

Abstract
Myasthenia gravis (MG) is an autoimmune disease mediated by antibodies to nicotinic acetylcholine receptor (AChR) interfering with the neuromuscular transmission. Experimental autoimmune MG serves as an excellent animal model to study possible therapeutic modalities for MG. This review will focus on the different ways to turn off the autoimmune response to AChR, which results in suppression of myasthenia. This paper will describe the use of fragments or peptides derived from the AChR, antigen-presenting cells and anti-T cell receptor antibodies, and will discuss the underlying mechanisms of action. Finally, the authors propose new promising therapeutic prospects, including treatment based on the modulation of regulatory T cells, which have recently been found to be functionally defective in MG patients.
Keywords: acetylcholine receptor, autoimmune, dendritic cells, myasthenia gravis, regulatory cells, T cells, tolerisation, vaccination
1. Introduction
Originally, the definition of vaccine was restricted to vaccinia preparations used for immunisation against smallpox. Over time the term has evolved to include all preparations used to generate protective immunity to microbial pathogens. More recently, the definition of vaccine has been further expanded to include the administration of antigenic material in order to tolerise or turn off antigen-specific immune responses to prevent or treat immune-mediated diseases.
Autoimmune diseases affect nearly 5% of adults in North America and Europe. These chronic relapsing disorders affect multiple organ systems and are characterised by substantial morbidity and mortality, and by their high social and economic costs. Vaccines that could prevent or treat these conditions would constitute an important addition to existing therapies. Moreover, the current treatment of autoimmune diseases usually involves general immunosuppression that may have severe side effects. The ‘holy grail’ of research in the field of autoimmune diseases is, therefore, the development of immunotherapies, namely ‘vaccines’ that would suppress the autoimmune response without affecting the entire immune system.
Myasthenia gravis (MG) is a neuromuscular disease caused in most patients by an autoimmune response against the nicotinic acetylcholine receptor (AChR) at the neuromuscular junction [1,2]. The autoantigen is clearly defined (Figure 1) and the anti-AChR antibodies induce antigenic modulation leading to accelerated degradation of the receptor at the neuromuscular junctions or blockade of the binding sites of AChR [3,4]. They also contribute to complement-dependent loss of the receptor at the neuromuscular junctions [5], leading to reduced AChR expression and characteristic symptoms of MG. However, autoreactive T cells contribute to the autoimmune response by recognising AChR epitopes in the context of major histocompatibility (MHC) class II molecules [6-8] and by playing a critical role in the synthesis of anti-AChR antibodies [9,10]. AChR-activated T cell lines inoculated into naive syngeneic mice are capable of inducing autoimmune manifestations in mice [11], further strengthening the important role of T cells in the anti-AChR autoimmune response.
Figure 1.
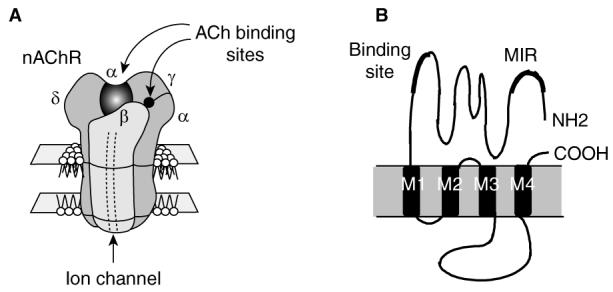
Structure of the fetal AChR, the main autoantigen in myasthenia gravis.A. AChR is a transmembrane glycoprotein of ∼300 kDa. Its stoichiometry is α2βγδ, and in the adult the γ-subunit is replaced by the ε-subunit [99]. B. Topology of the α-AChR subunit. This subunit contains residues involved in the ACh and the α-bungarotoxin binding sites, as well as the MIR recognised by most autoantibodies. In humans, there are 2 isoforms of the α-subunit produced by differential splicing of the exon P3A located between amino acids 58 and 59 [100]. Four regions of each subunit span the membrane. The M2 region is believed to be an α helix and lines the internal pore of the receptor. The M1, M3 and M4 regions contain hydrophobic sequences, but it is not known whether they traverse the membrane as α helices. The α-subunit includes two disulfide loops, at positions 128142 and 192193.
ACh: Acetylcholine; AChR: ACh receptor; MIR: Main immunogenic region.
The thymus plays a major role in human disease. Indeed, its involvement is strongly suggested by the beneficial effect of thymectomy, which is a common treatment for many patients at present [12]. However, thymectomy is not effective in all patients and its prognosis is best if it is performed early in the course of the disease [13]. Functional and morphological abnormalities of the thymus occur very frequently in patients as ∼ 50% present thymus hyperplasia with development of lymphoid follicles and 10 – 15% have an epithelial tumour of the thymus. Thymic hyperplasia is particularly common in young women and is often associated with a high level of anti-AChR antibodies [14] and with the human leukocyte antigen (HLA)-DR3 haplotype [15]. The anti-AChR antibody titre decreases after thymectomy in association with clinical improvement [16], indicating that the thymus plays a key role in anti-AChR antibody production. The main actors of the anti-AChR response are found in the hyperplastic thymus. A bias of intrathymic selection in favour of Vβ5.1 cells was observed [17]. Several activation features of B and T cells were demonstrated in the thymus from MG patients [18-21]. In addition, the AChR is present in the thymus in both epithelial and myoid cells [22,23], and its expression is regulated by pro-inflammatory cytokines [24,25]. The coincidence of these components in a genetically susceptible individual could explain the onset of the disease, although the triggering factor is still unknown (Figure 2).
Figure 2.
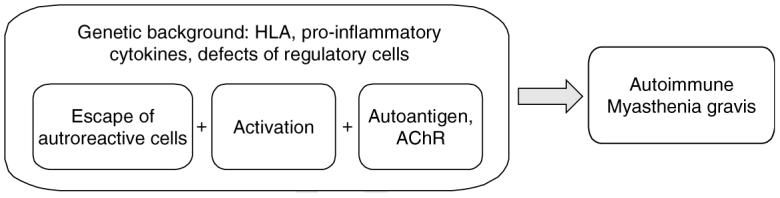
Pathogenesis of MG. The coincidence of several events could explain the onset of the anti-AChR autoimmune response that leads to MG disease: 1) the escape of autoreactive cells is evidenced by the bias of intrathymic selection [17]; 2) the activation status of the B and T cells were clearly shown [20,21], although the triggering event is not known. It could be an external agent (bacterial or viral antigens), an imbalance in production of hormones (such as in pregnancy or delivery) or a dysregulated production of neuropeptides (such as during emotional stress). This factor could be different for each patient; 3) The autoantigen (AChR) is present in the thymus in both normal controls and MG patients, but the inflammatory cytokines can upregulate its expression [24,25]. As indicated above, many arguments suggest that these events could occur in the thymus of the patients, but extrathymic events could also lead to an anti-AChR response, as shown in animal models of MG that do not present any thymic abnormalities.
AChR: Acetylcholine receptor; HLA: Human leukocyte antigen; MG: Myasthenia gravis.
Spontaneous myasthenia in animals was observed in dogs [26], an animal that is not easy to manipulate. Therefore, most of the research in animals has been performed in rodents immunised with AChR from Torpedo electric organ, a rich source of the receptor, in complete Freund's adjuvant [27]. Immunised animals show clinical signs of weakness similar to those described in humans except for the involvement of the thymus [28]. A variety of approaches have been attempted in experimental autoimmune MG (EAMG) to turn off antigen-specific immune responses and to affect the course of the disease (Figure 3). These include the use of denatured or native AChR, peptides or recombinant fragments derived from the AChR sequence, antibodies to the T cell receptor (TCR) or idiotypes, as well as manipulated dendritic cells (DCs).
Figure 3.
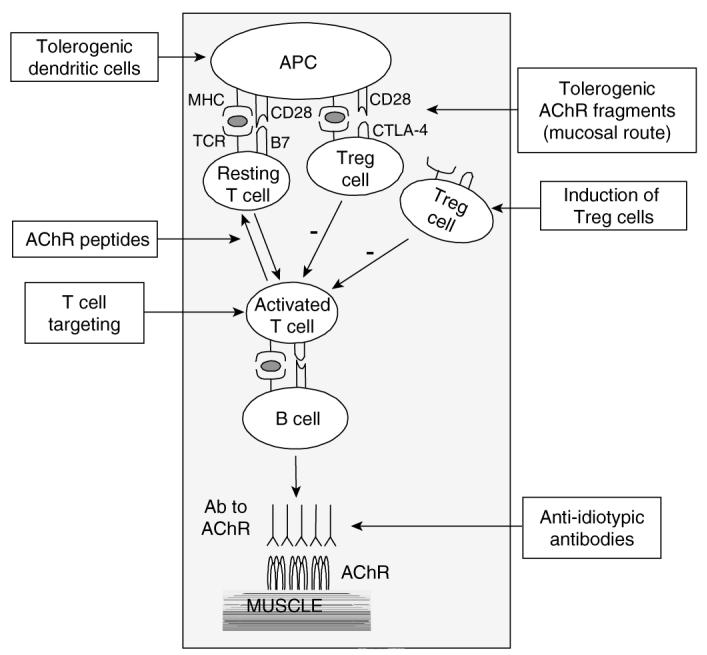
Vaccination for myasthenia gravis. The different possible ways of blocking the cell interactions to prevent anti-AChR antibody production are shown. Many attempts to inhibit the anti-AChR autoimmune response have been successful in EAMG models and some of them have been shown to be mediated through an increase of Treg activity. The direct induction or transfer of Treg cells has not yet been tested and is indicated in the figure as a possible new approach.
AChR: Acetylcholine receptor; APC: Antigen-presenting cell; EAMG: Experimental autoimmune myasthenia gravis; Treg: T regulatory.
This review highlights the status of recent efforts to develop tolerogenic vaccines for MG and identifies promising areas for future research.
2. Vaccination using AChR-derived sequences
EAMG serves as an excellent animal model to study possible therapeutic modalities to treat myasthenia in humans. The rat model, due to its chronic nature, is considered the most similar to the human form of the disease, although it also has its limitations because in rat myasthenia there is no involvement of the thymus [28]. Different derivatives of the autoantigen – AChR – have been tested as possible therapeutic agents in EAMG (Table 1). These have mainly been assayed for their ability to prevent the subsequent induction of EAMG. However, in recent years there have been several reports by the authors and others on the ability of AChR derivatives to suppress an already ongoing disease in animals. There are also many reports on the ability of AChR derivatives to affect the immune response to AChR either in vitro or in vivo. However, this review focuses mostly on procedures affecting the course of disease in experimental animal models of MG.
Table 1.
Different methods of vaccination used in experimental models of myasthenia gravis: putative mechanisms of action.
| Vaccines | Route | Species | Mechanism of action | Effects observed | References |
|---|---|---|---|---|---|
| Native AChR | Mucosal | Rats | Prevention | [32,33,35,36] | |
| Denatured AChR | Intradermal | Rabbits | Ongoing | [29] | |
| AChR recombinant fragments |
Mucosal (nasal and oral) |
Rats | Shift Th1 to Th2/Th3 Downregulation of costimulatory molecules |
Ongoing | [38-40] |
| AChR peptides | Oral | Rabbits Mice |
Anergy | Prevention Prevention and ongoing |
[43] [45,49,50] |
| AChR peptides | Intraperitoneal | Mice | Anergy, apoptosis Downregulation of IL-2, IFN-γ, IL-10 |
Prevention and ongoing |
[46,51] |
| Altered peptides | Oral | Mice | CD4+CD25+ Upregulation of IL-10, TGF |
Ongoing | [55] |
| Dendritic cells treated with IL-10 |
Intraperitoneal | Rat | Downregulation of costimulatory molecules |
Ongoing | [69] |
| Dendritic cells pulsed with AChR |
Subcutaneous | Rat | Reduced expression of BAFF |
Prevention | [67] |
| Anti-Vβ5.1 antibodies | Intraperitoneal | SCID mice | Shift Th1 to Th2 Deletion of Vβ5.1+ cells |
Biological parameters |
[70] |
AChR: Acetylcholine receptor; BAFF: B cell-activating factor; IL: Interleukin; IFN: Interferon; SCID: Severe combined immunodeficiency; TGF: Transforming growth factor; Th: T helper.
2.1 Native and denatured AChR
The authors' first successful attempt to modulate EAMG through an antigen-specific immunotherapy of EAMG was performed in 1978 using intradermal injection of a chemically modified Torpedo AChR [29]. This denatured AChR derivative (RCM-AChR) was shown not only to prevent the induction of EAMG in rabbits, but also to immunosuppress an ongoing disease.
The entire Torpedo AChR has also been used in its native form to modulate EAMG, but, as it is immunogenic when injected into animals, it has been administered via mucosal surfaces – a route of administration that is known to induce systemic regulation to the mucosally introduced antigen and bystander immunosuppression [30,31]. Most of these studies focused on attempts to prevent EAMG rather than treat an ongoing disease. It has been shown that mucosal (oral or nasal) administration of Torpedo AChR before immunisation with AChR prevented the clinical manifestation of the disease and suppressed cellular and humoral responses to the AChR [32-34]. However, when feeding with Torpedo AChR was performed during the acute phase of EAMG it led to the elicitation of antibodies to Torpedo AChR and to an increase in autoantibody titres to self-muscle AChR, although an inhibition of clinical manifestations could still be observed [35,36]. The priming effect on autoantibody levels induced by feeding with the xenogeneic and highly immunogenic Torpedo AChR and its limited availability hamper its application for therapeutic purposes, suggesting that a syngeneic, less immunogenic and more easily available tolerogen is required for immunotherapy of myasthenia in humans.
2.2 Recombinant allogeneic or syngeneic fragments
The cloning of the mammalian AChR in the early 1980s and the advances in genetic engineering have led to the mapping of regions within the AChR molecule that play a key role in the humoral and cellular autoimmune response in myasthenia. It was found that the extracellular domain of the receptor α-subunit is the main target of the autoimmune response. Moreover, a large portion of the antibodies to AChR is directed to a specific sequence within this domain (residues 67 – 76), which has accordingly been termed the main immunogenic region (MIR) [37] (Figure 1). These findings have paved the way for the use of fragments and peptides from selected regions of the AChR rather than the whole molecule for suppression of EAMG.
The authors' group have employed a recombinant fragment corresponding to the extracellular human AChR α-subunit (Hα1-205) to induce mucosal tolerance in rat EAMG. These fragments were shown to protect rats against EAMG when given prior to disease induction and, more importantly, to suppress an ongoing disease when administered either orally or nasally at the acute and even during the chronic phase of EAMG [38,39]. Disease suppression was monitored by clinical score and inhibition of weight loss, and was accompanied by decreased anti-AChR titres and reduced lymphocyte proliferation in response to the AChR fragment. Hα1-205 meets all the above-mentioned requirements for a tolerogen: it is of mammalian origin, available in large amounts, safe for use, as it does not induce any humoral anti-AChR response or signs of EAMG even when administered orally for a long time (3 months), and is conveniently administered by feeding. The mechanism of action of mucosal tolerance induction by these AChR recombinant fragments seems to be active suppression involving a shift from a T helper (Th)1 to a Th2/Th3 regulation of the anti-AChR response. This was demonstrated by a shift in the cytokine and anti-AChR IgG isotype profiles, by decreased levels of expression of costimulatory factors and by the ability of splenocytes from treated rats to protect recipients against EAMG. The authors have recently shown that a syngeneic fragment corresponding to the extracellular domain of the rat AChR α-subunit (Rα1-205) is as effective in rats as the allogeneic human fragment [40]. Nasal administration was also effective in suppressing ongoing EAMG by Hα1-205 and requires much smaller doses of tolerogen [41,42].
2.3 Peptides or altered peptides
Another antigen-specific approach for the suppression of EAMG by derivatives of AChR is based on the use of synthetic peptides. When considering the preparation of a future antigen-specific drug for therapy of human MG patients, peptides would have a clear advantage over recombinant fragments as they are not prepared in bacterial or mammalian cells and, therefore, their preparation is easier and more reproducible. The selection of peptides for immunomodulation is mostly based on the identification of highly immunogenic or functionally important regions within the AChR molecule and is mainly based on immunodominant T cell epitopes. Indeed, the authors have shown in the past that preimmunisation with peptides corresponding to selected extracellular and intra-cellular region within Torpedo and human AChR α and δ subunits protected rabbits against subsequent induction of EAMG [43,44]. This protection was correlated with delayed breakage of tolerance to all four receptor subunits. Atassi et al. showed that injection of mice with a peptide corresponding to residues 125 – 148 of Torpedo AChR α-subunit conjugated to mPEG had a protective effect against subsequent induction of EAMG when assessed by electrophysiological criteria [45].
The identification of immunodominant B and T cell epitopes on the AChR molecule has raised questions concerning their potential use as substitutes for the native antigen in tolerisation studies. EAMG has been prevented in B6 mice by the subcutaneous administration of a peptide corresponding to the Torpedo AChR T cell epitope in mice, residues 146 – 162 of the α-subunit [46-48], and by nasal delivery of a pool of Torpedo AChR α-subunit peptides containing this epitope [49]. Baggi et al. reported that oral administration of the T cell epitope α146-162 of the Torpedo AChR α-subunit suppressed T cell responses to AChR and ameliorated EAMG in C57Bl/6 (B6) mice [50]. Protection from EAMG was associated with reduced serum antibodies to self-mouse AChR and reduced AChR loss in muscle. The effect of Tα146-162 feeding was mediated by reduced production of interferon (IFN)-γ, interleukin (IL)-2 and IL-10 by AChR-reactive cells, suggesting T cell anergy. Transforming growth factor (TGF)-β-secreting Th3 cells did not seem to be involved in tolerance induction. Interestingly, high-dose systemic administration of the α146-162 peptide seems to represent another mechanism of action involving Fas-/Fas ligand-mediated apoptosis of CD4 cells [51].
The autoimmune response in myasthenia is polyclonal and peptides can only present one or two dominant epitopes, whereas fragments that encompass several B and/or T cell epitopes have a better chance of affecting the course of the polyclonal autoimmune response of the disease. The authors tried to overcome this problem by using long (50 – 60 amino acids), partially overlapping peptides. A mixture of four such peptides covering the entire extracellular portion of the mammalian AChR α-subunit was given to rats either nasally or orally prior to induction of EAMG. Nasal administration of the peptide mixture after disease induction (at the acute phase) had no significant effect on the course of EAMG [52]. Oral administration of 0.5 mg peptide mixture had no significant protective effect and 1.5 mg peptide mixture even led to exacerbation of clinical symptoms in treated rats. These observations seem to indicate that peptides cannot replace recombinant fragments in the suppression of ongoing EAMG via mucosal tolerance. As the authors have previously demonstrated the importance of tolerogen conformation for mucosal tolerance induction [53], the failure of peptides to suppress ongoing EAMG could stem from their inability to meet conformation requirements, even when they are relatively long.
Microbial peptides mimicking T cell epitopes were also shown to have suppressive effects on EAMG [54]. Pretreatment of rats by a peptide, derived from Haemophilus influenzae, selected from protein databases due to its partial homology (50%) to an identified T cell epitope of the human AChR α-subunit (residues 170 – 182) attenuated the induction and progression of EAMG. This may suggest that a non-pathogenic microbial mimicry peptide can serve as an immuno-modulator of the autoimmune attack on host antigens and can thereby affect the progression of antibody-mediated autoimmune diseases such as MG.
In addition to peptides that correspond to selected sequences of the autoantigen, there are also reports on the use of altered peptides bearing one or more mutations compared with the original immunodominant T cell AChR-specific epitopes in MG and/or EAMG. Two such peptides, representing sequences of the human AChR α-subunit, p195-212 and p259-271, were found to be immunodominant T cell epitopes in MG patients and in two strains of mice. An altered peptide ligand, composed of the tandemly arranged two analogues of these peptides each bearing single amino acid substitutions at important TCR contact residues, were shown to suppress ongoing EAMG in mice. The active suppression was mediated by CD4+CD25+ immunoregula-tory cells and was associated with the downregulation of Th1-type cytokines and the upregulation of the secretion of IL-10 and the immunosuppressive cytokine TGF-β [55].
2.4 Mechanisms of action
The approach of using self-antigen or autoantigen derivatives to suppress autoimmune diseases is not based on direct neutralisation of antibodies or cells reacting with the autoantigen; rather, it is rather based on the assumption that the administered antigen or its derivatives induce tolerance towards the self-autoantigen via direct or bystander suppression, anergy or apoptosis (Table 1). This has indeed been demonstrated for EAMG by shifts in the Th cell subpopulations regulating the AChR-specific response, as monitored by the levels of Th1-, Th2- and Th3-type cytokines and by the isotype profile of the anti-AChR humoral response. Such a shift depends on the nature of the tolerogen (i.e., its conformation, degree of denaturation, size) or the route of administration (i.e., mucosal exposure rather than injection). Indeed, the authors have shown that oral administration of a fragment corresponding to the extracellular domain of the human AChR α-subunit acts via active suppression mediated by a shift from Th1 to Th2/Th3 regulation of the anti-AChR response [38,39]. Interestingly, a syngeneic rat fragment, Rα1-205, administered orally to rats induced mucosal tolerance by a somewhat different mechanism involving a shift from Th1 to Th2 regulation, as evidenced by downregulated mRNA expression levels of IFN-γ and tumour necrosis factor-α, upregulated IL-10, changes in anti-AChR IgG isotypes and diminished Th1 signalling via CD28/CTLA-4:B7. However, unlike the xenogeneic fragment, the syngeneic Rα1-205 did not induce elevation in TGF-β and elicitation of autoregulatory cells [40].
Although IL-10 upregulation could be associated with clinical improvement in the experimental models, there is evidence that upregulation of IL-10 could be detrimental in MG. Indeed, IL-10 failed to abrogate EAMG at low doses (0.1 or 1 μg/day), and at the dose of 3 μg/day caused earlier onset and aggravated clinical signs of EAMG when compared with EAMG rats injected with PBS only [56]. In another study, IL-10 knockout mice had a lower incidence and severity of EAMG, with less muscle AChR loss [57]. Conversely, IL-10 transgenic mice developed EAMG and had higher levels of anti-AChR serum, compared with wild-type mice, after low-dose AChR immunisation [58]. These studies suggest that EAMG development is not strictly dependent on Th1 cell activity and implicates a critical precaution in planning immunotherapy for MG.
The disease-modulating effect of T cell epitopes such as Tα146-162 was seen with relatively high doses, and its mechanism of action appears to be the induction of anergy or deletion rather than active suppression, as no active cellular regulation or immune deviation was found in orally treated animals [50]. Thus, the suppression of EAMG by a T cell epitope given orally seems to be different from the mechanism underlying the suppression of EAMG by fragment Hα1-205, which involves active suppression and elicitation of regulatory cells (Table 1).
3. Vaccination using cells or molecules involved in the immune response
3.1 Anti-idiotypic antibodies
Autoreactive T cells and antibodies are found at low levels in normal individuals and are thought to be kept at bay by regulatory T cells and a network of idiotypic and anti-idiotype-bearing antigen receptors on lymphocytes, as well as idiotypic and anti-idiotypic antibodies. Disruption of this network by genetic, environmental and unknown factors is thought to result in autoimmune diseases. An obvious, ideal and specific therapy for such disorders would be to harness this regulatory network to re-establish immunological homeostasis. In practice, however, this is not an easy task, as most autoimmune diseases involve polyclonal responses to self-antigen as well as epitope spreading [59,60].
The authors have shown that ‘vaccination’ of mice with a certain idiotype prior to induction of EAMG by Torpedo AChR led to suppression of this particular idiotype in the treated mice, and their overall anti-AChR titres were reduced as compared with control mice [61]. However, as active immunisation would not be considered for the treatment of MG patients, the effect of passively transferred rabbit anti-idiotypes on EAMG was further analysed in an animal model. Passive transfer of mAb 5.5, which is directed to the AChR binding site, induces EAMG in chicken hatchings. Rabbit anti-idiotypic antibodies to mAb 5.5, which is directed to the AChR binding site, were shown to prevent EAMG passively transferred by mAb 5.5 in chickens and to reverse this disease [62].
Another model has been obtained by inducing anti-idiotypic antibodies by immunisation with a peptide (termed RhCA 67-16) encoded by RNA complementary to the Torpedo AChR MIR. This procedure prevented the development of EAMG in Lewis rats challenged with Torpedo AChR [63]. A monoclonal antibody (termed TCM 240) against the RhCA 67-16 peptide was found to recognise three different idiotypic antibodies (mAb 6, mAb 35 and mAb 198), which were previously reported to recognise the MIR and to cause EAMG [64]. The TCM 240 antibody was found to inhibit EAMG induced passively by mAb 35 or induced actively by immunisation with native Torpedo AChR [65]. Such findings suggest that the induction or use of anti-idiotypic antibodies could have therapeutic effects in MG, although Verschuuren et al. found that neonatal administration of polyclonal and monoclonal anti-idiotypic antibodies to AChR-specific antibodies did not significantly affect the course of EAMG [66].
3.2 Manipulated dendritic cells
DCs are usually regarded as antigen-presenting cells involved in T cell activation, but DCs also directly and indirectly affect B cell activation, antibody synthesis and isotype switching. Link and co-workers have used in vitro manipulated DCs to investigate their potential to induce tolerance and/or to influence the course of EAMG in rats.
DCs pulsed in vitro with AChR can mediate peripheral tolerance against rat EAMG, but do not affect ongoing EAMG [67]. This tolerance was associated with reduced expression of B cell-activating factor (BAFF), reduced numbers of B cells among splenic mononuclear cells (MNC) and a decrease in anti-AChR IgG antibody-secreting cells compared with rats injected with medium or unpulsed DCs. Similar results were observed with DCs exposed to TGF-β1 [68].
Most importantly, DCs from EAMG rats, exposed in vitro to IL-10 and then injected intraperitoneally, inhibited ongoing rat EAMG [69]. Rats that received such IL-10-modified DCs had lower clinical scores, reduced body weight loss, lower numbers of anti-AChR IgG antibody-secreting cells and lower affinity of anti-AChR antibodies, accompanied by lower expression of CD80 and CD86, and decreased lymphocyte proliferation among lymph node MNC as compared with control EAMG rats. Lower levels of IL-10 and IFN-γ were also found in the supernatants of AChR-stimulated lymph node MNC cultures from the treated rats. These results demonstrate that IL-10-modified DCs induced hyporesponsiveness by downregulating costimulatory molecules and reduced the production of anti-AChR antibodies possibly by inhibiting IL-10 production. These encouraging results with autologous DCs in suppressing ongoing EAMG should be tested in clinical trials for human MG.
3.3 Anti-TCR antibodies
Most early-onset MG (EOMG) patients are females, displaying the HLA-DR3 haplotype and presenting hyperplastic thymuses [14]. The authors demonstrated a selection bias of CD4+ T cells expressing the Vβ5.1 T cell receptor gene in the thymus of HLA-DR3 patients with MG [17]. Severe combined immunodeficiency (SCID) mice engrafted with MG thymic lymphocytes present several signs of MG pathogenicity (e.g., AChR loss and complement deposits at the muscle end plates of the chimeric mice) [70]. The treatment of these mice with anti-Vβ5.1 antibody prevented these signs of pathogenicity. Thymic cells depleted of Vβ5.1-positive cells in vitro before cell transfer were non-pathogenic, indicating that Vβ5.1-positive cells are involved in the production of pathogenic autoantibodies. AChR loss in SCID mice was prevented by targeting Vβ5.1 expressed on thymic cells from HLA-DR3 patients only, demonstrating specificity for HLA-DR3–peptide complexes. The action of the anti-Vβ5.1 antibody involved both the in vivo depletion of Vβ5.1-expressing cells and an increase in the IFN-γ:IL-4 ratio, pointing to an immune deviation-based mechanism. These results suggest that T cells expressing Vβ5.1 are involved in controlling pathogenic autoantibodies in MG patients presenting the HLA-DR3 haplotype.
In the next step, the authors searched for anti-TCR antibodies in the sera of MG patients [71]. Indeed, serum anti-TCR antibodies are involved in immune regulation directed against pathogenic T cells in homeostatic and pathological conditions [72]. It was found that EOMG patients had elevated IgG anti-Vβ5.1 antibodies. This increase was restricted largely to patients with HLA-DR3 and with less severe disease, and predicted clinical improvement in follow-up studies. Patient's sera containing anti-TCR antibodies bound specifically the native TCR on intact Vβ5.1-expressing cells and specifically inhibited the proliferation and IFN-γ production of purified Vβ5.1-expressing cells to alloantigens. The proliferation of a Vβ5.1-expressing T cell clone to an AChR peptide was also inhibited by patient's sera containing anti-Vβ5.1 antibodies, indicating a regulatory function for these antibodies. This evidence of spontaneously active anti-Vβ5.1 antibodies in EOMG patients suggests dynamic protective immune regulation directed against the excess of pathogenic Vβ5.1-expressing T cells.
Based on these data, one could consider the development of a peptide vaccination in HLA-DR3 MG patients, using Vβ5.1-derived peptides, which would boost the humoral response against autoaggressive cells. This approach is more practical and has longer term effects than those of mAbs to Vβ5.1 that would have to be humanised. Moreover, TCR peptide vaccines would be advantageously used in combination with thymectomy. Indeed, thymectomy should first eliminate the source of pathogenic autoreactive T cells, and the combined vaccination using Vβ5.1-derived peptides should rapidly eliminate the pathogenic T cells in the periphery resulting in accelerated regression of disease signs.
In the rat model of myasthenia, Blalock and co-workers [73] obtained reversal of EAMG by using a mAb (CTCR8) against AChR-specific T cells, which specifically recognised Vβ15-containing TCR on T cells specific for residues 100 – 116 of the AChR α-subunit. This antibody was found to label the cell surface of AChR100-116-specific T cell lines and clones, immunoprecipitate the TCR from such cells and block their proliferative responses to AChRα100-116. Interestingly, when AChR100-116-primed primary T cells were stimulated with peptide and treated with CTCR8, there was inhibition of IFN-γ and stimulation of IL-10 production. The change in the Th1/Th2 cytokine profile was accompanied by a reduction in AChR-specific IgG2a and IgM, with no effect on IgG1. Based on these results, CTCR8 was tested for prophylactic and therapeutic effects in EAMG induced by immunisation with purified native Torpedo AChR and was found to inhibit and reverse the disease. These findings suggest that mAbs to selected TCR sequences could in the future be used for the treatment of MG and other autoimmune diseases.
4. Vaccination using regulatory components
In the last few years, there has been a rebirth of suppressor cells as key players in immune regulation. These T regulatory (Treg) cells have been shown to possess an enormous potential for suppressing pathological immune responses in autoimmune diseases, transplantation and graft-versus-host disease [74-76]. This minor CD4+ T cell subset maintains the homeostatic equilibrium of immunity and tolerance, and expresses CD25, glucocorticoid-induced tumor necrosis factor receptor, cytotoxic T lymphocyte antigen (CTLA)-4 and CD62L [77,78]. A specific transcription factor, FoxP3 (Forkhead box P3), has been shown to control Treg cell development and the expression of their suppressive phenotype [79].
4.1 Natural regulatory CD4+CD25+ T cells
The proliferation of CD4+CD25- T cells in the presence of an antigen, mitogen or anti-TCR antibodies is suppressed when these cells are cocultured with Treg CD4+CD25+ cells [78]. Recent data indicate a functional defect of these cells in several autoimmune diseases, such as multiple sclerosis [80], polyglandular autoimmune disease [81] and Type I diabetes [82]. Several studies analysed the frequencies of Treg CD4+CD25+ cells in human MG patients. No defects in the number of these cells were observed [83-85]. To identify possible functional defects in the Treg cells in MG, the authors analysed the ability of CD4+CD25+ cells from thymuses of MG patients to inhibit the proliferation of CD4+CD25− cells. A severe functional defect in their regulatory activity was found together with a decreased expression of Foxp3, which is essential for Treg cell function [85]. The phenotypic analysis of CD4+CD25+ thymocytes revealed an increased number of cells with strong Fas expression in MG patients, thought to be effector cells [21]. However, when CD4+CD25+ cells were purified according to the level of Fas, both Fashi and Faslo thymocytes from MG patients remained unable to suppress the proliferation of responding cells, whereas both subsets were functional in healthy controls. These results indicate that the impaired Treg cell function is not due to contamination by activated effector T cells, and demonstrate a severe functional impairment of thymic Treg cells in MG disease. Thus, a vaccine to stimulate the function of Treg cells should be further investigated as a potential therapeutic modality in MG.
Although such a therapy has not been yet tested in MG, therapeutic vaccination using CD4+CD25+ regulatory T cells has been proposed for autoimmune diseases [86]. Induction of Treg activity by anti-CD3 mAb therapy was demonstrated in non-obese diabetic mice and patients with Type I diabetes [87]. Therefore, therapy based on expansion of Treg could be promising for MG, but there is still a need to understand the causes of Treg dysfunction in MG in order to determine the more appropriate way for Treg manipulation.
4.2 Activation molecules
As described in Table 1, the mechanisms mediating vaccination in MG models involve a Th1 to Th2 shift or an increase of Treg responses. Similar outcomes could be obtained by interfering with costimulatory molecules. One report shows that interference with the B7 costimulatory signal using CTLA4Ig (a soluble CD28 receptor analogue) resulted in a profound decrease in IL-2 production and significantly decreased lymphoproliferative responses and antibody responses by primed lymph node cells from rats with EAMG when stimulated with AChR in vitro, indicating that specific antigen stimulation in the presence of CTLA4Ig can induce certain features typical of anergy [88]. The authors demonstrated the capacity of CD40L blockade to modulate EAMG. Indeed, anti-CD40L antibodies given to rats at the chronic stage of EAMG suppressed the clinical progression of the autoimmune process and led to a decrease in the AChR-specific humoral response and in delayed-type hypersensitivity. The cytokine profile of treated rats suggests that the underlying mechanism involves downregulation of AChR-specific Th1-regulated responses accompanied by a significant upregulation of CTLA-4 expression [89].
Therefore, similar mechanisms could be obtained by immunomodulation of the immune system with a specific antigen or irrespective of an antigen, namely by modulating cytokine networks [90] or costimulation signalling [88,89,91]. These data raise the possibility that the mechanism underlying these observations is not necessarily a specific blocking of the MHC–TCR interaction, but could also result from the decrease in the activation state of the autoimmune cells by a bystander immunosuppression [92] or by the development of a T cell response directed to activation molecules. Such a concept has recently been proposed by Cohen in a review [93]. In this review, Cohen argues that T cell vaccination activates Treg cells of two different kinds:
anti-idiotypic Treg cells, recognising specific effector clones by their unique TCR CDR3 peptides
anti-ergotypic Treg cells, recognising the state of activation of target effector T cells, irrespective of their TCR specificity
It is clear that the role of Treg cells in T cell vaccination is to reduce the activation state of activated cells, and as these activated cells in patients are at least partially specific for the autoantigens, Treg cells would first affect these cells. Therefore, one can speculate that Treg cells that are specific of activation-associated molecules play a major role in the regulation of the immune and autoimmune responses. In the case of human MG, many signs of cell activation and inflammation have been demonstrated (Table 2). Indeed, the difference between the thymus in MG patients and in healthy controls is not due to the antigen, but appears to be mostly due to the differences in the activation state of the thymic cells. However, the triggering molecules inducing the high level of activation are not known (Figure 2). Even the presence of autoreactive T cells specific for the AChR has been demonstrated in both cases. These observations indicate that an efficient vaccine should target the activation molecules.
Table 2.
Factors involved in the anti-AChR autoimmune response in the MG thymus, the effector site of human MG disease.
| Normal thymus | MG thymus (hyperplasia) | References | |
|---|---|---|---|
| Expression of AChR | + | + | [22] |
| Inflammatory activity | − | + | [25] |
| Expression of class II Ag | + | ++ | [25] |
| Activated B cells | − | + | [20] |
| AChR-specific T cells | +/− | + | [7] |
| Activated T cells | − | + | [18,21] |
| Regulatory cells | + | defective | [85] |
AChR: Acetylcholine receptor; MG: Myasthenia gravis.
5. Expert opinion and conclusion
This discussion highlights the efforts made in the field of MG towards the development of therapeutic vaccines. Many possible ways to modulate the anti-AChR autoimmune response and to suppress ongoing disease have been tested in experimental animal models of MG. Some of these approaches yielded promising results, which should be further carefully tested in clinical trials. For example, the approach using DCs manipulated in vitro with cytokines demonstrates the efficiency of autologous DCs to treat ongoing EAMG. Clinical trials using this type of approach should be considered, taking into account the current use of DCs in other human pathologies such as cancers [94-96].
It should be kept in mind that not every strategy that is effective in suppressing an ongoing disease in experimental animal models would necessarily be successful in human patients. For example, mucosal tolerance by autoantigen-derived fragments or peptides, which was successful in experimental autoimmune encephalomyelitis, turned out not to be effective in human MS patients [97,98]. In spite of this failure in MS, it is worthwhile to test the potential of mucosal tolerance with AChR fragments in MG.
Autoimmune diseases are characterised by the chronic nature of the autoimmune response. In the case of MG, we believe that the immune response to AChR is due to chronic activation that may be due to the functional defect in the Treg cell subset observed in MG patients [85]. If, indeed, MG patients are not able to downregulate the activation status of their T cells, a therapy that is targeted at activation molecules could be effective.
The question is: what would be the ideal treatment for MG? Is it necessarily solely an autoantigen-specific approach? We believe that the ideal approach should combine antigen-specific immunotherapy together with direct, antigen-nonspecific modulation. Indeed, our data in EAMG suggest that mucosal tolerance by an antigen-specific approach would be most efficient when used in combination with direct modulators of the immune response, such as agents that affect the cytokine network or the expression of costimulatory or activation molecules.
Acknowledgements
These studies were supported by grants from the National Institutes of Health (NS39869), the European Commission (QLG1-CT-2001-10918 and QLRT-2001-00225), the Muscular Dystrophy Association (MDA 3826) and by the Association Française Contre les Myopathies. SBA was the recipient of a Weston Professorship from the Weizmann Institute.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1••.AHARONOV A, ABRAMSKY O, TARRAB-HAZDAI R, FUCHS S. Humoral antibodies to acetylcholine receptor in patients with myasthenia gravis. Lancet. 1975;2:340–342. doi: 10.1016/s0140-6736(75)92779-8. First evidence of the presence of antibodies against AChR in MG patients. [DOI] [PubMed] [Google Scholar]
- 2.LINDSTROM JM, SEYBOLD ME, LENNON VA, WHITTINGHAM S, DUANE DD. Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology. 1976;26:1054–1059. doi: 10.1212/wnl.26.11.1054. [DOI] [PubMed] [Google Scholar]
- 3.DRACHMAN DB, ADAMS RN, JOSIFEK LF, SELF SG. Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N. Engl. J. Med. 1982;307:769–775. doi: 10.1056/NEJM198209233071301. [DOI] [PubMed] [Google Scholar]
- 4.TZARTOS SJ, SOPHIANOS D, ZIMMERMAN K, STARZINSKI-POWITZ A. Antigenic modulation of human myotube acetylcholine receptor by myasthenic sera. Serum titer determines receptor internalization rate. J. Immunol. 1986;136:3231–3238. [PubMed] [Google Scholar]
- 5.ENGEL AG, LAMBERT EH, HOWARD FM. Immune complexes (IgG and C3) at the motor end-plate in myasthenia gravis: ultrastructural and light microscopic localization and electrophysiologic correlations. Mayo Clin. Proc. 1977;52:267–280. [PubMed] [Google Scholar]
- 6•.HOHLFELD R, TOYKA KV, TZARTOS SJ, CARSON W, CONTI-TRONCONI BM. Human T-helper lymphocytes in myasthenia gravis recognize the nicotinic receptor alpha subunit. Proc. Natl. Acad. Sci. USA. 1987;84:5379–5383. doi: 10.1073/pnas.84.15.5379. Demonstration that CD4+ T cells from MG patients proliferate in the presence of the AChR α-subunit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MELMS A, SCHALKE BC, KIRCHNER T, MULLER-HERMELINK HK, ALBERT E, WEKERLE H. Thymus in myasthenia gravis. Isolation of T-lymphocyte lines specific for the nicotinic acetylcholine receptor from thymuses of myasthenic patients. J. Clin. Invest. 1988;81:902–908. doi: 10.1172/JCI113401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8•.SUN JB, HARCOURT G, WANG ZY, et al. T cell responses to human recombinant acetylcholine receptor-alpha subunit in myasthenia gravis and controls. Eur. J. Immunol. 1992;22:1553–1559. doi: 10.1002/eji.1830220631. Quantification by enzme-linked immunospot of the number of peripheral blood mononuclear cells reacting to the AChR α-subunit. [DOI] [PubMed] [Google Scholar]
- 9.ASTHANA D, FUJII Y, HUSTON GE, LINDSTROM J. Regulation of antibody production by helper T cell clones in experimental autoimmune myasthenia gravis is mediated by IL-4 and antigen-specific T cell factors. Clin. Immunol. Immunopathol. 1993;67:240–248. doi: 10.1006/clin.1993.1071. [DOI] [PubMed] [Google Scholar]
- 10.ROSENBERG JS, OSHIMA M, ATASSI MZ. B-cell activation in vitro by helper T cells specific to region alpha 146-162 of Torpedo californica nicotinic acetylcholine receptor. J. Immunol. 1996;157:3192–3199. [PubMed] [Google Scholar]
- 11.KIRSHNER SL, KATZ LEVY Y, WIRGUIN I, ARGOV Z, MOZES E. Fine specificity of T cell lines and clones that are capable of inducing autoimmune manifestations in mice. Cell. Immunol. 1994;157:11–28. doi: 10.1006/cimm.1994.1201. [DOI] [PubMed] [Google Scholar]
- 12.VENUTA F, RENDINA EA, DE GIACOMO T, et al. Thymectomy for myasthenia gravis: a 27-year experience. Eur. J. Cardiothorac. Surg. 1999;15:621–624. doi: 10.1016/s1010-7940(99)00052-4. discussion 624-625. [DOI] [PubMed] [Google Scholar]
- 13.BEGHI E, ANTOZZI C, BATOCCHI AP, et al. Prognosis of myasthenia gravis: a multicenter follow-up study of 844 patients. J. Neurol. Sci. 1991;106:213–220. doi: 10.1016/0022-510x(91)90260-e. [DOI] [PubMed] [Google Scholar]
- 14•.BERRIH S, MOREL E, GAUD C, RAIMOND F, LE BRIGAND H, BACH JF. Anti-AChR antibodies, thymic histology, and T cell subsets in myasthenia gravis. Neurology. 1984;34:66–71. doi: 10.1212/wnl.34.1.66. Description of a clear association between thymic pathology and the anti-AChR antibody titre, showing that the highest titres are found in patients with thymic hyperplasia. [DOI] [PubMed] [Google Scholar]
- 15.SPURKLAND A, GILHUS NE, RONNINGEN KS, AARLI JA, VARTDAL F. Myasthenia gravis patients with thymus hyperplasia and myasthenia gravis patients with thymoma display different HLA associations. Tissue Antigens. 1991;37:90–93. doi: 10.1111/j.1399-0039.1991.tb01851.x. [DOI] [PubMed] [Google Scholar]
- 16.KUKS JB, OOSTERHUIS HJ, LIMBURG PC, THE TH. Anti-acetylcholine receptor antibodies decrease after thymectomy in patients with myasthenia gravis. Clinical correlations. J. Autoimmun. 1991;4:197–211. doi: 10.1016/0896-8411(91)90018-8. [DOI] [PubMed] [Google Scholar]
- 17.TRUFFAULT F, COHEN-KAMINSKY S, KHALIL I, LEVASSEUR P, BERRIH-AKNIN S. Altered intrathymic T-cell repertoire in human myasthenia gravis. Ann. Neurol. 1997;41:731–741. doi: 10.1002/ana.410410609. [DOI] [PubMed] [Google Scholar]
- 18.COHEN-KAMINSKY S, LEPRINCE C, GALANAUD P, ICHARDS Y, BERRIH-AKNIN S. B- and T-cell activation in the thymus of patients with myasthenia gravis. Thymus. 1989;14:187–193. [PubMed] [Google Scholar]
- 19.COHEN-KAMINSKY S, LEVASSEUR P, BINET JP, BERRIH-AKNIN S. Evidence of enhanced recombinant interleukin-2 sensitivity in thymic lymphocytes from patients with myasthenia gravis: possible role in autoimmune pathogenesis. J. Neuroimmunol. 1989;24:75–85. doi: 10.1016/0165-5728(89)90101-x. [DOI] [PubMed] [Google Scholar]
- 20••.LEPRINCE C, COHEN-KAMINSKY S, BERRIH-AKNIN S, et al. Thymic B cells from myasthenia gravis patients are activated B cells. Phenotypic and functional analysis. J. Immunol. 1990;145:2115–2122. B cells purified from MG thymic hyperplasia are activated and produce antibodies against AChR. [PubMed] [Google Scholar]
- 21•.MOULIAN N, BIDAULT J, TRUFFAULT F, YAMAMOTO AM, LEVASSEUR P, BERRIH-AKNIN S. Thymocyte Fas expression is dysregulated in myasthenia gravis patients with anti-acetylcholine receptor antibody. Blood. 1997;89:3287–3295. Description of Fas as a marker of activated cells in the MG thymus and correlations with anti-AChR antibody titres. [PubMed] [Google Scholar]
- 22••.WAKKACH A, GUYON T, BRUAND C, TZARTOS S, COHEN-KAMINSKY S, BERRIH-AKNIN S. Expression of acetylcholine receptor genes in human thymic epithelial cells: implications for myasthenia gravis. J. Immunol. 1996;157:3752–3760. Expression of AChR in human thymic epithelial cells at both the mRNA level and the protein level (α-subunit). [PubMed] [Google Scholar]
- 23.WAKKACH A, POEA S, CHASTRE E, et al. Establishment of a human thymic myoid cell line. Phenotypic and functional characteristics. Am. J. Pathol. 1999;155:1229–1240. doi: 10.1016/S0002-9440(10)65225-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24•.ZHENG Y, WHEATLEY LM, LIU T, LEVINSON AI. Acetylcholine receptor alpha subunit mRNA expression in human thymus: augmented expression in myasthenia gravis and upregulation by interferon-gamma. Clin. Immunol. 1999;91:170–177. doi: 10.1006/clim.1999.4689. AChR expression is increased in MG thymuses. [DOI] [PubMed] [Google Scholar]
- 25.POËA-GUYON S, CHRISTADOSS P, LE PANSE R, et al. Effects of cytokines on acetylcholine receptor expression: implications for myasthenia gravis. J. Immunol. 2005;174(10):5941–5949. doi: 10.4049/jimmunol.174.10.5941. [DOI] [PubMed] [Google Scholar]
- 26.SHELTON GD. Acquired myasthenia gravis: what we have learned from experimental and spontaneous animal models. Vet. Immunol. Immunopathol. 1999;69:239–249. doi: 10.1016/s0165-2427(99)00058-6. [DOI] [PubMed] [Google Scholar]
- 27.LINK H, XIAO BG. Rat models as tool to develop new immunotherapies. Immunol. Rev. 2001;184:117–128. doi: 10.1034/j.1600-065x.2001.1840111.x. [DOI] [PubMed] [Google Scholar]
- 28•.MEINL E, KLINKERT WE, WEKERLE H. The thymus in myasthenia gravis. Changes typical for the human disease are absent in experimental autoimmune myasthenia gravis of the Lewis rat. Am. J. Pathol. 1991;139:995–1008. The thymus is not involved in the experimental model of myasthenia in rats. [PMC free article] [PubMed] [Google Scholar]
- 29•.BARTFELD D, FUCHS S. Specific immunosuppression of experimental autoimmune myasthenia gravis by denatured acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 1978;75:4006–4010. doi: 10.1073/pnas.75.8.4006. This is the first demonstration that EAMG is suppressed by denatured Torpedo AChR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.WEINER HL. Oral tolerance: immune mechanisms and treatment of autoimmune diseases. Immunol. Today. 1997;18:335–343. doi: 10.1016/s0167-5699(97)01053-0. [DOI] [PubMed] [Google Scholar]
- 31.TOUSSIROT EA. Oral tolerance in the treatment of rheumatoid arthritis. Curr. Drug Targets Inflamm. Allergy. 2002;1:45–52. doi: 10.2174/1568010023344850. [DOI] [PubMed] [Google Scholar]
- 32.WANG ZY, QIAO J, LINK H. Suppression of experimental autoimmune myasthenia gravis by oral administration of acetylcholine receptor. J. Neuroimmunol. 1993;44:209–214. doi: 10.1016/0165-5728(93)90045-z. [DOI] [PubMed] [Google Scholar]
- 33.OKUMURA S, MCINTOSH K, DRACHMAN DB. Oral administration of acetylcholine receptor: effects on experimental myasthenia gravis. Ann. Neurol. 1994;36:704–713. doi: 10.1002/ana.410360504. [DOI] [PubMed] [Google Scholar]
- 34.MA CG, ZHANG GX, XIAO BG, LINK J, OLSSON T, LINK H. Suppression of experimental autoimmune myasthenia gravis by nasal administration of acetylcholine receptor. J. Neuroimmunol. 1995;58:51–60. doi: 10.1016/0165-5728(94)00187-s. [DOI] [PubMed] [Google Scholar]
- 35.DRACHMAN DB, OKUMURA S, ADAMS RN, MCINTOSH KR. Oral tolerance in myasthenia gravis. Ann. NY Acad. Sci. 1996;778:258–272. doi: 10.1111/j.1749-6632.1996.tb21134.x. [DOI] [PubMed] [Google Scholar]
- 36.SHI FD, BAI XF, LI HL, HUANG YM, VAN DER MEIDE PH, LINK H. Nasal tolerance in experimental autoimmune myasthenia gravis (EAMG): induction of protective tolerance in primed animals. Clin. Exp. Immunol. 1998;111:506–512. doi: 10.1046/j.1365-2249.1998.00521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.TZARTOS SJ, LINDSTROM JM. Monoclonal antibodies used to probe acetylcholine receptor structure: localization of the main immunogenic region and detection of similarities between subunits. Proc. Natl. Acad. Sci. USA. 1980;77:755–759. doi: 10.1073/pnas.77.2.755. Identification and localisation of the MIR of the AChR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.BARCHAN D, ASHER O, TZARTOS SJ, FUCHS S, SOUROUJON MC. Modulation of the anti-acetylcholine receptor response and experimental autoimmune myasthenia gravis by recombinant fragments of the acetylcholine receptor. Eur. J. Immunol. 1998;28:616–624. doi: 10.1002/(SICI)1521-4141(199802)28:02<616::AID-IMMU616>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 39•.IM SH, BARCHAN D, FUCHS S, SOUROUJON MC. Suppression of ongoing experimental myasthenia by oral treatment with an acetylcholine receptor recombinant fragment [see comments] J. Clin. Invest. 1999;104:1723–1730. doi: 10.1172/JCI8121. Demonstration that ongoing EAMG in rats is suppressed by oral or nasal tolerance with a xenogeneic or syngeneic recombinant fragment corresponding to the extracellular domain of the AChR α-subunit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•.MAITI PK, FEFERMAN T, IM SH, SOUROUJON MC, FUCHS S. Immunosuppression of rat myasthenia gravis by oral administration of a syngeneic acetylcholine receptor fragment. J. Neuroimmunol. 2004;152:112–120. doi: 10.1016/j.jneuroim.2004.04.010. See [39]. [DOI] [PubMed] [Google Scholar]
- 41•.BARCHAN D, SOUROUJON MC, IM SH, ANTOZZI C, FUCHS S. Antigen-specific modulation of experimental myasthenia gravis: nasal tolerization with recombinant fragments of the human acetylcholine receptor alpha-subunit. Proc. Natl. Acad. Sci. USA. 1999;96:8086–8091. doi: 10.1073/pnas.96.14.8086. See [39]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.IM S, BARCHAN D, FUCHS S, SOUROUJON MC. Mechanism of nasal tolerance induced by a recombinant fragment of acetylcholine receptor for treatment of experimental myasthenia gravis. J. Neuroimmunol. 2000;111:161–168. doi: 10.1016/s0165-5728(00)00395-7. See [39]. [DOI] [PubMed] [Google Scholar]
- 43.SOUROUJON MC, CARMON S, FUCHS S. Modulation of anti-acetylcholine receptor antibody specificities and of experimental autoimmune myasthenia gravis by synthetic peptides. Immunol. Lett. 1992;34:19–25. doi: 10.1016/0165-2478(92)90022-g. [DOI] [PubMed] [Google Scholar]
- 44.SOUROUJON MC, CARMON S, FUCHS S. Regulation of experimental autoimmune myasthenia gravis by synthetic peptides of the acetylcholine receptor. Ann. NY Acad. Sci. 1993;681:332–334. doi: 10.1111/j.1749-6632.1993.tb22910.x. [DOI] [PubMed] [Google Scholar]
- 45.ATASSI MZ, RUAN KH, JINNAI K, OSHIMA M, ASHIZAWA T. Epitope-specific suppression of antibody response in experimental autoimmune myasthenia gravis by a monomethoxypolyethylene glycol conjugate of a myasthenogenic synthetic peptide. Proc. Natl. Acad. Sci. USA. 1992;89:5852–5856. doi: 10.1073/pnas.89.13.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.WU B, DENG C, GOLUSZKO E, CHRISTADOSS P. Tolerance to a dominant T cell epitope in the acetylcholine receptor molecule induces epitope spread and suppresses murine myasthenia gravis. J. Immunol. 1997;159:3016–3023. [PubMed] [Google Scholar]
- 47.KARACHUNSKI PI, OSTLIE NS, OKITA DK, GARMAN R, CONTI-FINE BM. Subcutaneous administration of T-epitope sequences of the acetylcholine receptor prevents experimental myasthenia gravis. J. Neuroimmunol. 1999;93:108–121. doi: 10.1016/s0165-5728(98)00208-2. [DOI] [PubMed] [Google Scholar]
- 48.SHENOY M, OSHIMA M, ATASSI MZ, CHRISTADOSS P. Suppression of experimental autoimmune myasthenia gravis by epitope-specific neonatal tolerance to synthetic region alpha 146-162 of acetylcholine receptor. Clin. Immunol. Immunopathol. 1993;66:230–238. doi: 10.1006/clin.1993.1030. [DOI] [PubMed] [Google Scholar]
- 49.KARACHUNSKI PI, OSTLIE NS, OKITA DK, CONTI-FINE BM. Prevention of experimental myasthenia gravis by nasal administration of synthetic acetylcholine receptor T epitope sequences. J. Clin. Invest. 1997;100:3027–3035. doi: 10.1172/JCI119857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.BAGGI F, ANDREETTA F, CASPANI E, et al. Oral administration of an immunodominant T-cell epitope downregulates Th1/Th2 cytokines and prevents experimental myasthenia gravis. J. Clin. Invest. 1999;104:1287–1295. doi: 10.1172/JCI7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DENG C, GOLUSZKO E, CHRISTADOSS P. Fas/fas ligand pathway, apoptosis, and clonal anergy involved in systemic acetylcholine receptor t cell epitope tolerance. J. Immunol. 2001;166:3458–3467. doi: 10.4049/jimmunol.166.5.3458. [DOI] [PubMed] [Google Scholar]
- 52.SOUROUJON MC, MAITI PK, FEFERMAN T, IM SH, RAVEH L, FUCHS S. Suppression of myasthenia gravis by antigen-specific mucosal tolerance and modulation of cytokines and costimulatory factors. Ann. NY Acad. Sci. 2003;998:533–536. doi: 10.1196/annals.1254.069. [DOI] [PubMed] [Google Scholar]
- 53.IM SH, BARCHAN D, SOUROUJON MC, FUCHS S. Role of tolerogen conformation in induction of oral tolerance in experimental autoimmune myasthenia gravis. J. Immunol. 2000;165:3599–3605. doi: 10.4049/jimmunol.165.7.3599. [DOI] [PubMed] [Google Scholar]
- 54.IM SH, BARCHAN D, FEFERMAN T, RAVEH L, SOUROUJON MC, FUCHS S. Protective molecular mimicry in experimental myasthenia gravis. J. Neuroimmunol. 2002;126:99–106. doi: 10.1016/s0165-5728(02)00069-3. [DOI] [PubMed] [Google Scholar]
- 55.SELA M, MOZES E. Therapeutic vaccines in autoimmunity. Proc. Natl. Acad. Sci. USA. 2004;101(Suppl 2):14586–14592. doi: 10.1073/pnas.0404826101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56•.ZHANG GX, XIAO BG, YU LY, VAN DER MEIDE PH, LINK H. Interleukin 10 aggravates experimental autoimmune myasthenia gravis through inducing Th2 and B cell responses to AChR. J. Neuroimmunol. 2001;113:10–18. doi: 10.1016/s0165-5728(00)00411-2. Evidence for the deleterious role of IL-10 in the induction of EAMG. [DOI] [PubMed] [Google Scholar]
- 57•.POUSSIN MA, GOLUSZKO E, HUGHES TK, DUCHICELLA SI, CHRISTADOSS P. Suppression of experimental autoimmune myasthenia gravis in IL-10 gene-disrupted mice is associated with reduced B cells and serum cytotoxicity on mouse cell line expressing AChR. J. Neuroimmunol. 2000;111:152–160. doi: 10.1016/s0165-5728(00)00385-4. See [56]. [DOI] [PubMed] [Google Scholar]
- 58•.OSTLIE NS, KARACHUNSKI PI, WANG W, MONFARDINI C, KRONENBERG M, CONTI-FINE BM. Transgenic expression of IL-10 in T cells facilitates development of experimental myasthenia gravis. J. Immunol. 2001;166:4853–4862. doi: 10.4049/jimmunol.166.8.4853. See [56]. [DOI] [PubMed] [Google Scholar]
- 59.VANDERLUGT CL, MILLER SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 60.FEFERMAN T, IM SH, FUCHS S, SOUROUJON MC. Breakage of tolerance to hidden cytoplasmic epitopes of the acetylcholine receptor in experimental autoimmune myasthenia gravis. J. Neuroimmunol. 2003;140:153–158. doi: 10.1016/s0165-5728(03)00209-1. [DOI] [PubMed] [Google Scholar]
- 61.SOUROUJON MC, BARCHAN D, FUCHS S. Analysis and modulation of the immune response of mice to acetylcholine receptor by anti-idiotypes. Immunol. Lett. 1985;9:331–336. doi: 10.1016/0165-2478(85)90058-6. [DOI] [PubMed] [Google Scholar]
- 62.SOUROUJON MC, PACHNER AR, FUCHS S. The treatment of passively transferred experimental myasthenia with anti-idiotypic antibodies. Neurology. 1986;36:622–625. doi: 10.1212/wnl.36.5.622. [DOI] [PubMed] [Google Scholar]
- 63.ARAGA S, LEBOEUF RD, BLALOCK JE. Prevention of experimental autoimmune myasthenia gravis by manipulation of the immune network with a complementary peptide for the acetylcholine receptor [published erratum appears in Proc. Natl. Acad. Sci. USA (1994) 91(4):1598] Proc. Natl. Acad. Sci. USA. 1993;90:8747–8751. doi: 10.1073/pnas.90.18.8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.LOUTRARI H, KOKLA A, TZARTOS SJ. Passive transfer of experimental myasthenia gravis via antigenic modulation of acetylcholine receptor. Eur. J. Immunol. 1992;22:2449–2452. doi: 10.1002/eji.1830220939. [DOI] [PubMed] [Google Scholar]
- 65.ARAGA S, GALIN FS, KISHIMOTO M, ADACHI A, BLALOCK JB. Prevention of experimental autoimmune myasthenia gravis by a monoclonal antibody to a complementary peptide for the main immunogenic region of the acetylcholine receptors. J. Immunol. 1996;157:386–392. [PubMed] [Google Scholar]
- 66.VERSCHUUREN JJ, GRAUS YM, VAN BREDA VRIESMAN PJ, TZARTOS S, DE BAETS MH. In vivo effects of neonatal administration of antiidiotype antibodies on experimental autoimmune myasthenia gravis. Autoimmunity. 1991;10:173–179. doi: 10.3109/08916939109001887. [DOI] [PubMed] [Google Scholar]
- 67.XIAO BG, DUAN RS, LINK H, HUANG YM. Induction of peripheral tolerance to experimental autoimmune myasthenia gravis by acetylcholine receptor-pulsed dendritic cells. Cell. Immunol. 2003;223:63–69. doi: 10.1016/s0008-8749(03)00118-7. [DOI] [PubMed] [Google Scholar]
- 68.YARILIN D, DUAN R, HUANG YM, XIAO BG. Dendritic cells exposed in vitro to TGF-beta1 ameliorate experimental autoimmune myasthenia gravis. Clin. Exp. Immunol. 2002;127:214–219. doi: 10.1046/j.1365-2249.2002.01748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DUAN RS, ADIKARI SB, HUANG YM, LINK H, XIAO BG. Protective potential of experimental autoimmune myasthenia gravis in Lewis rats by IL-10-modified dendritic cells. Neurobiol. Dis. 2004;16:461–467. doi: 10.1016/j.nbd.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 70•.AISSAOUI A, KLINGEL-SCHMITT I, COUDERC J, et al. Prevention of autoimmune attack by targeting specific T-cell receptors in a severe combined immunodeficiency mouse model of myasthenia gravis [see comments] Ann. Neurol. 1999;46:559–567. doi: 10.1002/1531-8249(199910)46:4<559::aid-ana3>3.0.co;2-s. Evidence for a pathogenic role of Vβ5.1-expressing T cells from HLA-DR3 patients. [DOI] [PubMed] [Google Scholar]
- 71.JAMBOU F, ZHANG W, MENESTRIER M, et al. Circulating regulatory anti-T cell receptor antibodies in patients with myasthenia gravis. J. Clin. Invest. 2003;112:265–274. doi: 10.1172/JCI16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.OFFNER H, HASHIM GA, VANDENBARK AA. Immunity to T cell receptor peptides: theory and applications. Regul. Pept. 1994;51:77–90. doi: 10.1016/0167-0115(94)90197-x. [DOI] [PubMed] [Google Scholar]
- 73.XU L, VILLAIN M, GALIN FS, ARAGA S, BLALOCK JE. Prevention and reversal of experimental autoimmune myasthenia gravis by a monoclonal antibody against acetylcholine receptor-specific T cells. Cell. Immunol. 2001;208:107–114. doi: 10.1006/cimm.2001.1777. [DOI] [PubMed] [Google Scholar]
- 74.BENSINGER SJ, WALSH PT, ZHANG J, et al. Distinct IL-2 receptor signaling pattern in CD4+CD25+ regulatory T cells. J. Immunol. 2004;172:5287–5296. doi: 10.4049/jimmunol.172.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.HORWITZ DA, ZHENG SG, GRAY JD, WANG JH, OHTSUKA K, YAMAGIWA S. Regulatory T cells generated ex vivo as an approach for the therapy of autoimmune disease. Semin. Immunol. 2004;16:135–143. doi: 10.1016/j.smim.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 76.WOOD KJ, SAKAGUCHI S. Regulatory T cells in transplantation tolerance. Nat. Rev. Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 77.SHEVACH EM. Regulatory T cells in autoimmmunity*. Annu. Rev. Immunol. 2000;18:423–449. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- 78.BAECHER-ALLAN C, VIGLIETTA V, HAFLER DA. Human CD4+CD25+ regulatory T cells. Semin. Immunol. 2004;16:89–98. doi: 10.1016/j.smim.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 79••.FONTENOT JD, GAVIN MA, RUDENSKY AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003;4:330–336. doi: 10.1038/ni904. Convincing demonstration of the role of FoxP3 in the regulatory function of CD4+CD25+ cells. [DOI] [PubMed] [Google Scholar]
- 80.VIGLIETTA V, BAECHER-ALLAN C, WEINER HL, HAFLER DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J. Exp. Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.KRIEGEL MA, LOHMANN T, GABLER C, BLANK N, KALDEN JR, LORENZ HM. Defective suppressor function of human CD4+ CD25+ regulatory T cells in autoimmune polyglandular syndrome Type II. J. Exp. Med. 2004;199:1285–1291. doi: 10.1084/jem.20032158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.LINDLEY S, DAYAN CM, BISHOP A, ROEP BO, PEAKMAN M. TREE TI: Defective suppressor function in CD4(+)CD25(+) T-cells from patients with Type 1 diabetes. Diabetes. 2005;54:92–99. doi: 10.2337/diabetes.54.1.92. [DOI] [PubMed] [Google Scholar]
- 83.HUANG YM, PIRSKANEN R, GISCOMBE R, LINK H, LEFVERT AK. Circulating CD4CD25 and CD4CD25 T cells in myasthenia gravis and in relation to thymectomy. Scand J. Immunol. 2004;59:408–414. doi: 10.1111/j.0300-9475.2004.01410.x. [DOI] [PubMed] [Google Scholar]
- 84.SUN Y, QIAO J, LU CZ, ZHAO CB, ZHU XM, XIAO BG. Increase of circulating CD4+CD25+ T cells in myasthenia gravis patients with stability and thymectomy. Clin. Immunol. 2004;112:284–289. doi: 10.1016/j.clim.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 85••.BALANDINA A, LECART S, DARTEVELLE P, SAOUDI A, BERRIH-AKNIN S. Functional defect of regulatory CD4(+)CD25+ T cells in the thymus of patients with autoimmune myasthenia gravis. Blood. 2005;105:735–741. doi: 10.1182/blood-2003-11-3900. Clear demonstration of a severe defect in the regulatory function of CD4+CD25+ cells in MG patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.BLUESTONE JA, TANG Q. Therapeutic vaccination using CD4+CD25+ antigen-specific regulatory T cells. Proc. Natl. Acad. Sci. USA. 2004;101(Suppl 2):14622–14626. doi: 10.1073/pnas.0405234101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.CHATENOUD L. CD3-specific antibody-induced active tolerance: from bench to bedside. Nat. Rev. Immunol. 2003;3:123–132. doi: 10.1038/nri1000. [DOI] [PubMed] [Google Scholar]
- 88.MCINTOSH KR, LINSLEY PS, DRACHMAN DB. Immunosuppression and induction of anergy by CTLA4Ig in vitro: effects on cellular and antibody responses of lymphocytes from rats with experimental autoimmune myasthenia gravis. Cell. Immunol. 1995;166:103–112. doi: 10.1006/cimm.1995.0012. [DOI] [PubMed] [Google Scholar]
- 89.IM SH, BARCHAN D, MAITI PK, FUCHS S, SOUROUJON MC. Blockade of cd40 ligand suppresses chronic experimental myasthenia gravis by down-regulation of th1 differentiation and up-regulation of ctla-4. J. Immunol. 2001;166:6893–6898. doi: 10.4049/jimmunol.166.11.6893. [DOI] [PubMed] [Google Scholar]
- 90.IM SH, BARCHAN D, MAITI PK, RAVEH L, SOUROUJON MC, FUCHS S. Suppression of experimental myasthenia gravis, a B cell-mediated autoimmune disease, by blockade of IL-18. FASEB J. 2001;15:2140–2148. doi: 10.1096/fj.01-0072com. [DOI] [PubMed] [Google Scholar]
- 91.STEINMAN L. Immune therapy for autoimmune diseases. Science. 2004;305:212–216. doi: 10.1126/science.1099896. [DOI] [PubMed] [Google Scholar]
- 92.KOMAGATA Y, WEINER HL. Oral tolerance. Rev. Immunogenet. 2000;2:61–73. [PubMed] [Google Scholar]
- 93.COHEN IR, QUINTANA FJ, MIMRAN A. Tregs in T cell vaccination: exploring the regulation of regulation. J. Clin. Invest. 2004;114:1227–1232. doi: 10.1172/JCI23396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.FIGDOR CG, DE VRIES IJ, LESTERHUIS WJ, MELIEF CJ. Dendritic cell immunotherapy: mapping the way. Nat. Med. 2004;10:475–480. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 95.LESTERHUIS WJ, DE VRIES IJ, ADEMA GJ, PUNT CJ. Dendritic cell-based vaccines in cancer immunotherapy: an update on clinical and immunological results. Ann. Oncol. 2004;15(Suppl 4):iv145–iv151. doi: 10.1093/annonc/mdh919. [DOI] [PubMed] [Google Scholar]
- 96.RIESER C, RAMONER R, HOLTL L, et al. Mature dendritic cells induce T-helper type-1-dominant immune responses in patients with metastatic renal cell carcinoma. Urol. Int. 1999;63:151–159. doi: 10.1159/000030438. [DOI] [PubMed] [Google Scholar]
- 97.WIENDL H, HOHLFELD R. Therapeutic approaches in multiple sclerosis: lessons from failed and interrupted treatment trials. BioDrugs. 2002;16:183–200. doi: 10.2165/00063030-200216030-00003. [DOI] [PubMed] [Google Scholar]
- 98.WARDROP RM, 3rd, WHITACRE CC. Oral tolerance in the treatment of inflammatory autoimmune diseases. Inflamm. Res. 1999;48:106–119. doi: 10.1007/s000110050433. [DOI] [PubMed] [Google Scholar]
- 99.MISHINA M, TAKAI T, IMOTO K, et al. Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nature. 1986;321:406–411. doi: 10.1038/321406a0. [DOI] [PubMed] [Google Scholar]
- 100.BEESON D, MORRIS A, VINCENT A, NEWSOM-DAVIS J. The human muscle nicotinic acetylcholine receptor alpha-subunit exist as two isoforms: a novel exon. EMBO J. 1990;9:2101–2106. doi: 10.1002/j.1460-2075.1990.tb07378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]