29.Paliga K, Peraus G, Kreger S, Durrwang U, Hesse L, Multhaup G, Masters CL, Beyreuther K, Weidemann A. Human amyloid precursor-like protein 1—cDNA cloning, ectopic expression in COS-7 cells and identification of soluble forms in the cerebrospinal fluid. Eur J Biochem. 1997;250:354–363. doi: 10.1111/j.1432-1033.1997.0354a.x. [DOI] [PubMed] [Google Scholar]
- 30.Slunt HH, Thinakaran G, Vonkoch C, Lo ACY, Tanzi RE, Sisodia SS. Expression of a ubiquitous, cross-reactive homolog of the mouse beta-amyloid precursor protein (App) J Biol Chem. 1994;269:2637–2644. [PubMed] [Google Scholar]
- 31.Scheinfeld MH, Ghersi E, Laky K, Fowlkes BJ, D’Adamio L. Processing of beta-amyloid precursor-like protein-1 and-2 by gamma-secretase regulates transcription. J Biol Chem. 2002;277:44195–44201. doi: 10.1074/jbc.M208110200. [DOI] [PubMed] [Google Scholar]
- 32.Soriano S, Lu DC, Chandra S, Pietrzik CU, Koo EH. The amyloidogenic pathway of amyloid precursor protein (APP) is independent of its cleavage by caspases. J Biol Chem. 2001;276:29045–29050. doi: 10.1074/jbc.M102456200. [DOI] [PubMed] [Google Scholar]
- 33.Kim TW, Pettingell WH, Jung YK, Kovacs DM, Tanzi RE. Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protease. Science. 1997;277:373–376. doi: 10.1126/science.277.5324.373. [DOI] [PubMed] [Google Scholar]
- 34.Lu DC, Soriano S, Bredesen DE, Koo EH. Caspase cleavage of the amyloid precursor protein modulates amyloid beta-protein toxicity. J Neurochem. 2003;87:733–741. doi: 10.1046/j.1471-4159.2003.02059.x. [DOI] [PubMed] [Google Scholar]
- 35.Heredia L, Lin R, Vigo FS, Kedikian G, Busciglio J, Lorenzo A. Deposition of amyloid fibrils promotes cell-surface accumulation of amyloid beta precursor protein. Neurobiol Dis. 2004;16:617–629. doi: 10.1016/j.nbd.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 36.Melchor JP, Van Nostrand WE. Fibrillar amyloid beta-protein mediates the pathologic accumulation of its secreted precursor in human cerebrovascular smooth muscle cells. J Biol Chem. 2000;275:9782–9791. doi: 10.1074/jbc.275.13.9782. [DOI] [PubMed] [Google Scholar]
- 37.Wang YC, Ha Y. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell. 2004;15:343–353. doi: 10.1016/j.molcel.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 38.Dumanchin-Njock C, da Costa CA, Mercken L, Pradier L, Checler F. The caspase-derived C-terminal fragment of beta APP induces caspase-independent toxicity and triggers selective increase of A beta 42 in mammalian cells. J Neurochem. 2001;78:1153–1161. doi: 10.1046/j.1471-4159.2001.00513.x. [DOI] [PubMed] [Google Scholar]
- 39.Kinoshita A, Whelan CM, Berezovska O, Hyman BT. The gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein induces apoptosis via Tip60 in H4 cells. J Biol Chem. 2002;277:28530–28536. doi: 10.1074/jbc.M203372200. [DOI] [PubMed] [Google Scholar]
- 40.Kim HS, Kim EM, Lee JP, Park CH, Kim S, Seo JH, Chang KA, Yu E, Jeong SJ, Chong YH, Suh YH. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3 beta expression. FASEB J. 2003;17:1951–1953. doi: 10.1096/fj.03-0106fje. Epub 2003 Aug 15. [DOI] [PubMed] [Google Scholar]
- 41.Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C. Presenilin dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001;2:835–841. doi: 10.1093/embo-reports/kve180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su JH, Kesslak JP, Head E, Cotman CW. Caspase-cleaved amyloid precursor protein and activated caspase-3 are co-localized in the granules of granulovacuolar degeneration in Alzheimer’s disease and Down’s syndrome brain. Acta Neuropathol. 2002;104:1–6. doi: 10.1007/s00401-002-0548-2. [DOI] [PubMed] [Google Scholar]
- 43.Levites Y, Das P, Price RW, Rochette MJ, Kostura LA, McGowan EM, Murphy MP, Golde TE. Anti-A beta(42)- and anti-A beta(40)-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J Clin Invest. 2006;116:193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]