

Abstract
Summary: Accurate and complete replication of the genome in every cell division is a prerequisite of genomic stability. Thus, both prokaryotic and eukaryotic replication forks are extremely precise and robust molecular machines that have evolved to be up to the task. However, it has recently become clear that the replication fork is more of a hurdler than a runner: it must overcome various obstacles present on its way. Such obstacles can be called natural impediments to DNA replication, as opposed to external and genetic factors. Natural impediments to DNA replication are particular DNA binding proteins, unusual secondary structures in DNA, and transcription complexes that occasionally (in eukaryotes) or constantly (in prokaryotes) operate on replicating templates. This review describes the mechanisms and consequences of replication stalling at various natural impediments, with an emphasis on the role of replication stalling in genomic instability.
INTRODUCTION
Accurate replication of the whole genome with every cell division is a prerequisite for maintaining genomic stability. Multilevel control mechanisms, including numerous means of fork restart as well as checkpoint controls, ensure completion and accuracy of replication (for recent reviews on restart, see references 43, 91, 179, 185, 194, 195, and 257; for checkpoint controls, see references 134, 149, 164, and 186; for bacterial replication forks, see references 135 and 220; and for eukaryotic replication forks, see references 55 and 291). However, there is never a guarantee that these mechanisms are absolute. Thus, factors that interfere with DNA replication can jeopardize genomic integrity. Such factors can be categorized into three groups: exogenous, genetic, and intrinsic.
Exogenous factors that affect DNA replication do so either by damaging the DNA template (for example, UV light, gamma irradiation, DNA-modifying agents, and topoisomerase poisons) or by depleting nucleotide pools (for example, hydroxyurea and methotrexate) (71). In the first case, replication is blocked at the sites of damage because of the inability of the replication fork to pass through a corrupted DNA template. In the second case, replication is inhibited throughout the genome because of the lack of all or some of the deoxynucleoside triphosphates (dNTPs) necessary for DNA synthesis. Technically speaking, chemically induced DNA lesions can also be considered an intrinsic impediment to replication, since they abound under physiological conditions in the absence of any external damaging agents. For example, an average Escherichia coli cell has about 100 such lesions in the DNA, each with a 30-min half-life. When unrepaired, these lesions represent a challenge to replication (276).
Genetic determinants of replication inhibition are mutations in genes that affect the accuracy and speed of DNA synthesis (for example, components of the replication apparatus and nucleotide pool control).
Intrinsic, or natural, impediments of replication include DNA binding proteins, transcription units, unusual DNA structures, and replication slow zones (recent reviews include references 109 and 251). A growing body of evidence accumulated over the last decade indicates that replication inhibition by natural impediments can lead to genomic instability, drawing additional attention to this issue (for example, see references 15, 32, 33, 150, 201, 211, 242, 279, and 289 and below). In this review, evidence of replication inhibition at natural impediments is summarized and discussed, with emphasis on the genome's function and stability.
NATURAL IMPEDIMENTS TO DNA REPLICATION
Studies of attenuation of DNA replication by natural impediments were accelerated by the invention of neutral/alkaline (162) and neutral/neutral (20) two-dimensional gel electrophoresis of replication intermediates, which allow for investigation of progression of the replication fork in vivo. The neutral/neutral version has rapidly gained enormous popularity.
Stalling of replication forks at natural impediments can either occur “on purpose” or be “accidental” (if inhibition of replication is a side effect of another biological process). Sometimes, the distinction between the two scenarios is not obvious. The first instance includes termination of replication at bacterial termini and in eukaryotic ribosomal operons; replication slow zones in budding yeast; both the replication termination site and the replication pause site in the mating-type locus in fission yeast; and, probably, EBNA-1 of Epstein-Barr virus (EBV). The latter instance probably includes unusual DNA structures, collisions with transcription, and certain proteins (such as TraY in E. coli and certain nonhistone-DNA complexes in yeast). Typically, replication arrest in the first instance is nearly complete; hence, such sites are usually called RFBs, for “replication fork barriers” (the exceptions are replication slow zones in budding yeast and the replication pause site in the fission yeast mating-type locus). In the second instance, forks are usually capable of bypassing the obstacle after temporarily pausing. Such sites are therefore called RFPs, for “replication fork pausing” sites. Pausing can last from several seconds, as it does in yeast centromeres, to almost half an hour, as in EBV.
DNA Binding Proteins
Termination of replication in prokaryotes.
In E. coli, bidirectional replication starts from the origin, oriC, and the two forks travel in the opposite direction until they meet at the terminus, from which they cannot escape (52, 98). Proper termination is determined by Ter sequences (95, 101). The E. coli terminus contains at least ten Ter sites: TerA, TerD, TerE, TerI, and TerH trap the counterclockwise fork, and TerB, TerC, TerF, TerG, and TerJ trap the clockwise fork (Fig. 1A, left; for reviews, see references 5, 28, and 97 and, most recently, reference 214). Discovery of a trans-acting factor required for replication arrest at Ter sites in vitro (99, 131, 233, 270) was soon followed by cloning of the tus gene, which encodes the 35.8-kDa protein Tus (102). Recombinant Tus was necessary and sufficient to arrest replication in vitro, and the leading strand arrest site was mapped right at the edge of the Tus binding site (100). Tus binds Ter sites as a monomer. It blocks the helicase activity of the main replicative E. coli helicase DnaB in vitro in an orientation-specific manner (93, 127, 155), providing an explanation for the orientation specificity of fork arrest in the E. coli chromosome. The term contrahelicase was coined to account for this newly discovered activity (127).
FIG. 1.

Termination of replication in E. coli and B. subtilis. (A) Organization of replication termini in E. coli (left) and B. subtilis (right). In circular bacterial chromosomes, two replication forks (white arrow, clockwise fork; black arrow, counterclockwise fork) start from the bidirectional origin of replication (oriC). Arrest sites are located at the terminus. In E. coli, TerC, TerB, TerF, TerG, and TerJ arrest the clockwise fork (white squares), and TerA, TerD, TerE, TerI, and TerH arrest the counterclockwise fork (black squares). In B. subtilis, TerI (also called IR I), TerIII, and TerV arrest the clockwise fork (white squares), and TerII (also called IR II), TerIV, and TerVI arrest the counterclockwise fork (black squares). Each Ter site contains core (C) and auxiliary (A) sites. TerI (IR I) is the most frequently used termination site. Checkpoint replication arrest sites in the B. subtilis chromosome are indicated by stars: the black star shows the position of arrest of the counterclockwise fork, and the white star shows the position of arrest of the clockwise fork. The positions of sites in both chromosomes are not to scale. (B) Replication termination proteins: Tus of E. coli (left) and RTP of B. subtilis (right) bound to the corresponding Ter sites. In the case of Tus (which binds Ter as a monomer), three models were proposed to explain the asymmetry of fork blocking. (i) The fork-blocking side of Tus contains protruding α helices (gray star), which protect the DNA binding domain from displacement. (ii) Certain amino acid residues on the fork-blocking side of Tus (white star) make protein-protein contacts with the helicase, blocking it. (iii) When the helicase approaches the Ter-Tus complex from the blocking side, it unwinds a part of the Ter site, exposing a particular cytosine residue (black star), which makes contact with Tus, leading to an increased stability of the Ter-Tus complex. In the case of RTP, the asymmetry of fork blocking comes from the different strengths of binding of the RTP dimers to the core and auxiliary sites. Within one Ter site, each core and auxiliary site binds a dimer of RTP (therefore, each Ter site binds four RTP monomers). If the core site is met by the fork first, the fork will be arrested, but if the auxiliary site is met first, both dimers will be displaced and the fork will proceed.
Several explanations of the nature of the polarity were put forward. (i) Crystallization of the Ter-Tus complex (120) revealed protruding α-helical segments on the fork-blocking side of Tus (Fig. 1B, left). Therefore, it was suggested that when the DnaB helicase approaches the Ter-Tus complex from its blocking end, it encounters these segments and cannot reach the DNA binding domain to disrupt the protein-DNA interactions. In contrast, when the helicase approaches from the passage end, it can reach the DNA binding domain, disrupt Tus-DNA contacts, and proceed further. Thus, the presence of these protruding α-helical segments on one side of Tus could determine the orientation specificity of fork arrest. (ii) It was further suggested that the contrahelicase activity of Tus could depend on specific protein-protein contacts between Tus and DnaB helicase rather than resulting from “passive roadblock” (210). The region of this protein-protein interaction was mapped to the L1 loop of Tus, located on its helicase-blocking side. It was therefore proposed that the localization of the L1 loop could be the determinant of polarity as well. (iii) Recently, it was suggested that when DnaB approaches the Ter-Tus complex from the blocking side, it unwinds the corresponding part of the Ter site, exposing a particular cytosine residue. The contact between this flipped cytosine and Tus stabilizes the complex to such an extent that it dissociates 40 times more slowly than the regular Ter-Tus complex. This locking behavior can occur only when DnaB approaches from the blocking side of Tus (209). Note that model i implies simple steric hindrance, model ii implies specific protein-protein contacts, and model iii implies asymmetrical DNA unwinding but not protein-protein interactions.
The mechanism of replication termination in Bacillus subtilis is generally similar to that in E. coli in terms of the existence of a trap opposite from the origin of replication in the chromosome, consisting of two clusters of sequences that arrest replication forks of opposite polarities (Fig. 1A, right). Yet both protein and DNA components differ in sequence as well as architecture, and the explanation of the polar nature of arrest seems to be different (for reviews, see references 5, 27, and 97). There are at least six termination sites: TerI through TerVI, with TerI being the most frequently used (TerI and TerII were discovered first; until recently, they were referred to in the literature as IRI and IRII, for inverted repeat I and inverted repeat II, respectively). Each Ter site is bipartite, i.e., it consists of two elements, core and auxiliary sites, and each of those elements binds one homodimer of the replication termination protein, RTP (the molecular weight of a monomer is 14.5 kDa). RTP was purified (158) and shown to arrest replication in vitro in an orientation-dependent manner when bound to the TerI (IRI) and TerII (IRII) sites (124, 271). RTP and Tus share very little sequence similarity, and the crystal structure of RTP revealed a lack of structural similarity and confirmed that RTP exists as a symmetrical dimer (28).
Two symmetrical RTP homodimers bind one Ter site. How can a symmetrical protein structure arrest replication in an orientation-dependent manner? The answer comes from the asymmetry of protein-DNA contacts between RTP dimers and core and auxiliary sites (Fig. 1B, right) (27, 151, 178, 252). RTP binds the core site much more strongly than the auxiliary site. It can bind the core site in the absence of the auxiliary site but not vice versa. The replication fork is arrested only if it approaches the core site first, and yet the presence of the auxiliary site is required. These data, taken together, suggest the following explanation. When the replication fork approaches the auxiliary site first, it displaces the RTP dimer bound to it, and the RTP dimer at the core site can be displaced as well. However, if the replication fork approaches the core site first, the RTP dimer at the core site cannot be displaced, owing to the cooperative action of the dimer bound to the auxiliary site. This explanation is supported by the fact that bidirectional Ter sites from B. subtilis plasmids consist of two core sites (191).
The RTP dimer has not been crystallized together with its binding sites. Thus, it is theoretically possible that the structure of the RTP dimer bound to the core site might differ from that of the RTP dimer bound to the auxiliary site, which could add to the asymmetry of the complex. Like Tus, RTP is probably not a simple roadblock either, since specific interactions between the contrahelicase surface of RTP and the helicase appear to be important for replication arrest (178).
Why is it important to block the progression of the forks at the terminus rather than allowing them to meet and terminate at random? One possible explanation comes from the observation that in vitro, Ter sites positioned in the oriC-replicated plasmid the same way they are positioned in the chromosome prevent overreplication of the plasmid DNA (94). In the presence of Tus, the replication fork that reached a Ter site in the blocking orientation first was arrested, and the second replication fork stopped when it reached the first one. In contrast, in the absence of Tus, replication failed to cease when the two forks met each other, leading to overreplication. Therefore, the authors suggested that the role of the Ter/Tus system might be in preventing chromosomal overreplication. Although it is not obvious why the replication fork arrested at the Ter-Tus site would stop the oppositely moving fork whereas a simple collision between the two forks would not, this explanation seems very appealing. Supporting it is the finding that the absence of Tus in vivo leads to the switch to rolling circle replication instead of termination and unstable maintenance of the R1 plasmid (138). It would be interesting to know how eukaryotes get around this problem, since it is believed that termination of replication in eukaryotic chromosomes occurs by random meeting of the two converging forks (or by their reaching the end of the chromosome). Perhaps eukaryotic forks are different in the sense that they properly terminate upon meeting each other. Replication slow zones in yeast (33; also see below), which lead to periodic pausing of forks in the genome, might also provide a clue.
Another explanation could be that the existence of the terminus as a spatial chromosomal macrodomain helps coordinate the completion of replication, resolution of accidental chromosome dimers (by the XerCD/dif system), chromosomal segregation, and cell division in bacteria. Unlike eukaryotes, bacteria do not separate these processes temporally and therefore require tight coordination between them (see, for example, reference 216 and references therein). In E. coli, coordination between cell division and chromosome segregation is facilitated by FtsK, a septum-localized DNA translocase, which assists the resolution of chromosome dimers by the XerCD recombinase, and decatenation of sister chromatids by interaction with the DNA topoisomerase IV (232).
In both gram-positive and gram-negative bacteria, environmental stresses (such as amino acid starvation) induce accumulation of a signaling molecule, ppGpp (guanosinetetraphosphate), which activates a stringent response—a pleiotropic reaction aimed at energy saving—that includes shutting down transcription of rRNAs and replication (31). During the stringent response in E. coli, replication is blocked at the level of initiation. In B. subtilis, DNA synthesis starts but both forks become arrested at around 200 kb from the origin (Fig. 1A, right) (157). This phenomenon was called “replication checkpoint of B. subtilis,” and its functioning was shown to depend on the presence of RTP (156); however, the mechanism of regulation of the replication arrest is not known. The RTP binding site was located around one of the two arrest sites (4). It was further shown that this site was bipartite, like Ter, and bound two RTP dimers (although much more weakly than Ter sites from the terminus). Surprisingly, however, when placed in a plasmid, it was capable of arresting replication in vivo regardless of orientation and the stringent response (75). Therefore, it is not clear whether this site is actually involved in the checkpoint fork arrest and, if it is, what makes it active only during the stringent response. One elegant answer to the last question could be that RTP binding to the checkpoint sites is disrupted by transcription through them, which is shut down by ppGpp during the stringent response (27). It is also not known whether replication arrest during the checkpoint possesses directionality (because the sites of arrest are located close to the origin, there might be no need for it).
Interestingly, elevation of ppGpp levels dramatically improves the survival of UV-irradiated E. coli cells deficient in the Holliday junction resolvase RuvABC. This is likely owing to ppGpp-mediated destabilization of transcription complexes stalled at UV-induced lesions in DNA (184; also see below). Therefore, although the stringent response inhibits replication initiation in E. coli, it also helps in the completion of already-started rounds of replication.
Studies of the bacterial replication termination proteins Tus and RTP showed that the strength of binding to DNA does not necessarily determine whether the protein blocks the fork. Instead, the architecture of the protein and/or its interaction with the components of the replication machinery matters (27, 75, 120, 210).
Eukaryotic ribosomal barriers.
The eukaryotic rRNA RFB was first discovered in Saccharomyces cerevisiae, in the nontranscribed spacer of the rRNA genes (21, 162). The rRNA locus in S. cerevisiae is arranged as a cluster of tandem repeats of the following composition: 35S RNA (precursor of the 18S, 5.8S, and 25S rRNAs), transcribed by RNA polymerase I, followed by the first nontranscribed spacer and 5S rRNA, transcribed by the RNA polymerase III in the opposite direction, followed by the second nontranscribed spacer; the latter contains a bidirectional origin of replication (Fig. 2A). It was demonstrated that the first nontranscribed spacer contains a replication barrier (21, 162). The RFB function does not depend on transcription per se (22) but is caused by the DNA binding protein Fob1 (132), which wraps the FRB DNA around itself by binding two sites in the target sequence (129). One site, RFB1, is the major site of replication arrest, and the other site, FRB2, is the minor site (22, 129). Fob1 possesses potent polar contrahelicase activity (208), arresting only the forks that are about to enter the transcription units in the 3′-to-5′ direction. It was therefore suggested that a possible role of ribosomal barriers was to protect the rRNA genes from the head-on transcription-replication collisions (see below). Deletion of the fob1 gene in S. cerevisiae (along with a reduction in the copy number of rRNA gene repeats) leads to transcription-mediated replication inhibition along the whole transcribed area (279). Two proteins which are known to act upon stalled replication forks, Tof1 and Csm3, have been shown to affect forks arrested at the Fob1-caused barrier: in their absence, the replication blockage becomes less prominent (29). Tof1 is the budding yeast homologue of the fission yeast Swi1, and Csm3 is the budding yeast homologue of the fission yeast Swi3 (see below).
FIG. 2.

Ribosomal RFBs. (A) Organization of rRNA locus in S. cerevisiae. The locus consists of multiple units of direct repeats, each of which contains a 35S RNA transcription unit (which after processing gives rise to 18S, 5.8S, and 25S rRNAs) transcribed by RNA polymerase I, a 5S RNA gene transcribed by RNA polymerase III, and two nontranscribed spacers. One nontranscribed spacer (NTS2) contains a bidirectional origin of replication (ARS), whereas the other nontranscribed spacer (NTS1) contains an RFB that arrests forks that are about to enter the 35S transcription unit (i.e., forks that move from right to left). (B) Comparison of ribosomal RFBs in budding yeast, mammals, and fission yeast. In budding yeast, the RFB is caused by the Fob1 protein; in mammals, the RFB is caused by the TTF-1 protein, which is also a transcription termination factor for rRNAs. Fission yeast combines both scenarios: RFB1 is caused by Sap1, which, like Fob1, is not involved in termination of rRNA transcription, and RFB2 and RFB3 are caused by Reb1, which, like TTF-1, is a transcription termination factor. Arrows, rRNA transcription units. The fork that moves from right to left is blocked.
In mice, RFBs at the 3′ ends of ribosomal genes colocalize with the rRNA transcription terminators (172) and are caused by the TTF-1 protein (transcription termination factor 1) (76). TTF-1 happens to be a polar contrahelicase (as is Fob1), as it is able to inhibit the helicase activity of the simian virus 40 (SV40) large T antigen in vitro (243). However, unlike Fob1, TTF-1 is also the rRNA transcription termination factor that facilitates termination of rRNA transcription (in the opposite direction).
Schizosaccharomyces pombe seems to combine both aforementioned scenarios. There are three pause sites in the S. pombe rRNA gene that are caused by protein binding (the fourth site, RFP4, is minor and becomes prominent in the absence of RFB1 to RFB3; it is most likely caused by collisions with transcription [145]). RFB1 is the strongest arrest site. It is located distal to the rRNA transcription unit and is therefore met by the replication fork first. Inhibition of replication at RFB1 is mediated by the Sap1 protein (144, 192). Like Fob1, Sap1 is not involved in the termination of transcription of rRNA, but unlike Fob1, it possesses another important function, most likely in chromatin organization, which makes it essential for yeast viability. Sap1 is also involved in another event associated with replication stalling, fork pausing in the mating-type switch locus mat1, although in that case, Sap1 binding is not required for the fork stalling but rather is involved in the imprinting (see below). Replication fork stalling at RFB2 and RFB3 (located proximal to the rRNA transcription unit) is caused by the transcription termination protein Reb1, as in the mammalian scenario (256). Replication stalling at all three sites is dependent on the products of two mating-type switching genes, swi1 and swi3 (145), just as the stalling in the mating-type switch locus mat1 (see below). Swi1 (homolog of the budding yeast Tof1) was shown to stabilize stalled forks genome-wide and to activate checkpoint in response to genotoxic stress (217). Interestingly, forks stalled at RFP4 did not depend on Swi1 and Swi3 (145). A comparison of RFBs from budding yeast, mammals, and fission yeast is shown in Fig. 2B.
Ribosomal RFBs have also been observed in Pisum sativum (171), Tetrahymena thermophila (174, 310), Xenopus laevis (181, 300), and humans (165). In Tetrahymena (310) and Xenopus (181), they appear to be developmentally regulated, being most evident at the stages of maximal amplification and transcription, respectively, of the rRNA genes. Unlike all of the other organisms examined so far, the ribosomal RFB in human rRNA locus was found to be nonpolar (bidirectional), while still acting to make transcription and replication proceed in the same direction (165). Interestingly, in T. thermophila, the RFB was shown to predominantly inhibit forks moving in the direction of transcription (174).
Epstein-Barr virus protein EBNA-1.
EBNA-1 (for “Epstein-Barr nuclear antigen 1”) is the only viral protein essential for latent replication of the EBV circular genome from the latent origin of replication, OriP. (In contrast, lytic EBV replication uses two other origins, orilytL and orilytR, and virus-encoded replication machinery [reviewed in reference 292].) OriP consists of two functional elements: FR (for “family of repeats”), which has 20 strong EBNA-1 binding sites, and DS (for “dyad symmetry”), which has 4 weaker EBNA-1 binding sites. EBNA-1 binding to the DS element facilitates initiation of replication (presumably by recruitment of ORC [for “origin recognition complex”]). Two replication forks assemble at the DS element and proceed in opposite directions. Subsequently, one of them is arrested in the FR element. Thus, initially bidirectional replication is converted essentially into a unidirectional mode (Fig. 3). The replication barrier in the EBV DNA (74) was shown to be EBNA-1 dependent: EBNA-1 significantly enhances replication pausing at this barrier, but, interestingly, replication pausing occurs even in the absence of EBNA-1 (although to a much lesser extent) (59). In vitro, EBNA-1 binding to the DNA substrate inhibits the helicase activity of both SV40 large T antigen and the E. coli main replicative helicase DnaB in an orientation-independent manner (unlike Tus, RTP, and Fob-1) (64).
FIG. 3.
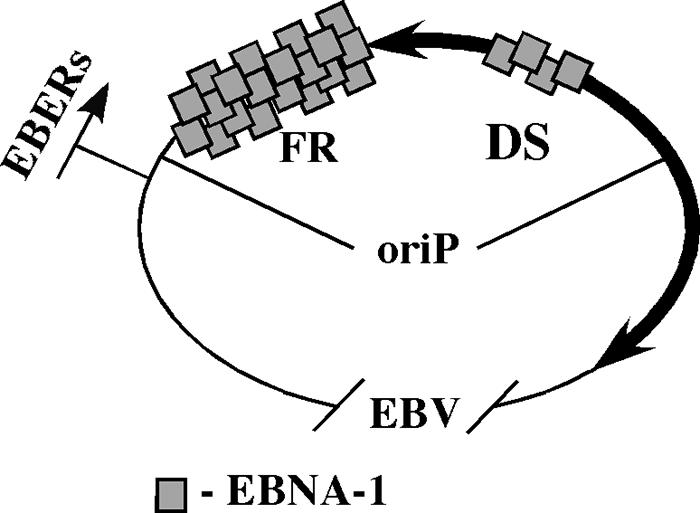
Asymmetrical replication of EBV. OriP, the latent viral origin of replication, consists of the dyad symmetry element (DS) and the family of repeats (FR). Bidirectional replication starts at the DS, which contains four weak binding sites for EBNA-1, but one of the forks is halted at the FR, which contains 20 strong binding sites for EBNA-1. This effectively converts bidirectional replication to unidirectional replication. OriP is a fraction of the viral genome; the picture is not to scale. EBNA-1, EBV nuclear antigen 1; EBERs (EBER-1 and EBER-2), nontranslated, small latent phase-specific EBV-encoded RNAs, transcribed by RNA polymerase III.
The timing of EBNA-1-caused replication inhibition was quantified (219). The speed of replication in the portion of EBV DNA without the replication pause site was measured as 1.3 kb/min, whereas it took 36 min to replicate a 10-kb fragment containing the EBNA-1-dependent pause site (instead of the expected 8 min). Therefore, the time of replication fork pausing was estimated to be 28 min on average. Interestingly, replication stalling was not complete, and so the two forks met (and terminated) at random positions, albeit with a very strong bias toward the pausing area.
The biological meaning of the EBNA-1-mediated replication barrier and the resulting asymmetrical replication of the viral genome could be the prevention of the head-on transcription-replication collisions at the EBERs. EBERs (EBER-1 and EBER-2, two nontranslated EBV-encoded RNAs) are latent phase-specific small RNAs transcribed by RNA polymerase III. They are located close to the FR element and are replicated codirectionally with transcription as a result of replication arrest (Fig. 3). Alternatively, replication inhibition at the sites of EBNA-1 binding to FR might be a mere consequence of its strong binding. The FR element is required for the proper segregation of viral DNA in the host cells after mitosis, and strong binding of EBNA-1 to FR ensures the segregation.
Nonhistone protein-DNA complexes in budding yeast.
Extensive studies of chromosomal replication in the budding yeast S. cerevisiae detected replication stalling at a number of genomic loci where nonhistone proteins were bound to DNA beside the ribosomal barrier. The first example of inhibition of replication by tightly bound protein-DNA complexes was described when replication forks were shown to pause at centromeres in the S. cerevisiae genome (82). By altering the direction of replication through the centromeric region of the genome, the authors were able to conclude that the pausing was orientation independent. Using yeast plasmids, they confirmed the orientation independence of the pause site and showed that this pausing strongly correlated with the ability of the centromeric DNA to bind nonhistone proteins that form a tightly packed nuclease-resistant structure of centromeres. Comparison of the amount of stalled versus normal replication intermediates allowed them to estimate the time of pausing as 0.1 to 0.2 min.
Replication stalling was detected at inactive origins of replication located near one of the transcriptionally silent mating-type loci, the HML locus (293). The authors suggested that it is most likely caused by the presence of origin-specific proteins, such as ORC (for “origin recognition complex”).
Replication stalling was also observed in telomeric and subtelomeric regions (115). It was shown to be orientation independent, and the only candidate protein responsible for it was the telomeric DNA binding protein Rap1 (the Reb1, Tbf1, Rif1 and Rif2, and Sir2, Sir3, and Sir4 proteins were not required) (177).
The Rrm3 helicase was identified because its absence increased recombination in the rRNA locus in yeast (126). When replication in the rrm3-deficient yeast strain was studied meticulously (114-116), it appeared that replication forks pause at about 1,400 discrete sites in the S. cerevisiae genome. Breakage of forks and high levels of recombination were also observed at these sites. These sites include tRNA genes, centromeres, telomeres, silent mating-type loci, and inactive origins of replication as well as the rRNA locus (specifically, the 5S rRNA genes, the beginnings and ends of the 35S rRNA genes, and inactive origins of replication), i.e., positions of tightly bound nonhistone protein-DNA complexes. Confirming the involvement of the protein-DNA complexes, disruption of the binding sites and/or elimination of the corresponding proteins abolished replication stalling. The fact that some replication stalling at the same loci was observed in the wild-type yeast but was greatly increased in the absence of Rrm3 suggests that the function of Rrm3 is to remove such tightly bound protein conglomerates from the DNA to assist replication of the genome. The existence of at least one helicase, which helps the replication forks to pass tightly bound protein-DNA complexes without stalling and to avoid subsequent fork breakage at numerous sites in the genome, indicates the importance and the genome-wide scale of the problem of protein-caused impediments of replication (114). In vertebrates, the Williams syndrome transcription factor-imitation-switch protein (WSTF-ISWI) chromatin remodeling complex (ISWI is a nucleosome-dependent ATPase) might be involved in assisting the replication of heterochromatin (18).
Replication stalling in the mating-type switch locus of fission yeast.
The fission yeast S. pombe has two mating types: plus and minus. The current type of a cell is encoded in the mat1 locus. The switch occurs by means of transferring the genetic information from one of the silent donor loci, mat2-P or mat3-M, into the active mat1 locus (Fig. 4). The switch is regulated in a very interesting way: it occurs after two consecutive divisions, with only one of the four cells undergoing it. When a newly switched cell divides, it generates a “nonswitchable” cell and a “switchable” cell; in the next round of division, the switchable cell gives rise to a nonswitched cell and a switched cell. Since all of the cells are identical in terms of genetic information, epigenetic regulation, i.e., a strand-specific imprinting that marks the DNA strand that is segregated to the switchable cell, has been proposed to explain the asymmetry (reviewed in reference 46).
FIG. 4.

Organization of the mating-type locus in S. pombe. The current mating type of a cell is determined by the mat1 locus. The switch occurs by means of transferring the genetic information from one of the silent donor loci, mat2-P or mat3-M, into the active mat1 locus. RTS1 (for “replication termination sequence 1”) blocks the fork that moves from left to right, making sure that replication of the mat1 locus occurs by the fork that moves from right to left. The latter pauses at MPS1 (for “mat1 pause site 1”). This pausing leads to the proper placement of the RNA primer, which is the mark of imprinting that regulates the switch.
An RNA primer left behind on the lagging strand and sealed with DNA was suggested to be this strand-specific label. When this strand is used as a leading-strand template in the next round of replication, leading-strand synthesis stops at the RNA primer, which induces the recombination event in this chromatid. After the division, the cell that inherited this switched chromatid becomes switched. According to this model, the direction of replication through the mat1 locus is crucial for proper switch regulation (47). A polar replication termination site (called RTS1, for “replication termination site 1” [Fig. 4]) was observed near the site of imprinting. This site ensures a unidirectional mode of replication through the mat1 locus (45).
Orientation-specific pausing of the replication fork was observed at the site of imprinting (called MPS1, for “mat1 pause site 1” [Fig. 4]). It was suggested that this pausing facilitates placement of the primer at a specific position of the lagging strand (48).
The Swi1 and Swi3 proteins, which are necessary for the imprinting, were shown to also be necessary for replication stalling at both sites (48). Swi1 (a homologue of budding yeast Tof1) is involved in the stabilization of stalled forks and activation of checkpoint in response to genotoxic stress (217), and both Swi1 and Swi3 are involved in replication stalling at the ribosomal locus in S. pombe (see above). Interestingly, the Sap1 protein, which is responsible for replication stalling in the ribosomal locus, is involved here as well. However, Sap1 is not necessary here for the fork pausing itself but rather is involved in the establishment and/or maintenance of the imprinted state (48). Another two proteins, Rtf1 and Rtf2 (replication termination factors 1 and 2, respectively), contribute to the replication stalling at RTS1 (41). Rtf1 has sequence similarity with Reb1 of budding yeast and TTF-1 of mammals, which cause replication stalling in rRNA genes (see above).
Although it is presumed that protein-DNA complexes are responsible for the replication stalling at MPS1 and replication termination at RTS1, the cause of neither has been identified. The Swi1 and Swi3 proteins are not known to possess sequence specificity; thus, although they play a major role in replication stalling, there should be another factor that is responsible for the sequence specificity (Rtf1 and Rtf2 are good candidates).
Note that the mechanism used by fission yeast is different from that used by the budding yeast S. cerevisiae, which can switch mating types as often as every generation by a mechanism that involves the introduction of a double-stranded break by a site-specific endonuclease (reviewed in reference 83).
Artificially constructed, tightly bound protein-DNA complexes in E. coli.
Controlled, site-specific replication stalling was achieved by using tight binding of multiple molecules of TetR-YFP (tetracycline repressor fused to yellow fluorescent protein) to an array of 240 copies of its operator, tetO, inserted into the E. coli chromosome (239). Induction of the expression of TetR-YFP caused replication stalling independent of the fork's polarity. The stalling was observed on two-dimensional gels and prevented cell proliferation, which indicated that even after a prolonged period of time, this barrier could not be overcome by the replication fork. Addition of anhydrotetracycline, which abolishes the binding of TetR to tetO, resulted in resumption of replication and disappearance of the stalled intermediates in just 5 min. This, together with the fact that the SSB protein was associated with the tetO array for several hours, suggests that the replication fork was paused in a physiological state and resumed replication immediately after the obstacle was removed. Supporting this explanation is the finding that replication restart did not require recombination proteins.
“Infinite” replication stalling at the TetR-tetO array resulted in unviability of cells, so why did the cells not deal with this situation by homologous recombination? One explanation could be that the bulky, 240-copy protein-DNA complex prevented the accessibility of DNA for the recombination machinery.
Another example of replication stalling at artificially constructed, tightly bound, bulky protein-DNA complexes in E. coli comes from a study of replication of the d(GA)n/d(TC)n minisatellite. When cloned in a plasmid, the d(GA)n/d(TC)n repeats caused a length-dependent and orientation-independent replication inhibition in E. coli (142). This turned out to be owing to the binding of multiple molecules of the TraY protein. TraY belongs to the family of ribbon-helix-helix DNA-binding proteins (246) and is essential for F factor conjugal transfer (152). Binding of TraY to DNA introduces substantial bending (173); thus, binding of multiple TraY protomers to d(GA)n/d(TC)n repeats results in the formation of nucleosome-like structures wherein the DNA is wrapped around the core of TraY molecules. The existence of such a complex is supported by the observation of TraY-induced topological changes in circular DNA molecules carrying the d(GA)n/d(TC)n repeats.
Transcription
Transcription-replication collisions.
The replication fork and the RNA polymerase share the same DNA template; thus, occasional collisions between the two machineries are inevitable and can interfere with replication fork progression (19, 218). Given that both processes are polar, they can collide either head-on or codirectionally (Fig. 5). In the case of head-on collisions, the front edge of the RNA polymerase collides with the lagging-strand synthesis components of the replication fork. In the codirectional case, by contrast, the rear edge of the RNA polymerase meets the leading-strand synthesis components of the replication fork. The speed of the replication fork in prokaryotes is approximately 20-fold faster than is transcription (∼800 nucleotides [nt]/s versus 20 to 50 nt/s [reviewed in reference 136]). Therefore, both in the head-on and in the codirectional scenarios, there would be a collision. However, in eukaryotes, the speed of replication is comparable to that of transcription, making codirectional collisions unlikely.
FIG. 5.

Schematic representation of the head-on and codirectional collisions between replication and transcription in bacteria. The replication fork (trombone view), which moves from left to right, is on the left (leading- and lagging-strand DNA polymerases are shown as ovals; the DNA helicase DnaB is shown as a hexagon). The RNA polymerase with the nascent transcript is on the right. In the case of head-on collision (A), the RNA polymerase transcribes the lagging-strand template; in the case of codirectional collision (B), the RNA polymerase transcribes the leading-strand template.
Experimental data on transcription-replication collisions demonstrated that various replication machineries are inhibited to different extents by transcription complexes present on the same templates. Two sets of studies of the interactions between bacteriophage replisomes and their host bacterial RNA polymerases were carried out in vitro. The phage T4 replication fork appeared to pause more strongly when it encountered stalled E. coli RNA polymerase head-on than codirectionally (166, 167). When the RNA polymerase was allowed to move slowly, replication fork pausing became less pronounced. In contrast, the phage φ29 replisome halted upon encountering stalled B. subtilis RNA polymerase in both orientations (62, 63). Once RNA polymerase movement was resumed, DNA synthesis continued at its normal speed in the head-on case but was much slower for the codirectional alignment.
The first evidence of transcription-replication collisions in vivo came from studies of an inducible replication origin placed on either side of the rrnB ribosomal operon in the E. coli chromosome (67). Replication codirectional with the ribosomal operon proceeded at its typical high speed but was significantly slower when faced with head-on transcription. In eukaryotes, transcription-replication collisions were detected at two genomic loci: tRNA (58) and rRNA (279). In the first case, polar RFPs were observed at tRNA genes in S. cerevisiae (58). The replication fork stalled when it encountered tRNA genes transcribed head-on but not those transcribed codirectionally. The dependence of RFP activity on the functionality of both the gene promoter and RNA polymerase III argued for the direct involvement of transcription. It was proposed that the transcription elongation complex was responsible for the RFP sites at the tRNA genes. Recently, however, it was suggested that the transcription initiation complex, rather than elongating RNA polymerase, could be responsible for replication slowing at these genes (114). In the second case, transcription-replication collisions at the ribosomal locus in S. cerevisiae were detected when the fob1 gene (which encodes the Fob1 protein, responsible for the replication fork barrier, RFB, at the 3′ end of the rRNA genes; see above) was deleted and the number of rRNA gene repeats was reduced to increase the ratio of transcribed to nontranscribed rRNA (279).
Although the general consensus was that head-on collisions with the transcription machinery are much more detrimental for replication fork progression than are the codirectional collisions, the mechanism responsible for replication slowing in the head-on versus codirectional scenarios was under debate. Two possible scenarios of replication inhibition in the case of head-on collision with the transcription machinery have been proposed: physical interaction with the transcription machinery or excessive positive superhelicity generated by the two head-on processes. Both elongating RNA polymerase (168, 303) and the replication fork (237) generate positive supercoils in the downstream DNA. Consequently, frontal movement of the two machineries could generate a highly positively supercoiled DNA domain restraining both processes prior to direct encounter (19, 58, 67). Supporting the formation of such positively supercoiled domains, the fraction of DNA knots appeared to be greater in those plasmids where replication collided with transcription head-on than codirectionally (222). This was explained by the migration of positive supercoils, accumulated between the replisome and RNA polymerase, to the newly synthesized DNA behind the fork. Recently, a study was set up to distinguish between the two scenarios (199). It confirmed that codirectional transcription had no (or little) effect on replication fork progression in E. coli cells compared with that of head-on transcription, which severely impeded replication fork progression in ColE1-based plasmids. It also proved that in the head-on scenario, the replication fork was slowed as the result of direct physical interaction with the transcription machinery rather than by propagation of superhelical stress, since replication pausing zones were, in fact, strictly limited to the transcribed DNA areas. Note that these data do not refute the accumulation of positive supercoils upon the head-on transcription-replication collisions (222) but exclude these topological constraints as the cause of replication inhibition.
There is more to transcription than elongation. Recently, replication stalling caused by the transcription initiation complex as well as the RNA polymerase present at the transcription terminator was observed in E. coli (198). These findings might be important given that the majority of genes are not actively transcribed during DNA replication. The replication fork stalls upon head-on encounters with the transcription initiation complex and codirectional encounters with the RNA polymerase present at the transcription terminator. (The latter is likely owing to the existence of some trapped form of the RNA polymerase that cannot be readily displaced from DNA by the replication fork, as in the course of codirectional collision with the normal transcription elongation complex).
Notably, in both instances, the replication fork stalled after passing the transcribed region from either direction (Fig. 6). It is therefore plausible to speculate that transcription initiation and termination elements could serve as polar “punctuation marks” for DNA replication, i.e., attenuating replication fork progression as it traverses the transcribed areas. One possible role of such “punctuation marks” could be to provide extra time for the mismatch repair or gene conversion machineries to clear the coding areas of any newly acquired mutations, thereby helping to maintain the integrity of transcribed regions.
FIG. 6.
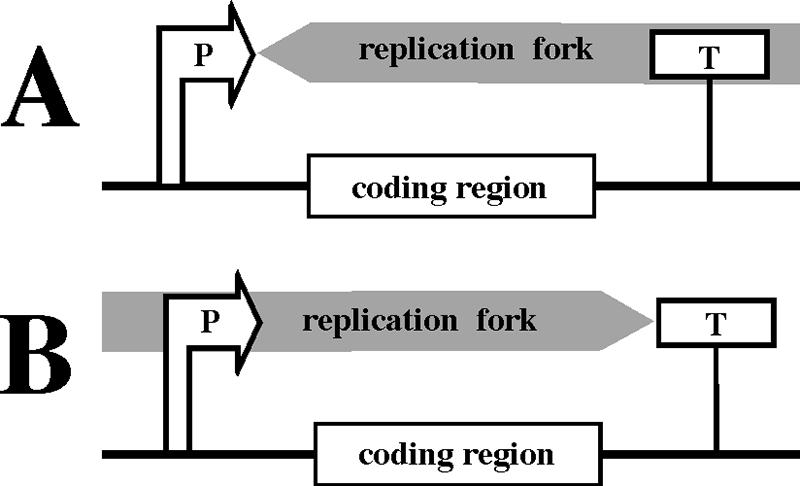
Transcription regulatory elements are punctuation marks for DNA replication. P, promoter; T, transcription terminator. The replication fork is slowed down by a head-on collision with the transcription initiation complex at the promoter (A) or a codirectional passage through the transcription terminator (B), i.e., when it passes the coding region from either direction.
Another possibility is that stalling of the replication fork at promoters and terminators could promote recombination outside of the coding areas. It was shown that mitotic recombination events induced by DNA damage often occur in intergenic regions containing promoters and/or terminators of transcription (49). Note that this is different from meiotic recombination. In meiotic recombination, although double-stranded breaks in DNA that initiate recombination are also more often localized in the intergenic promoter-containing regions than in the coding sequences, they do not result from stalled forks. Instead, they are introduced by Spo11 (reviewed in reference 125), and their preferential positioning is explained by the increased accessibility of DNA, owing to more open chromatin in those areas (see references 9, 77, 159, 304, and 305 and references therein).
Transcription-replication collisions and organization of genomes.
The data on the organization of bacterial, plasmid, and bacteriophage genomes point to selection against head-on collisions (19, 218). Sequencing of the E. coli genome confirmed that there was a bias toward codirectional alignment of transcription units with the direction of replication (16). Most strikingly, all seven ribosomal operons face the direction of their replichores. For other genes, however, this bias is much less pronounced: approximately 62% of tRNA genes and only approximately 55% of protein-coding genes are aligned codirectionally with replication. Similar principles of gene arrangement were observed for other bacteria, such as B. subtilis, Borrelia burgdorferi, Treponema pallidum, Haemophilus influenzae, Helicobacter pylori, Mycoplasma genitalium, and Mycoplasma pneumoniae (187), as well as for bacteriophages T7 and lambda (19).
While both DNA replication and transcription in bacteria continue throughout the life cycle, the situation is different in eukaryotes, owing to the existence of the phases in the cell cycle and temporal separation of DNA replication from most of transcription. It is well known, however, that at least some genes, including histone genes and genes coding for components of the protein synthesis machinery, such as tRNA and rRNA genes, are being transcribed during S phase (37, 235, 273). In the case of rRNA genes, the RFB at their 3′ end assures codirectional alignment of transcription and replication (see above). The existence of such barriers suggests a requirement to protect important genes from head-on transcription-replication collisions.
Particularly highly expressed protein-coding genes, which represent only a small fraction of all genes, are spaced along the chromosome, oriented codirectionally with replication, and form their own topological domains (53, 54). At the same time, genome-wide analysis of gene distribution in bacteria suggested that it is the “essentiality” rather than the “expressiveness” of a gene that likely determines its orientation relative to replication (250). About 90% of essential genes in B. subtilis and 70% of essential genes in E. coli are transcribed codirectionally with replication. The authors believe that the deleterious consequences of the head-on transcription-replication collisions could come from the displacement of the RNA polymerase, formation of truncated transcripts, and consequently, truncated proteins that would serve as dominant-negative forms of essential proteins. Note, however, that the amounts of such truncated peptides should be negligible given the short time that the replication fork spends at any given gene. Also, this model implies that RNA polymerase is displaced during head-on collisions but not during codirectional collisions, which might not be the case.
The issue of RNA polymerase displacement is somewhat contradictory. In vitro, RNA polymerases from both E. coli and B. subtilis were shown to stay bound to the DNA template and to continue elongation after the passage of the replication fork in both orientations (62, 63, 166, 167), but in an in vivo study carried out with E. coli, RNA polymerase was shown to be dislodged from DNA when it encountered the replication fork in both orientations (67). An alternative explanation for the preferential alignment of essential genes with the direction of replication could therefore be that the head-on transcription-replication collisions may be avoided at essential genes to prevent their inhibitory effects on replication. Blockage of DNA replication and subsequent restart occasionally might lead to genomic instability. In the case of head-on transcription-replication collisions, there is a fair chance that the restarted fork would encounter another elongating RNA polymerase, increasing the chances of inaccurate restart. These multiple replication restarts could thus be avoided within essential genes to protect their loci from instability.
The study (184) strongly supports the idea that stalled RNA polymerase could be a major challenge for replication in vivo and could require replication restart. The authors observe that an increase in the amount of the stringent response messenger ppGpp dramatically improves the survival rate of UV-irradiated E. coli cells deficient in the Holliday junction resolvase RuvABC and that this survival depends on the RecG helicase. These data are explained in the following way. UV irradiation leads to the appearance of lesions in DNA, which cause RNA polymerase stalling; the subsequent collisions between the replication fork and stalled RNA polymerase molecules require replication restart. In the wild-type cells, replication restart can be carried out by the action of RuvABC on stalled replication forks. Other pathways of restart are probably less prominent, as the cells die due to the inability to cope with the amount of required restart events in the absence of RuvABC. At high levels of ppGpp, which destabilizes transcription complexes stalled at UV-induced lesions, the amount of stalled RNA polymerases becomes smaller and RuvABC-deficient cells can survive UV irradiation, because other pathways become sufficient to deal with the small number of stalled forks. This observation proves that the presence of stable stalled transcription complexes creates a requirement for the replication restart and, if this requirement is not met, leads to loss of viability. The viability of UV-irradiated RuvABC-deficient cells at high levels of ppGpp depends on the RecG helicase, which implicates RecG in the other pathways of restart. RecG can facilitate regression of the fork, formation of the four-way junction, and, upon repair or bypass of the lesion, reannealing of the nascent strands.
Direct evidence of genomic instability caused by head-on transcription-replication collisions is described below.
Strong RNA-DNA hybrids.
Two other transcription-related examples of replication inhibition have been described in vivo: R loops (259, 260, 288) and transcription-dependent replication inhibition at the poly(G)/poly(C) repeat (141).
An R loop is a structure formed when cRNA hybridizes with one of the strands in the DNA duplex, displacing the other DNA strand and making it form a loop. Short, transient R loops are normal intermediates of transcription. Long and unusually stable R loops form when transcribed RNA is meant to be a primer for DNA replication; i.e., it needs to stay attached to DNA after the RNA polymerase is gone. This is the case for certain bacterial plasmids, for example, ColE1-based plasmids (113), and for the mitochondrial genomes of eukaryotic cells from yeasts to humans (7, 35, 223). (In ColE1-based plasmids, the transcript that forms the R loop and primes replication is called RNA II).
R loops have been shown to interfere with the movement of the replication fork in the study of a peculiar derivative of the pBR322 plasmid with two ColE1 origins of replication facing each other. (This is different from the majority of spontaneously formed plasmid dimers that are always organized “head-to-tail” so the two origins face the same direction.) As with other plasmid dimers, both origins in this one were potentially functional, but only one was active in any replicating molecule. The silent origin was capable of stalling the replication fork moving from the active origin, and the mutual orientation of the origins was crucial for this effect (288). This effect was completely dependent on the presence of the promoter within the silent origin (259). Given that the main replicative helicase, DnaB, is not capable of dissociating RNA-DNA hybrids in vitro, it was suggested that DnaB is unable to unwind the RNA-DNA hybrid at the silent origin. Since DnaB moves in the 5′-to-3′ direction along the lagging-strand template, it encounters hybrids transcribed from the head-on-oriented promoters but not from the codirectional ones. In accordance with this explanation, the site of the replication blockage mapped exactly to the 3′ end of RNA II (260).
The ColE1 promoter transcript RNA II is different from regular transcripts. It forms a long hybrid with the template DNA, thus allowing for the priming of initiation of replication. Since it is this hybrid that arrests replication, there is strong doubt that this situation could be similar to the conventional collision of replication and transcription from regular promoters because RNA normally does not form such long, stable hybrids with DNA. Long R loops at highly transcribed regions become evident only in the absence of one of three cellular enzymes: topoisomerase I, RNase H, or RecG. Thus, these enzymes normally take care of long R loops by preventing their formation, hydrolyzing them, or unwinding them, respectively (61, 103, 290).
Topoisomerase I relaxes negative superhelicity formed behind the elongating RNA polymerase (60). In its absence, R-loop formation becomes favorable, owing to the increased negative superhelicity that promotes DNA unwinding and thus the formation of the RNA-DNA hybrid. Growth of the topA strain is impaired, especially at low temperatures (when the RNA-DNA hybrids are particularly stable) (182). It is completely abolished in the absence of RNase H (which degrades RNA in the RNA-DNA hybrids) and is partially rescued when it is overexpressed (61). Inactivation of RecG, a structure-specific helicase that promotes branch migration of Holliday junctions, is also synthetically lethal with topoisomerase I deficiency (103). In vitro, RecG can efficiently dissociate R loops (290). It is therefore believed that the deleterious effects of R loops in the topA strain are responsible for its phenotype. RNase H overproduction was also shown to correct the main known defect of the topA strain, a reduced rate of transcription elongation (106). Therefore, it is believed that transcriptional blocks at R loops (when RNA polymerase encounters an R loop formed by a preceding RNA polymerase) result in the low rate of transcription and, ultimately, poor growth. It is feasible, however, that since R loops affect replication progression, replication stalling at R loops could account for their toxic effect as well. Alternatively, R loops can first lead to the formation of stalled ternary transcription complexes, and those in turn can block replication progression. As discussed above, stalled ternary transcription complexes were shown to affect replication progression of the bacteriophage DNA polymerases in vitro to various extents (62, 63, 166, 167).
Transcription-dependent replication inhibition at the poly(G)/poly(C) repeats in DNA, particularly when the poly(G) strand was in the nascent RNA, was observed in E. coli cells (141). The strength of this block did not depend on the direction of replication through the complex. Although the exact nature of this phenomenon is unknown, the following explanations are plausible. The replication fork could be inhibited by the stable RNA-DNA hybrid (which is unusually strong, owing to its 100% GC content). Alternatively, it could be owing to the formation of unusual structures composed either of the duplex portion of DNA and the displaced DNA strand or of the RNA-DNA hybrid and the displaced DNA strand (the so-called “collapsed R loop” [206, 247, 248]). Yet another scenario could be that these structures first trap the RNA polymerase, leading to the formation of the stalled ternary transcription complex and in turn arresting the replication fork.
Unusual DNA Structures
Brief overview of unusual DNA structures.
Although DNA usually exists in the form of a right-handed double helix (B form), alternative DNA structures can occasionally form. They include cruciforms, H-DNA (triplex), G quartets, Z-DNA, and S-DNA (slipped-strand DNA) (Fig. 7). The ability of a sequence to undergo structural transition depends on its symmetry, base composition, DNA supercoiling, and ambient conditions, etc. (269). To undergo structural transitions, DNA sequences must possess some sort of symmetry or structural regularity. Based on sequence arrangement and symmetry, three major types of simple DNA repeats are usually considered: inverted repeats (IRs), mirror repeats (MRs), and direct tandem repeats (DTRs) (Fig. 7). IRs are DNA sequences in which DNA bases that are equidistant from the symmetry center in a DNA strand are Watson-Crick (WC) complements to each other. MRs are also symmetrical, but here, equidistant DNA bases are identical to each other. Finally, DTRs are simple, noninterrupted iterations of a core repeat unit along the DNA strand. The distinction between different repeat types is not absolute. For example, the d(AT)n/d(TA)n sequence can be looked at as an IR, MR, or DTR.
FIG. 7.

Unusual DNA structures and the types of repeats that form them. Structures that can be formed in double-stranded DNA (left) and the types of repetitive sequences that can form them (right) are shown. R, purines; Y, pyrimidines. Identical repetitive units are in black, and the cDNA strands are in white.
IRs are capable of forming cruciform structures in double-stranded DNA or hairpins in single-stranded DNA (reviewed in reference 160). Homopurine-homopyrimidine MRs have been shown to adopt an intramolecular triple-helical DNA conformation called H-DNA (reviewed in reference 203). Depending on the chemical nature of the strand donated to the triplex, pyrimidine or purine, the resultant structures are called H-y or H-r, respectively. The H-y form is favorable under acidic pH whereas the H-r form is stable at physiological pH in the presence of bivalent cations. DTRs can adopt a variety of conformations depending on their base composition. A structure called a G quartet, or quadruplex, can be formed by DTRs containing tandemly arranged runs of guanines (reviewed in reference 78). It is built from stacked G-4 blocks that are additionally stabilized in the presence of monovalent ions (301). DTRs consisting of regularly alternating purines and pyrimidines can flip into a left-handed Z-DNA conformation (reviewed in reference 249). Finally, DTRs of various base compositions can adopt a conformation called slipped-strand DNA (S-DNA) (reviewed in reference 269): upon denaturing and renaturing, the complementary repeats can mispair, resulting in a peculiar combination of double-helical stretches intervened by single-stranded loops. For repeated units of certain base compositions, the loops can be additionally stabilized by hydrogen bonds of both WC and non-WC nature (230).
Natural DNAs, particularly in eukaryotes, are greatly enriched in simple DNA repeats (44, 262). During the last decade, these repeats attracted very broad attention owing to two major scientific developments. First, it was discovered that more than two dozen human hereditary disorders are caused by progressive expansions of simple microsatellites (see below) (229, 298). Second, a drastic increase in the length polymorphism of mono- and dinucleotide repeats was observed in certain human cancers, such as hereditary nonpolyposis colorectal cancer (207) (although different from the “expansion diseases,” the microsatellite instability in this case is likely the consequence, not the cause, of the disease's progression). Consequently, the replication and stability of various DNA repeats have attracted very broad attention, revitalizing interest in unusual DNA structures.
Inhibition of replication by unusual DNA structures.
There is a growing body of evidence of replication inhibition due to the formation of hairpins, triplexes, and G quartets both in vitro (with both single-stranded and double-stranded templates) and in vivo. In vitro, a variety of DNA polymerases have been shown to be inhibited by IRs (most likely owing to the formation of hairpins) (10, 34, 107, 119, 267, 295), G-quartet-forming repeats in the presence of potassium ions (strongly indicating G-quartet formation) (285, 296, 302), and homopurine-homopyrimidine MRs (probably owing to the formation of triplexes) (8, 50, 139, 153, 196, 228, 254). In the latter case, it turned out that the mechanisms for replication inhibition by triplex-forming sequences were different for the single-stranded and double-stranded templates. In the single-stranded DNA templates, DNA polymerization was shown to stop in the middle of H motifs, presumably because when the newly synthesized DNA chain reached the center of a repeat, its remaining single-stranded segment folded back, forming a triplex behind the polymerase and in turn trapping the latter (Fig. 8A) (8, 153). DNA polymerization through H motifs in double-stranded nicked circular DNAs (designed in such a way that the DNA supercoiling would be irrelevant) progressed smoothly when the purine strand served as a template and the pyrimidine strand was displaced. In contrast, when the pyrimidine strand served as a template and the purine strand was displaced, DNA polymerization was completely blocked at the middle of an H motif (Fig. 8B) (254). The proposed mechanism was that during DNA synthesis, DNA polymerase ran into the H motif, displacing its nontemplate DNA strand; the displaced homopurine strand could then fold back, forming a stable triplex downstream from the polymerase that in turn blocked further polymerization. If the displaced strand was homopyrimidine, a stable triplex was not formed, since cytosines were not protonated, and polymerization proceeded normally.
FIG. 8.

Two models of triplex-caused DNA polymerization arrest in vitro. (A) On a single-stranded template, a triplex forms behind the polymerase. The template strand folds back on the newly synthesized strand. (B) On a double-stranded (nicked circular) template, a triplex forms in front of the polymerase. The nontemplate strand folds on the duplex in front when displaced by DNA synthesis. Arrows indicate the direction of DNA synthesis. A triplex is formed within a homopurine/homopyrimidine mirror repeat; the pyrimidine strand is white, and the purine strand is black. Note that a pyrimidine triplex is shown in panel A, whereas a purine triplex is shown in panel B, according to the cited studies (see the text).
Replication inhibition due to the formation of unusual DNA structures in vivo involves the following paradox. Although unusual structures can easily form without much constraint in single-stranded DNA, they are not energetically favorable (or, better put, they are less energetically favorable than the continuous double helix) in double-stranded DNA; thus, they should not be stable unless there is a substantially high degree of negative supercoiling that facilitates DNA unwinding. This is not the case for eukaryotic genomes, and even though the level of negative superhelicity of bacterial DNAs could support formation of unusual DNA structures, those structures should no longer be favorable upon approach of the replication fork owing to the fact that the replication fork generates a high degree of positive superhelicity in front of itself by unwinding the two strands of the duplex (237). Thus, encounter of the replication fork with preformed unusual DNA structures seems unlikely in vivo. However, in the course of replication, portions of the lagging-strand template transiently become single stranded, providing a window of opportunity for the formation of unusual structures such as perfect or imperfect hairpins, triplexes, and G quartets. These structures could in turn interfere with the progression of the lagging-strand polymerase, ultimately leading to blockage of the whole fork since the two polymerases are linked. To account for the fact that formation of the structures on the lagging-strand template that block progression of the replication fork is triggered by the fork itself, the term suicidal sequences was proposed (254). Preferential deletion of repeats on the lagging strand was observed, supporting the formation of these structures on the lagging-strand template (282).
In vivo, replication inhibition due to the formation of unusual DNA structures was first proposed for the (GA)n/(TC)n repeat (244, 245) and the GA-rich repeat from the hamster dhfr gene (23) in mammalian cells. Later on, this issue was mostly studied for trinucleotide repeats that are involved in a variety of expansion diseases (see below). Note that there is not a single instance wherein all alternative explanations, such as involvement of a protein's binding in the replication blockage by a structure-forming DNA sequence, have been ruled out.
The best-studied trinucleotide repeats are (CAG)n/(CTG)n, (CGG)n/(CCG)n, and (GAA)n/(TTC)n. Early studies of expandable repeats suggested their unusual structural potential (133, 205, 230, 231, 238, 307, 308). Single-stranded d(CGG)n, d(CCG)n, d(CTG)n, and d(CAG)n stretches can fold into hairpin-like structures stabilized by both WC and non-WC base pairs (36, 72, 180, 311). Hairpins formed by the above four repeats differ in the nature of non-WC base pairs. Thus, their stability varies owing to a differential contribution from different mismatches in the following way: CGG > CCG ≈ CTG > CAG (72). Recently, formation of folded structures by extended d(GAA)n and d(TTC)n repeats has also been postulated (88). Individual strands of expandable repeats can fold into other DNA conformations as well. For example, single-stranded d(CGG)n repeats fold into a stable quadruplex structure (283). The (GAA)n/(TTC)n repeat can form triple-helical H-DNA. Different labs proposed H-y (73), H-r (253), or a composite triplex structure called sticky DNA (253, 287). Finally, an AT-rich repeat implicated in SCA10, (ATTCT)n/(AGAAT)n, apparently forms a peculiar unwound structure while under superhelical stress (241). Formation of unusual structures by expandable repeats was shown to stall DNA polymerization in vitro (73, 122, 285), and there is a growing body of evidence showing that the same is true in vivo.
The (CAG)n/(CTG)n and (CGG)n/(CCG)n repeats were shown to inhibit the replication fork in E. coli (255), S. cerevisiae (234), and mammalian cells (I. Voineagu, unpublished observations), presumably due to the formation of imperfect hairpins [and/or G quartets in the case of (CGG)n/(CCG)n]. In both prokaryotes and eukaryotes, repeat-caused replication inhibition was length dependent. Although all of the systems produced qualitatively similar results, substantial quantitative differences were observed between different cell types. For example, a relatively short (CGG)10/(CCG)10 repeat inhibited DNA replication in yeast whereas a four-times-longer repeat was required for similar inhibition in bacterial and mammalian cells. One possible explanation could be that for the very AT-rich S. cerevisiae genome, GC-rich sequences such as the (CGG)n/(CCG)n repeat are extremely foreign. Furthermore, replication stalling by the (CGG)n/(CCG)n repeat in yeast did not depend on its orientation in the replicon (234), which is different from the orientation-dependent replication blockage in bacteria (255) and mammals (I. Voineagu, unpublished). The (GAA)n/(TTC)n repeat was found to inhibit replication in S. cerevisiae in an orientation-dependent manner, specifically when the repeat's homopurine strand serves as the lagging-strand template, strongly implicating the formation of a triplex (140). There are indications that the (GAA)n/(TTC)n repeat also inhibits replication in mammalian cells (221; also M. Krasilnikova, unpublished observations). The strength of inhibition in all systems depended on the repeat's base composition in the following order: (CGG)n/(CCG)n > (GAA)n/(TTC)n > (CAG)n/(CTG)n, correlating with the repeat's propensity to form unusual DNA structures.
Studies of replication inhibition by trinucleotide repeats are of special importance since it is most likely involved in the mechanism of the repeats' expansion—a phenomenon that causes a number of human diseases (200; also see below).
Replication Slow Zones in Budding Yeast
Analysis of Mec1 (yeast homologue of mammalian ATR)-depleted yeast revealed chromosomal breakage at positions that corresponded to simultaneously discovered replication slow zones (RSZs)—novel chromosomal determinants regularly positioned between origins of replication (33). Importantly, whereas chromosomal breakage was evident only in Mec1-deficient cells, the RSZs were also evident in wild-type yeast (yet they did not lead to breakage if Mec1 was active). Chromosomal breakage that resulted from replication stalling in the absence of Mec1 was not a consequence of fork collapse, as the forks were paused in the normal physiological state. Instead, it resulted from some other unidentified factors (one discussed possibility was mitotic spindle forces). Replication stalling (and consequent chromosomal breakage) in the mec1 mutant was suppressed by the inactivation of Sml1 (an inhibitor of the ribonucleotide reductase Rnr1), suggesting that the mechanism by which replication was made slow in the slow zones was the lack of dNTPs. The lack of dNTPs alone, however, cannot explain pausing of replication at specific positions in the genome, since RSZs neither had a nonrandom base composition nor were simple consequences of meeting of the two converging forks. Therefore, the RSZs were proposed to be genetically programmed and probably play an as-yet-unknown role in chromosomal metabolism. It is also not known what the determinant(s) of the RSZs is. There is a clear analogy between RSZs in yeast and common fragile sites in mammals (278; also see below). This analogy is extended by the observation that ATR deficiency in mammalian cells results in expression of common fragile sites with and, more importantly, even without replication inhibitors (32).
GENOMIC INSTABILITY CAUSED BY REPLICATION STALLING AT NATURAL IMPEDIMENTS
Replication stalling that requires fork restart and/or activation of checkpoint mechanisms in the absence of any external damage is a common event in both bacteria (43, 176, 179, 258) and eukaryotes (32, 33, 211). It could result from either endogenous DNA damage (e.g., oxidation) or replication barriers.
In eukaryotes, replication forks are stabilized in the competent state and protected from the regression and subsequent restart pathways, which could lead to genomic instability. In budding yeast, Mrc1 and Tof1 stabilize stalled forks (40, 123, 169), probably through their interaction with the Cdc45-MCM2-7 complex (213). Mrc1 and Tof1 also activate the intra-S-phase checkpoint kinases Mec1 (homologue of mammalian ATR) and Rad53 (homologue of mammalian CHK2) (2, 66, 123). Activation of the intra-S-phase checkpoint helps further stabilize the forks and finish replication of the genome by allowing conserved forks to wait until replication conditions improve without disassembling (33, 57, 149, 161, 272, 280). In the absence of the intra-S-phase checkpoint, spontaneous accumulations of double-stranded breaks and chromosomal rearrangements occur without external DNA damage, which suggests that DNA replication per se is the source of genomic instability (32, 33, 211).
In fission yeast, genome-wide fork stabilization is carried out by Swi1 (homologue of Tof1), Cds1 (homologue of Rad53), and Rad3 (homologue of Mec1) (217).
In bacteria, stabilization of stalled forks has not been detected. Unlike eukaryotes, in which there is a chance that if one fork stalls, another fork from the opposite direction will reach the same place in a reasonable time, there is no such chance in bacteria (because of the large size of the replicon and terminators that prevent or at least enormously delay the fork's escape from the terminus; see above). Since replication timing is the rate-limiting step in the bacterial cell cycle, it seems beneficial to disassemble and restart stalled forks right away instead of passively waiting (although “infinite” replication stalling, which resulted in unviability of cells, has been reported [239]; see above).
It is interesting to consider whether the cell can distinguish between a temporarily stalled replication fork (which is worth preserving in the anticipation of removal of the factor that caused the stalling) versus an indefinitely stalled one (which is worth disassembling). The concept of “gratuitous” transcription-coupled repair was proposed to predict recruitment of transcription-coupled repair machinery to the temporarily stalled RNA polymerase (84, 85). It assumes that it is nearly impossible to distinguish between a temporarily stalled RNA polymerase and an RNA polymerase indefinitely stalled at a DNA lesion. The challenge of distinguishing between a temporarily stalled replication fork and a terminally stalled fork seems to be a very similar task. Unless a special mechanism is developed to distinguish between the two scenarios, any stalling site could potentially become a “decision-making” site. It is not clear how the decision is made.
Temporarily paused forks do not pose any danger to the genome's integrity. A permanently stalled fork can be processed in several ways. Figure 9 schematically shows several pathways for restarting a replication fork halted at a corrupted template. It represents the authors' compilation of a large number of studies, too numerous to cite in their entirety.
FIG. 9.

Replication restart. A schematic representation of pathways that act to restart a replication fork, stalled at a “corrupted” template, is shown. See the text for details. *, reversal and rewinding of the fork are most likely assisted by proteins; DNA supercoiling may also contribute to both processes. **, generally, two models that explain replication restart by homologous recombination exist; they differ in the sequence of events. (i) Formation of a D loop (the short arm of the four-way junction invades the parental duplex downstream of the junction) and reestablishment of the replication fork (i.e., reloading of the polymerases and other fork components and resumption of DNA synthesis) are followed by cleavage. (ii) Cleavage (which releases a full chromosomal arm) is followed by the formation of a D loop and reestablishment of the replication fork.
In a replication fork halted at a lesion, both lagging and, as recently became evident, leading DNA polymerase can “jump” (i.e., be reloaded onto DNA) after repriming downstream of a lesion, leaving behind a gap (89). Gaps can later be filled in by translesion polymerases or initiate homologous recombination (170). Translesion polymerases can temporarily replace the normal replicative polymerase when the fork stalls (110). Regression of the fork and formation of the so-called chicken foot structure (96, 240) can allow the lesion to be repaired. Alternatively, it was proposed that the lesion can be bypassed by the template switch, a mechanism that temporarily uses one of the newly synthesized strands as a template (184). Upon bypass, the two nascent strands can rewind back together to reestablish the fork's structure (80). Homologous recombination (accompanied by cleavage of DNA) can be involved in restart (43, 92, 137, 179, 185, 266). Cleavage of the four-way junction can release a chromosomal arm, which would require homologous recombination to form a D loop and reestablish the fork. Alternatively, homologous recombination can occur first between the nascent arm and the parental arm, which would lead to formation of the D loop and reestablishment of the fork (reloading of the fork proteins) and subsequently would require a cleavage to reestablish the normal forked DNA structure.
While the switch to translesion polymerases could be error prone, owing to their low fidelity, the other pathways are generally error free. Importantly, however, restart by homologous recombination can lead to chromosomal rearrangements if recombination occurs between ectopic rather than allelic homologous sequences. Therefore, as it was beautifully put by Lambert et al., “Recombination is a ‘double-edged sword,’ preventing cell death when the replisome disassembles at the expense of genetic stability” (150). The fork reversal-template switch pathways have also been implicated in instability of repetitive sequences (80, 128, 140, 202). The fact that virtually all of the pathways for replication restart can, under certain circumstances, result in DNA rearrangements makes replication stalling a powerful source of genomic instability (42, 43, 195, 204, 255). A growing number of studies show that replication stalling at natural impediments can be a source of genomic instability, such as chromosomal rearrangements, chromosomal fragility, and instability of various repetitive sequences.
Genomic Instability Caused by Protein Binding
A direct link between replication stalling and recombination-caused chromosomal rearrangements was observed in fission yeast when an inducible replication stall site was introduced into the S. pombe genome (150). The RTS1 sequence (polar replication termination site located near the imprinting site in the mating-type locus mat1; see above) was inserted on both sides of the ura4 gene in an inverted orientation. This made the total number of RTS1 sequences in the genome three: one on each side of the ura4 gene on chromosome II and one in the mat1 locus on chromosome III. Swi1- and Rtf1-dependent replication stalling (see above) was observed at these ectopic RTS1 sequences. Induction of either Swi1 or Rtf1 expression resulted in replication stalling and an elevated level of recombination events. Intrachromosomal recombination between the two RTS1 sequences that flanked the ura4 gene resulted in ura4 inversions. Ectopic recombination between one of the RTS1 sequences around the ura4 gene and the RTS1 sequence in the mat1 locus led to either noncrossover gene conversions or chromosomal translocations. The correlation between the induction of the proteins required for replication stalling, the accumulation of stalled forks, and the increase in the recombination frequencies strongly suggests that genomic instability is caused by replication stalling at a protein-caused natural impediment. Interestingly, replication stalling at the very same Rts1 sequence in the mat1 locus does not induce recombination (as judged by the absence of association with recombination proteins compared with the presence of such association in the case of the ectopic RTS1). This might be an illustration of decision making at the stalled replication fork. Fast recruitment of recombination proteins to the ectopic RTS1 sequences argues in favor of the absence of fork conservation there. However, Swi1 acts on stalled forks at the RTS1 sequence at both mat1 and ectopic loci, indicating that the difference should arise from some other fork-associated protein.
Another example of genomic instability caused by protein-mediated replication arrest came from a study that showed that Ter sites in E. coli plasmids were deletion hotspots (15). Although it was not clear exactly how these deletions occurred, mutational analysis indicated the involvement of topoisomerase I and recD (14). On the other hand, Ter sites are also hotspots for homologous recombination in plasmids (105) as well as the chromosome (104). Therefore, deletions could result from an aberrant recombinational repair of collapsed replication forks. However, it was suggested that the forks, arrested at the ectopic Ter site in the E. coli chromosome, are not broken; instead, recombination is initiated when the second round of replication arrives at the site of arrest (13).
Genomic Instability Caused by Transcription
Transcription plays a major role in genomic stability. On the one hand, it promotes repair of the template DNA strand by recruiting the transcription-coupled repair system (193). On the other hand, it causes increased frequency of mutations of the nontemplate DNA strand, a phenomenon called transcription-associated mutation (TAM). Finally, transcription stimulates recombination, and this phenomenon is called transcription-associated recombination (TAR). TAR between ectopic homologous sequences leads to genomic instability (for example, see references 275 and 281). TAM and TAR are reviewed in reference 1. TAM most likely occurs because during transcription, the nontemplate DNA strand transiently becomes single stranded and thus more susceptible to external and internal damage. The mechanism(s) of TAR is much less clear. Among proposed explanations of TAR are the “opening” of chromatin, generation of supercoiling in DNA, formation of unusual DNA structures, appearance of single-stranded DNA regions, and formation of RNA-DNA hybrids. In fact, the opening of chromatin was shown to be the major mechanism of TAR in several systems: mating-type switch (226) and meiotic recombination (304) in budding yeast and site-specific V(D)J recombination of immunoglobulin genes (190, 261, 306). In a recent study of budding yeast, TAR (scored as formation of deletions between two direct repeats) was observed only when simultaneous replication and transcription occurred. Moreover, TAR was dependent on the relative direction of transcription and replication, being evident only when the transcription unit was facing replication head-on, i.e., when detectable replication stalling was observed on two-dimensional gels. These data raise a possibility that transcription-replication collisions (replication stalling and subsequent restart via recombination) can be another reason for TAR (242).
Transcription was also shown to cause illegitimate recombination and formation of deletions in E. coli plasmids, in an orientation-dependent manner, when a nick was introduced into the plasmid. This nick served as one end point of deletions while various positions in the transcribed region served as the other end points. The proposed explanation was that replication stalling in the transcribed area was the source of a free DNA end that was joined to the nick (289).
Another example of transcription-replication collisions and genomic instability came from the analysis of Fob1-deficient budding yeast. The Fob1 protein serves as a barrier in the ribosomal locus of S. cerevisiae and prevents replication forks from entering transcription units in the head-on orientation (see above). The ribosomal locus of S. cerevisiae possesses remarkable instability: a hyper-recombination activity, termed HOT1, is responsible for the appearance of extrachromosomal ribosomal circles (ERCs), the hallmark of aging in yeast. Furthermore, expansions and contractions of the rRNA gene array can be observed under certain conditions. In the absence of Fob1, the amount of expansions, contractions, and ERCs is reduced and the yeast's life span is concurrently increased (51, 130). It was first suggested that the main source of rRNA locus instability and ERC formation was an aberrant repair of replication forks stalled at the Fob1 barrier, i.e., that HOT1 is a consequence of replication blocking. It turned out, however, that while the hyper-recombination activity of HOT1 was dependent on Fob1, it was independent of replication, implying that Fob1 has another recombination-associated function (26, 294). In the absence of Fob1, head-on transcription-replication collisions are permitted, so why did they not lead to rRNA instability? The answer is that the ribosomal genes are multicopy, and the fraction of simultaneously replicated and transcribed genes is low. When the number of rRNA repeats was reduced in the Fob1 knockout strain (to increase the amount of simultaneously replicated and transcribed rRNA gene copies), replication stalling caused by the head-on collisions with transcription became evident and an increased amount of extrachromosomal circles was detected (279). The appearance of extrachromosomal circles in the yeast strain that lacks Fob1 and carries a decreased amount of rRNA gene copies proves that head-on transcription-replication collisions are a source of genomic instability in eukaryotes.
Altogether, replication stalling and restart upon head-on collisions with the transcription machinery are a risk factor for genomic instability. Consequently, as described above, transcription-replication interplay shapes the organization of genomes that are simultaneously replicated and transcribed: a head-on orientation of transcription units relative to the direction of replication is avoided, especially for essential genes.
Genomic Instability Caused by Unusual DNA Structures
Replication stalling at unusual structures in the DNA (see above) was connected to the instability of various repeats that form these structures. Most studies were done of the repeats whose expansions cause human diseases. More than two dozen human hereditary disorders are now attributed to expansions of simple DNA repeats (298); for the most up-to-date list of the repeat-caused diseases, see reference 229. Trinucleotide repeats, specifically (CGG)n/(CCG)n, (CTG)n/(CAG)n, and (GAA)n/(TTC)n, account for the majority of these diseases; thus, they attract the most interest. Expansion of trinucleotide repeats causes such debilitating diseases as fragile X mental retardation (143, 286), Huntington's disease (108), myotonic dystrophy (25, 175), Friedreich's ataxia (30), and many others. It has now become clear, however, that expansions are not limited to triplet repeats. Tetrameric (163), pentameric (183), dodecameric (148), and other repeats can undergo expansions as well. Besides microsatellites, minisatellites appear to be able to undergo expansions/contractions as well (11, 212, 309). Expandable repeats vary in sequence, are situated in different genes, and affect gene expression in various ways (229, 298). Still, there is one underlying similarity: repeats are stably inherited and cause no harm until the total length of a repetitive stretch exceeds approximately 100 bp. For trinucleotide repeats, this number translates into roughly 30 repeats. If this threshold length is exceeded, repeats start expanding during intergenerational transmissions. A remarkable feature of expansions is that the longer a repeat is, the more likely its subsequent expansion will be. In a few generations, this results in the addition of thousands of repeats, disruption of gene(s) expression, and disease (3). This mode of repeat expansions is responsible for the long-established anticipation in the inheritance of associated diseases, i.e., an increase in the probability, onset, and severity of a disease as it passes through generations.
Repeat expansions should not be confused with a different phenomenon, called microsatellite instability (MIN), characteristic of mono- and dinucleotide repeats. In normal human cells, the number of such repeats within a given gene is maintained with high accuracy. In several hereditary and sporadic human tumors, however, a 1,000-fold increase in length polymorphism was observed for these microsatellites (38, 111, 236). Unlike expandable repeats, repeat length in MIN changes in small increments. MIN is caused by mutations in mismatch repair genes (24, 65, 154, 215, 225, 227). Deficiency in mismatch repair makes it difficult to correct small insertions and deletions that accumulate upon slippage of the template and daughter DNA strands within a repeat (146).
The mechanism of large-scale repeat expansions involved in human diseases is not known. However, several facts make it tempting to speculate that anomalous DNA replication of the repeats could be responsible for their expansions in human diseases. These facts are as follows. (i) The propensity for expansions correlates with the ability of repeats to form unusual secondary structures (188). (ii) Formation of these structures by expandable repeats has been shown to stall DNA polymerization in vitro and in vivo (see above). (iii) There is a massive accumulation of repeated DNA in the process of expansions. (iv) In model systems, replication is clearly involved in repeat expansion, since the stability of trinucleotide repeats in bacterial, yeast, and cultured mammalian cells appears to depend on their orientation relative to replication origins (6, 39, 70, 121, 197, 224, 268), and mutations in the replication apparatus of bacteria and yeast increased instability of trinucleotide repeats (12, 69, 112, 117, 128, 234, 264, 265, 274, 299). (v) Normal alleles of repeat-containing genes in humans contain multiple interruptions that disappear in expandable alleles, resulting in homogenous repetitive runs (147) [in normal FMR1 alleles, for example, (CGG)n runs are interrupted by AGG triplets; remarkably, these AGG interruptions abolished the replication blockage in bacteria (255)]. (vi) The expansion threshold in humans and replication inhibition thresholds in the model systems generally are the same for bacteria (255), yeast (140, 234), and mammalian cells (I. Voineagu, unpublished results). (vii) This threshold is usually around 30 to 40 uninterrupted units (297), which roughly corresponds to the size of the eukaryotic Okazaki fragments (55, 56).
Indeed, among the variety of models of repeat expansions (reviewed in reference 201), replication models were the first to emerge (121, 189). They implicate both lagging (17, 81, 90, 189, 284) and leading (87, 118) strands.
One of the current models suggests that repeat expansions and contractions occur during leading-strand synthesis after replication fork stalling and restart within the repetitive run (Fig. 10) (140, 202). Specifically, when the leading DNA polymerase runs into an expandable repeat, the Okazaki initiation zone on the lagging-strand template becomes repetitive; thus, it can fold into a stable conformation (one of the unusual DNA structures described above). This structure would inhibit progression of the lagging DNA polymerase, resulting in replication fork stalling and dissociation. For replication to restart, some accessory proteins should unwind the lagging-strand template. Meanwhile, the newly synthesized leading strand and its template could misalign within the repeat. Such misalignment can obviously generate repetitive slip-outs on either the template or nascent DNA strand. Although spontaneous misalignment between the leading DNA strand and its template should be energetically unfavorable and unstable, it could happen in the following way. Collapse of the stalled replication fork commonly leads to its reversal (96, 240). Reversal of a fork stalled within an expandable repeat would lead to the formation of a four-way junction with the single-stranded repetitive extension at the 3′ end of the leading strand. Such a single-stranded repeat would tend to acquire a secondary structure. To restart replication, the reversed fork should be flipped back by the eukaryotic homologues of RuvAB(C) or RecG. The structure that has formed on the leading strand could still be intact. Resumption of DNA synthesis would then lead to repeat expansions. The model suggests that although the initial block occurs during lagging-strand synthesis, repeat expansions occur on the leading strand. Replication stalling at unusual structures formed by the expandable repeats is paramount for their instability and most likely their involvement in the generation of a variety of the expansion diseases.
FIG. 10.

Replication model of trinucleotide repeat expansion. (A) Replication stalling caused by the formation of an unusual structure on the lagging-strand template. (B) Fork reversal (possibly assisted by proteins). The 3′ end of the nascent leading strand is single stranded; thus, it tends to fold into a hairpin-like structure. (C) The fork rewinds back to restart (again, assisted by proteins not shown), but the 3′ end of the nascent leading strand is still structured, potentially leading to the repeat's expansion. Ovals, two DNA polymerases in the replication fork; arrows, direction of DNA synthesis; black, structure-prone strand of the repetitive tract; white, complementary strand.
Chromosomal Fragility
Chromosomal fragility is a cytogenetic term used to describe human metaphase chromosomes with gaps or breaks that occur under conditions of replication stress (reviewed in references 68, 79, 263, and 278). The fragile sites are not random and are classified as common, i.e., present in all individuals, and rare, i.e., present in fewer than 5% of the population (see below). “Expression” of a fragile site is the frequency with which it can be observed on metaphase spreads. Expression of fragile sites is induced by various (sometimes specific for a particular site or group of sites) inhibitors of replication (for example, aphidicolin, folate, thymidylate). This provides strong evidence that inhibition of replication can be a source of chromosomal fragility. Needless to say, the consequences of chromosome fragility (and subsequent loss of a chromosomal arm or translocation with another chromosome) can be the most devastating.
Cytologically observed chromosomal fragility could mean two things at the molecular level: either chromatin is decondensed and therefore is not observed by staining or the chromosome is really broken. Chromosome breakage can be a consequence of two events: when the cell enters mitosis having in its genome either unreplicated regions or unhealed double-strand breaks. Unreplicated regions can break under the tension generated by spindle forces, when the two sister chromatids are pulled to the opposite poles. Alternatively, an unreplicated region in the parental chromosome can become single-stranded regions (gaps) in the daughter chromosomes, and these gaps can be converted into double-stranded breaks by nucleases. A double-strand break in DNA makes a chromosome discontinous; therefore, the two arms are no longer held together and can segregate to different daughter cells. The sources of double-strand breaks can be (i) processing of stalled replication forks by Holliday junction resolvase or by nucleases, mechanical forces, etc., and (ii) processing of single-strand breaks or gaps (which can result from DNA damage or can sometimes be left behind the replication fork) by replication, nucleases, and mechanical forces, etc.
Obviously, cells possess checkpoint mechanisms that prevent progression of the cell cycle in both cases (unreplicated regions or unhealed double-stranded breaks); however, the fact that chromosomal fragility is observed points to the fact that these mechanisms are not absolute. For example, in humans, ATR checkpoint kinase prevents the expression of common fragile sites (32). When ATR is inhibited, expression of common fragile sites increases 5- to 20-fold and, amazingly, becomes prominent even in the absence of replication inhibitors. These data point out the importance of replication checkpoints for the prevention of chromosomal fragility and argue that unreplicated regions in the genome likely cause chromosomal fragility. Also supporting this is the finding that fragile sites replicate late in the normal S phase. As discussed above, RSZs—a phenomenon recently discovered in budding yeast—resemble features of common fragile sites in humans (33; also see above). Just as expression of human fragile sites is stimulated in the absence of ATR, chromosome breakage at RSZs is evident in the absence of Mec1 (the yeast homologue of ATR), although slow replication is evident even in the presence of Mec1. Unlike human fragile sites, though, there is no AT-rich bias in the RSZs.
Rare fragile sites are caused by expansions of repetitive elements such as di- and trinucleotide microsatellites and some AT-rich minisatellites beyond the normal length. These expansions are abnormal and can also cause various diseases. For example, the first rare fragile site was discovered in patients with fragile X mental retardation caused by expanded (CGG)n/(CCG)n repeats in the 5′ untranslated region of the FMR gene. The expanded (CGG)n/(CCG)n repeats are capable of the formation of secondary structures, such as hairpins and G quartets, that stall replication (see above). This replication inhibition, however, might not be the only cause of chromosomal fragility. Instead, DNA methylation and the subsequent heterochromatinization of a large region of the DNA around the expanded repeat that results in delayed replication of hundreds of kilobases likely contributes to the fragility observed in cells derived from patients (86, 277).
There are at least 80 common fragile sites in the human genome [20 of the most prominent include 3p14.2 (FRA3B), 16q23 (FRA16D), 6q26 (FRA6E), 7q32.3 (FRA7H), and Xp22.3 (FRAXB)]. They are present in all individuals; therefore, they are normal components of chromosomes. Common fragile sites do not have di- or trinucleotide repeats. They are hundreds of kilobases of relatively AT-rich DNA. They are proposed to be highly flexible (on the DNA level) and to be prone to formation of secondary structures such as multiple hairpins. The reason for the replication inhibition that leaves behind unreplicated and/or single-stranded regions at common fragile sites is unknown. It might be unusual chromatin structure, base composition, or formation of secondary structures that are obstacles for the replication fork.
CONCLUSION
Replication stalling at natural impediments is a universal phenomenon observed in a variety of prokaryotic and eukaryotic genomes. Over the last decade, enormous progress has been made in identifying both the precedents of such stalling and their consequences. It appears that replication stalling at natural impediments can occur either on purpose or accidentally (if inhibition of replication is a side effect of another biological process). A growing body of evidence indicates that a consequence of replication stalling at natural impediments could be genomic instability, such as chromosomal rearrangements, chromosomal fragility, and the instability of various repetitive sequences, drawing additional attention to this issue. We believe that further investigation of replication stalling at natural impediments might provide invaluable insights into DNA replication, fork restart, checkpoint control, fork conservation, and maintenance of genomic stability. It should also answer one of the most intriguing questions in the field: what determines the fate of replication forks stalled at different barriers and how that fate is decided on a case-by-case basis.
Acknowledgments
We thank Lucia Rothman-Denes for valuable advice and support, Carl Schildkraut for sharing his expertise on EBV replication, and anonymous reviewers for constructive suggestions.
This work was supported by NIH grant GM60987 to S.M.M.
REFERENCES
- 1.Aguilera, A. 2002. The connection between transcription and genomic instability. EMBO J. 21:195-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alcasabas, A. A., A. J. Osborn, J. Bachant, F. Hu, P. J. Werler, K. Bousset, K. Furuya, J. F. Diffley, A. M. Carr, and S. J. Elledge. 2001. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 3:958-965. [DOI] [PubMed] [Google Scholar]
- 3.Ashley, C., Jr., and S. T. Warren. 1995. Trinucleotide repeat expansion and human disease. Annu. Rev. Genet. 29:703-728. [DOI] [PubMed] [Google Scholar]
- 4.Autret, S., A. Levine, F. Vannier, Y. Fujita, and S. J. Seror. 1999. The replication checkpoint control in Bacillus subtilis: identification of a novel RTP-binding sequence essential for the replication fork arrest after induction of the stringent response. Mol. Microbiol. 31:1665-1679. [DOI] [PubMed] [Google Scholar]
- 5.Baker, T. A. 1995. Replication arrest. Cell 80:521-524. [DOI] [PubMed] [Google Scholar]
- 6.Balakumaran, B. S., C. H. Freudenreich, and V. A. Zakian. 2000. CGG/CCG repeats exhibit orientation-dependent instability and orientation-independent fragility in Saccharomyces cerevisiae. Hum. Mol. Genet. 9:93-100. [DOI] [PubMed] [Google Scholar]
- 7.Baldacci, G., and G. Bernardi. 1982. Replication origins are associated with transcription initiation sequences in the mitochondrial genome of yeast. EMBO J. 1:987-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baran, N., A. Lapidot, and H. Manor. 1991. Formation of DNA triplexes accounts for arrests of DNA synthesis at d(TC)n and d(GA)n tracts. Proc. Natl. Acad. Sci. USA 88:507-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baudat, F., and A. Nicolas. 1997. Clustering of meiotic double-strand breaks on yeast chromosome III. Proc. Natl. Acad. Sci. USA 94:5213-5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bedinger, P., M. Munn, and B. M. Alberts. 1989. Sequence-specific pausing during in vitro DNA replication on double stranded DNA templates. J. Biol. Chem. 264:16880-16886. [PubMed] [Google Scholar]
- 11.Bell, G. I., S. Horita, and J. H. Karam. 1984. A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes 33:176-183. [DOI] [PubMed] [Google Scholar]
- 12.Bhattacharyya, S., and R. Lahue. 2004. Saccharomyces cerevisiae Srs2 DNA helicase selectively blocks expansions of trinucleotide repeats. Mol. Cell. Biol. 24:7324-7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bidnenko, V., S. D. Ehrlich, and B. Michel. 2002. Replication fork collapse at replication terminator sequences. EMBO J. 21:3898-3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bierne, H., S. D. Ehrlich, and B. Michel. 1997. Deletions at stalled replication forks occur by two different pathways. EMBO J. 16:3332-3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bierne, H., S. D. Ehrlich, and B. Michel. 1991. The replication termination signal terB of the Escherichia coli chromosome is a deletion hot spot. EMBO J. 10:2699-2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blattner, F. R., G. Plunkett, III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 17.Bowater, R. P., and R. D. Wells. 2001. The intrinsically unstable life of DNA triplet repeats associated with human hereditary disorders. Prog. Nucleic Acid Res. Mol. Biol. 66:159-202. [DOI] [PubMed] [Google Scholar]
- 18.Bozhenok, L., P. A. Wade, and P. Varga-Weisz. 2002. WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J. 21:2231-2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brewer, B. J. 1988. When polymerases collide: replication and the transcriptional organization of the E. coli chromosome. Cell 53:679-686. [DOI] [PubMed] [Google Scholar]
- 20.Brewer, B. J., and W. L. Fangman. 1987. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51:463-471. [DOI] [PubMed] [Google Scholar]
- 21.Brewer, B. J., and W. L. Fangman. 1988. A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell 55:637-643. [DOI] [PubMed] [Google Scholar]
- 22.Brewer, B. J., D. Lockshon, and W. L. Fangman. 1992. The arrest of replication forks in the rDNA of yeast occurs independently of transcription. Cell 71:267-276. [DOI] [PubMed] [Google Scholar]
- 23.Brinton, B. T., M. S. Caddle, and N. H. Heintz. 1991. Position and orientation-dependent effects of a eukaryotic Z-triplex DNA motif on episomal DNA replication in COS-7 cells. J. Biol. Chem. 266:5153-5161. [PubMed] [Google Scholar]
- 24.Bronner, C. E., S. M. Baker, P. T. Morrison, G. Warren, L. G. Smith, M. K. Lescoe, M. Kane, C. Earabino, J. Lipford, A. Lindblom, P. Tannergard, R. J. Bollag, A. R. Godwin, D. C. Ward, M. Nordenskjold, R. Fishel, R. Kolodner, and R. M. Liskay. 1994. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 368:258-261. [DOI] [PubMed] [Google Scholar]
- 25.Brook, J. D., M. E. McCurrach, H. G. Harley, A. J. Buckler, D. Church, H. Aburatani, K. Hunter, V. P. Stanton, J. P. Thirion, T. Hudson, R. Sohn, B. Zemelman, R. G. Snell, S. A. Rundle, S. Crow, J. Davies, P. Shelbourne, J. Buxton, C. Jones, V. Juvonen, K. Johnson, P. S. Harper, D. J. Shaw, and D. E. Housman. 1992. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68:799-808. [DOI] [PubMed] [Google Scholar]
- 26.Burkhalter, M. D., and J. M. Sogo. 2004. rDNA enhancer affects replication initiation and mitotic recombination: Fob1 mediates nucleolytic processing independently of replication. Mol. Cell 15:409-421. [DOI] [PubMed] [Google Scholar]
- 27.Bussiere, D. E., and D. Bastia. 1999. Termination of DNA replication of bacterial and plasmid chromosomes. Mol. Microbiol. 31:1611-1618. [DOI] [PubMed] [Google Scholar]
- 28.Bussiere, D. E., D. Bastia, and S. W. White. 1995. Crystal structure of the replication terminator protein from B. subtilis at 2.6 A. Cell 80:651-660. [DOI] [PubMed] [Google Scholar]
- 29.Calzada, A., B. Hodgson, M. Kanemaki, A. Bueno, and K. Labib. 2005. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 19:1905-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campuzano, V., L. Montermini, M. D. Molto, L. Pianese, M. Cossee, F. Cavalcanti, E. Monros, F. Rodius, F. Duclos, A. Monticelli, F. Zara, J. Canizares, H. Koutnikova, S. I. Bidichandari, C. Gellera, A. Brice, P. Trouillas, G. De Michele, A. Filla, R. De Frutos, F. Palau, P. I. Patel, S. Di Donato, J.-L. Mandel, S. Cocozza, M. Koenig, and M. Pandolfo. 1996. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271:1423-1427. [DOI] [PubMed] [Google Scholar]
- 31.Cashel, M., D. R. Gentry, V. J. Hernandez, and D. Vinella. 1996. The stringent response, p. 1410-1438. In F. C. Neidhardt, J. L. Ingraham, K. B. Low, B. Magasanik, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella typhimurium: cellular and molecular biology, 2nd ed. American Society for Microbiology, Washington, DC.
- 32.Casper, A. M., P. Nghiem, M. F. Arlt, and T. W. Glover. 2002. ATR regulates fragile site stability. Cell 111:779-789. [DOI] [PubMed] [Google Scholar]
- 33.Cha, R. S., and N. Kleckner. 2002. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297:602-606. [DOI] [PubMed] [Google Scholar]
- 34.Chalberg, M. D., and P. T. Englund. 1979. The effect of template secondary structure on vaccinia DNA polymerase. J. Biol. Chem. 254:7820-7826. [PubMed] [Google Scholar]
- 35.Chang, D. D., and D. A. Clayton. 1985. Priming of human mitochondrial DNA replication occurs at the light-strand promoter. Proc. Natl. Acad. Sci. USA 82:351-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen, X., S. V. Mariappan, P. Catasti, R. Ratliff, R. K. Moyzis, A. Laayoun, S. S. Smith, E. M. Bradbury, and G. Gupta. 1995. Hairpins are formed by the single DNA strands of the fragile X triplet repeats: structure and biological implications. Proc. Natl. Acad. Sci. USA 92:5199-5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho, R. J., M. J. Campbell, E. A. Winzeler, L. Steinmetz, A. Conway, L. Wodicka, T. G. Wolfsberg, A. E. Gabrielian, D. Landsman, D. J. Lockhart, and R. W. Davis. 1998. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell 2:65-73. [DOI] [PubMed] [Google Scholar]
- 38.Claij, N., and H. te Riele. 1999. Microsatellite instability in human cancer: a prognostic marker for chemotherapy? Exp. Cell Res. 246:1-10. [DOI] [PubMed] [Google Scholar]
- 39.Cleary, J. D., K. Nichol, Y. H. Wang, and C. E. Pearson. 2002. Evidence of cis-acting factors in replication-mediated trinucleotide repeat instability in primate cells. Nat. Genet. 31:37-46. [DOI] [PubMed] [Google Scholar]
- 40.Cobb, J. A., L. Bjergbaek, K. Shimada, C. Frei, and S. M. Gasser. 2003. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 22:4325-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Codlin, S., and J. Z. Dalgaard. 2003. Complex mechanism of site-specific DNA replication termination in fission yeast. EMBO J. 22:3431-3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Courcelle, J., and P. C. Hanawalt. 2003. RecA-dependent recovery of arrested DNA replication forks. Annu. Rev. Genet. 37:611-646. [DOI] [PubMed] [Google Scholar]
- 43.Cox, M. M., M. F. Goodman, K. N. Kreuzer, D. J. Sherratt, S. J. Sandler, and K. J. Marians. 2000. The importance of repairing stalled replication forks. Nature 404:37-41. [DOI] [PubMed] [Google Scholar]
- 44.Cox, R., and S. M. Mirkin. 1997. Characteristic enrichment of DNA repeats in different genomes. Proc. Natl. Acad. Sci. USA 94:5237-5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dalgaard, J. Z., and A. J. Klar. 2001. A DNA replication-arrest site RTS1 regulates imprinting by determining the direction of replication at mat1 in S. pombe. Genes Dev. 15:2060-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dalgaard, J. Z., and A. J. Klar. 2001. Does S. pombe exploit the intrinsic asymmetry of DNA synthesis to imprint daughter cells for mating-type switching? Trends Genet. 17:153-157. [DOI] [PubMed] [Google Scholar]
- 47.Dalgaard, J. Z., and A. J. Klar. 1999. Orientation of DNA replication establishes mating-type switching pattern in S. pombe. Nature 400:181-184. [DOI] [PubMed] [Google Scholar]
- 48.Dalgaard, J. Z., and A. J. Klar. 2000. swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell 102:745-751. [DOI] [PubMed] [Google Scholar]
- 49.Dardalhon, M., B. de Massy, A. Nicolas, and D. Averbeck. 1998. Mitotic recombination and localized DNA double-strand breaks are induced after 8-methoxypsoralen and UVA irradiation in Saccharomyces cerevisiae. Curr. Genet. 34:30-42. [DOI] [PubMed] [Google Scholar]
- 50.Dayn, A., G. M. Samadashwily, and S. M. Mirkin. 1992. Intramolecular DNA triplexes: unusual sequence requirements and influence on DNA polymerization. Proc. Natl. Acad. Sci. USA 89:11406-11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Defossez, P. A., R. Prusty, M. Kaeberlein, S. J. Lin, P. Ferrigno, P. A. Silver, R. L. Keil, and L. Guarente. 1999. Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol. Cell 3:447-455. [DOI] [PubMed] [Google Scholar]
- 52.de Massy, B., S. Bâejar, J. Louarn, J. M. Louarn, and J. P. Bouchâe. 1987. Inhibition of replication forks exiting the terminus region of the Escherichia coli chromosome occurs at two loci separated by 5 min. Proc. Natl. Acad. Sci. USA 84:1759-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deng, S., R. A. Stein, and N. P. Higgins. 2005. Organization of supercoil domains and their reorganization by transcription. Mol. Microbiol. 57:1511-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng, S., R. A. Stein, and N. P. Higgins. 2004. Transcription-induced barriers to supercoil diffusion in the Salmonella typhimurium chromosome. Proc. Natl. Acad. Sci. USA 101:3398-3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DePamphilis, M. L. 2002. Eukaryotic DNA replication fork. Chemtracts Biochem. Mol. Biol. 15:313-325. [Google Scholar]
- 56.DePamphilis, M. L., and P. M. Wassarman. 1980. Replication of eukaryotic chromosomes: a close-up of the replication fork. Annu. Rev. Biochem. 49:627-666. [DOI] [PubMed] [Google Scholar]
- 57.Desany, B. A., A. A. Alcasabas, J. B. Bachant, and S. J. Elledge. 1998. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 12:2956-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deshpande, A. M., and C. S. Newlon. 1996. DNA replication fork pause sites dependent on transcription. Science 272:1030-1033. [DOI] [PubMed] [Google Scholar]
- 59.Dhar, V., and C. L. Schildkraut. 1991. Role of EBNA-1 in arresting replication forks at the Epstein-Barr virus oriP family of tandem repeats. Mol. Cell. Biol. 11:6268-6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Drolet, M., X. Bi, and L. F. Liu. 1994. Hypernegative supercoiling of the DNA template during transcription elongation in vitro. J. Biol. Chem. 269:2068-2074. [PubMed] [Google Scholar]
- 61.Drolet, M., P. Phoenix, R. Menzel, E. Massâe, L. F. Liu, and R. J. Crouch. 1995. Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl. Acad. Sci. USA 92:3526-3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elias-Arnanz, M., and M. Salas. 1997. Bacteriophage phi29 DNA replication arrest caused by codirectional collisions with the transcription machinery. EMBO J. 16:5775-5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elias-Arnanz, M., and M. Salas. 1999. Resolution of head-on collisions between the transcription machinery and bacteriophage phi29 DNA polymerase is dependent on RNA polymerase translocation. EMBO J. 18:5675-5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ermakova, O. V., L. Frappier, and C. L. Schildkraut. 1996. Role of the EBNA-1 protein in pausing of replication forks in the Epstein-Barr virus genome. J. Biol. Chem. 271:33009-33017. [DOI] [PubMed] [Google Scholar]
- 65.Fishel, R., M. K. Lescoe, M. R. S. Rao, N. G. Copeland, N. A. Jenkins, J. Garber, M. Kane, and R. Kolodner. 1993. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 75:1027-1038. [DOI] [PubMed] [Google Scholar]
- 66.Foss, E. J. 2001. Tof1p regulates DNA damage responses during S phase in Saccharomyces cerevisiae. Genetics 157:567-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.French, S. 1992. Consequences of replication fork movement trough transcription units in vivo. Science 258:1362-1365. [DOI] [PubMed] [Google Scholar]
- 68.Freudenreich, C. H. 2005. Molecular mechanisms of chromosomal fragility. Chemtracts Biochem. Mol. Biol. 18:141-152. [Google Scholar]
- 69.Freudenreich, C. H., S. M. Kantrow, and V. A. Zakian. 1998. Expansion and length-dependent fragility of CTG repeats in yeast. Science 279:853-856. [DOI] [PubMed] [Google Scholar]
- 70.Freudenreich, C. H., J. B. Stavenhagen, and V. A. Zakian. 1997. Stability of a CTG/CAG trinucleotide repeat in yeast is dependent on its orientation in the genome. Mol. Cell. Biol. 17:2090-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Friedberg, E. C., G. C. Walker, W. Siede, R. D. Wood, R. A. Schultz, and T. Ellenberger. 2005. DNA repair and mutagenesis, 2nd ed. ASM Press, Washington, DC.
- 72.Gacy, A. M., G. Goellner, N. Juranic, S. Macura, and C. T. McMurray. 1995. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 81:533-540. [DOI] [PubMed] [Google Scholar]
- 73.Gacy, A. M., G. M. Goellner, C. Spiro, X. Chen, G. Gupta, E. M. Bradbury, R. B. Dyer, M. J. Mikesell, J. Z. Yao, A. J. Johnson, A. Richter, S. B. Melancon, and C. T. McMurray. 1998. GAA instability in Friedreich's ataxia shares a common, DNA-directed and intraallelic mechanism with other trinucleotide diseases. Mol. Cell 1:583-593. [DOI] [PubMed] [Google Scholar]
- 74.Gahn, T. A., and C. L. Schildkraut. 1989. The Epstein-Barr virus origin of plasmid replication, oriP, contains both the initiation and termination sites of DNA replication. Cell 58:527-535. [DOI] [PubMed] [Google Scholar]
- 75.Gautam, A., and D. Bastia. 2001. A replication terminus located at or near a replication checkpoint of Bacillus subtilis functions independently of stringent control. J. Biol. Chem. 276:8771-8777. [DOI] [PubMed] [Google Scholar]
- 76.Gerber, J. K., E. Gogel, C. Berger, M. Wallisch, F. Mèuller, I. Grummt, and F. Grummt. 1997. Termination of mammalian rDNA replication: polar arrest of replication fork movement by transcription termination factor TTF-I. Cell 90:559-667. [DOI] [PubMed] [Google Scholar]
- 77.Gerton, J. L., J. DeRisi, R. Shroff, M. Lichten, P. O. Brown, and T. D. Petes. 2000. Inaugural article: global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 97:11383-11390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gilbert, D. E., and J. Feigon. 1999. Multistranded DNA structures. Curr. Opin. Struct. Biol. 9:305-314. [DOI] [PubMed] [Google Scholar]
- 79.Glover, T. W., M. F. Arlt, A. M. Casper, and S. G. Durkin. 2005. Mechanisms of common fragile site instability. Hum. Mol. Genet. 14:197-205. [DOI] [PubMed] [Google Scholar]
- 80.Goldfless, S. J., A. S. Morag, K. A. Belisle, V. A. Sutera, Jr., and S. T. Lovett. 2006. DNA repeat rearrangements mediated by DnaK-dependent replication fork repair. Mol. Cell 21:595-604. [DOI] [PubMed] [Google Scholar]
- 81.Gordenin, D. A., T. A. Kunkel, and M. A. Resnick. 1997. Repeat expansion—all in a flap? Nat. Genet. 16:116-118. [DOI] [PubMed] [Google Scholar]
- 82.Greenfeder, S. A., and C. S. Newlon. 1992. Replication forks pause at yeast centromeres. Mol. Cell. Biol. 12:4056-4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haber, J. E. 1998. Mating-type gene switching in Saccharomyces cerevisiae. Annu. Rev. Genet. 32:561-599. [DOI] [PubMed] [Google Scholar]
- 84.Hanawalt, P. C. 2001. Controlling the efficiency of excision repair. Mutat. Res. 485:3-13. [DOI] [PubMed] [Google Scholar]
- 85.Hanawalt, P. C. 1994. Transcription-coupled repair and human disease. Science 266:1957-1958. [DOI] [PubMed] [Google Scholar]
- 86.Hansen, R. S., T. K. Canfield, M. M. Lamb, S. M. Gartler, and C. D. Laird. 1993. Association of fragile X syndrome with delayed replication of the FMR1 gene. Cell 73:1403-1409. [DOI] [PubMed] [Google Scholar]
- 87.Hashem, V. I., and R. R. Sinden. 2005. Duplications between direct repeats stabilized by DNA secondary structure occur preferentially in the leading strand during DNA replication. Mutat. Res. 570:215-226. [DOI] [PubMed] [Google Scholar]
- 88.Heidenfelder, B. L., A. M. Makhov, and M. D. Topal. 2003. Hairpin formation in Friedreich's ataxia triplet repeat expansion. J. Biol. Chem. 278:2425-2431. [DOI] [PubMed] [Google Scholar]
- 89.Heller, R. C., and K. J. Marians. 2006. Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439:557-562. [DOI] [PubMed] [Google Scholar]
- 90.Henricksen, L. A., S. Tom, Y. Liu, and R. A. Bambara. 2000. Inhibition of flap endonuclease 1 by flap secondary structure and relevance to repeat sequence expansion. J. Biol. Chem. 275:16420-16427. [DOI] [PubMed] [Google Scholar]
- 91.Heyer, W. D. 2004. Damage signaling: RecQ sends an SOS to you. Curr. Biol. 14:895-897. [DOI] [PubMed] [Google Scholar]
- 92.Heyer, W. D., and R. Kanaar. 2004. Recombination mechanisms. Fortieth anniversary meeting of the Holliday model. Mol. Cell 16:1-9. [DOI] [PubMed] [Google Scholar]
- 93.Hiasa, H., and K. J. Marians. 1992. Differential inhibition of the DNA translocation and DNA unwinding activities of DNA helicases by the Escherichia coli Tus protein. J. Biol. Chem. 267:11379-81135. [PubMed] [Google Scholar]
- 94.Hiasa, H., and K. J. Marians. 1994. Tus prevents overreplication of oriC plasmid DNA. J. Biol. Chem. 269:26959-26968. [PubMed] [Google Scholar]
- 95.Hidaka, M., M. Akiyama, and T. Horiuchi. 1988. A consensus sequence of three DNA replication terminus sites on the E. coli chromosome is highly homologous to the terR sites of the R6K plasmid. Cell 55:467-475. [DOI] [PubMed] [Google Scholar]
- 96.Higgins, N. P., K. Kato, and B. Strauss. 1976. A model for replication repair in mammalian cells. J. Mol. Biol. 101:417-425. [DOI] [PubMed] [Google Scholar]
- 97.Hill, T. M. 1992. Arrest of bacterial DNA replication. Annu. Rev. Microbiol. 46:603-633. [DOI] [PubMed] [Google Scholar]
- 98.Hill, T. M., J. M. Henson, and P. L. Kuempel. 1987. The terminus region of the Escherichia coli chromosome contains two separate loci that exhibit polar inhibition of replication. Proc. Natl. Acad. Sci. USA 84:1754-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hill, T. M., B. J. Kopp, and P. L. Kuempel. 1988. Termination of DNA replication in Escherichia coli requires a trans-acting factor. J. Bacteriol. 170:662-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hill, T. M., and K. J. Marians. 1990. Escherichia coli Tus protein acts to arrest the progression of DNA replication forks in vitro. Proc. Natl. Acad. Sci. USA 87:2481-2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hill, T. M., A. J. Pelletier, M. L. Tecklenburg, and P. L. Kuempel. 1988. Identification of the DNA sequence from the E. coli terminus region that halts replication forks. Cell 55:459-466. [DOI] [PubMed] [Google Scholar]
- 102.Hill, T. M., M. L. Tecklenburg, A. J. Pelletier, and P. L. Kuempel. 1989. tus, the trans-acting gene required for termination of DNA replication in Escherichia coli, encodes a DNA-binding protein. Proc. Natl. Acad. Sci. USA 86:1593-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hong, X., G. W. Cadwell, and T. Kogoma. 1995. Escherichia coli RecG and RecA proteins in R-loop formation. EMBO J. 14:2385-2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Horiuchi, T., and Y. Fujimura. 1995. Recombinational rescue of the stalled DNA replication fork: a model based on analysis of an Escherichia coli strain with a chromosome region difficult to replicate. J. Bacteriol. 177:783-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Horiuchi, T., Y. Fujimura, H. Nishitani, T. Kobayashi, and M. Hidaka. 1994. The DNA replication fork blocked at the Ter site may be an entrance for the RecBCD enzyme into duplex DNA. J. Bacteriol. 176:4656-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hraiky, C., M. A. Raymond, and M. Drolet. 2000. RNase H overproduction corrects a defect at the level of transcription elongation during rRNA synthesis in the absence of DNA topoisomerase I in Escherichia coli. J. Biol. Chem. 275:11257-11263. [DOI] [PubMed] [Google Scholar]
- 107.Huang, C. C., and J. E. Hearst. 1980. Pauses at positions of secondary structure during in vitro replication of single-stranded fd bacteriophage DNA by T4 DNA polymerase. Anal. Biochem. 103:127-139. [DOI] [PubMed] [Google Scholar]
- 108.Huntington's Disease Collaborative Research Group. 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971-983. [DOI] [PubMed] [Google Scholar]
- 109.Hyrien, O. 2000. Mechanisms and consequences of replication fork arrest. Biochimie 82:5-17. [DOI] [PubMed] [Google Scholar]
- 110.Indiani, C., P. McInerney, R. Georgescu, M. F. Goodman, and M. O'Donnell. 2005. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol. Cell 19:805-815. [DOI] [PubMed] [Google Scholar]
- 111.Ionov, Y., M. A. Peinado, S. Malkhosyan, D. Shibata, and M. Perucho. 1993. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 363:558-561. [DOI] [PubMed] [Google Scholar]
- 112.Ireland, M. J., S. S. Reinke, and D. M. Livingston. 2000. The impact of lagging strand replication mutations on the stability of CAG repeat tracts in yeast. Genetics 155:1657-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Itoh, T., and J. Tomizawa. 1980. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proc. Natl. Acad. Sci. USA 77:2450-2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ivessa, A. S., B. A. Lenzmeier, J. B. Bessler, L. K. Goudsouzian, S. L. Schnakenberg, and V. A. Zakian. 2003. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell 12:1525-1536. [DOI] [PubMed] [Google Scholar]
- 115.Ivessa, A. S., J. Q. Zhou, V. P. Schulz, E. K. Monson, and V. A. Zakian. 2002. Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes Dev. 16:1383-1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ivessa, A. S., J. Q. Zhou, and V. A. Zakian. 2000. The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell 100:479-489. [DOI] [PubMed] [Google Scholar]
- 117.Iyer, R. R., A. Pluciennik, W. A. Rosche, R. R. Sinden, and R. D. Wells. 2000. DNA polymerase III proofreading mutants enhance the expansion and deletion of triplet repeat sequences in Escherichia coli. J. Biol. Chem. 275:2174-2184. [DOI] [PubMed] [Google Scholar]
- 118.Iyer, R. R., and R. D. Wells. 1999. Expansion and deletion of triplet repeat sequences in Escherichia coli occur on the leading strand of DNA replication. J. Biol. Chem. 274:3865-3877. [DOI] [PubMed] [Google Scholar]
- 119.Kaguni, L. S., and D. A. Clayton. 1982. Template-directed pausing in in vitro DNA synthesis by DNA polymerase a from Drosophila melanogaster embryos. Proc. Natl. Acad. Sci. USA 79:983-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kamada, K., T. Horiuchi, K. Ohsumi, N. Shimamoto, and K. Morikawa. 1996. Structure of a replication-terminator protein complexed with DNA. Nature 383:598-603. [DOI] [PubMed] [Google Scholar]
- 121.Kang, S., A. Jaworski, K. Ohshima, and R. D. Wells. 1995. Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat. Genet. 10:213-218. [DOI] [PubMed] [Google Scholar]
- 122.Kang, S., K. Ohshima, M. Shimizu, S. Amirhaeri, and R. D. Wells. 1995. Pausing of DNA synthesis in vitro at specific loci in CTG and CGG triplet repeats from human hereditary disease genes. J. Biol. Chem. 270:27014-27021. [DOI] [PubMed] [Google Scholar]
- 123.Katou, Y., Y. Kanoh, M. Bando, H. Noguchi, H. Tanaka, T. Ashikari, K. Sugimoto, and K. Shirahige. 2003. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424:1078-1083. [DOI] [PubMed] [Google Scholar]
- 124.Kaul, S., B. K. Mohanty, T. Sahoo, I. Patel, S. A. Khan, and D. Bastia. 1994. The replication terminator protein of the gram-positive bacterium Bacillus subtilis functions as a polar contrahelicase in gram-negative Escherichia coli. Proc. Natl. Acad. Sci. USA 91:11143-11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Keeney, S. 2001. Mechanism and control of meiotic recombination initiation. Curr. Top. Dev. Biol. 52:1-53. [DOI] [PubMed] [Google Scholar]
- 126.Keil, R. L., and A. D. McWilliams. 1993. A gene with specific and global effects on recombination of sequences from tandemly repeated genes in Saccharomyces cerevisiae. Genetics 135:711-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Khatri, G. S., T. MacAllister, P. R. Sista, and D. Bastia. 1989. The replication terminator protein of E. coli is a DNA sequence-specific contra-helicase. Cell 59:667-674. [DOI] [PubMed] [Google Scholar]
- 128.Kim, S.-H., M. J. Pytlos, and R. R. Sinden. 2006. Replication restart: a pathway for (CTG) · (CAG) repeat deletion in Escherichia coli. Mutat. Res. 595:5-22. [DOI] [PubMed] [Google Scholar]
- 129.Kobayashi, T. 2003. The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol. Cell. Biol. 23:9178-9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kobayashi, T., D. J. Heck, M. Nomura, and T. Horiuchi. 1998. Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev. 12:3821-3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kobayashi, T., M. Hidaka, and T. Horiuchi. 1989. Evidence of a ter specific binding protein essential for the termination reaction of DNA replication in Escherichia coli. EMBO J. 8:2435-4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kobayashi, T., and T. Horiuchi. 1996. A yeast gene product, Fob1 protein, required for both replication fork blocking and recombinational hotspot activities. Genes Cells 1:465-474. [DOI] [PubMed] [Google Scholar]
- 133.Kohwi, Y., H. Wang, and T. Kohwi-Shigematsu. 1993. A single trinucleotide, 5′AGC3′/5′GCT3′, of the triplet-repeat disease genes confers metal ion-induced non-B DNA structure. Nucleic Acids Res. 21:5651-5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kolodner, R. D., C. D. Putnam, and K. Myung. 2002. Maintenance of genome stability in Saccharomyces cerevisiae. Science 297:552-557. [DOI] [PubMed] [Google Scholar]
- 135.Kornberg, A. 2000. Ten commandments: lessons from the enzymology of DNA replication. J. Bacteriol. 28:3613-3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kornberg, A., and T. Baker. 1992. DNA replication, 2nd ed. W. H. Freeman and Co., New York, NY.
- 137.Kowalczykowski, S. C. 2000. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 25:156-165. [DOI] [PubMed] [Google Scholar]
- 138.Krabbe, M., J. Zabielski, R. Bernander, and K. Nordstrèom. 1997. Inactivation of the replication-termination system affects the replication mode and causes unstable maintenance of plasmid R1. Mol. Microbiol. 24:723-735. [DOI] [PubMed] [Google Scholar]
- 139.Krasilnikov, A. S., I. G. Panyutin, G. M. Samadashwily, R. Cox, Y. S. Lazurkin, and S. M. Mirkin. 1997. Mechanisms of triplex-caused polymerization arrest. Nucleic Acids Res. 25:1339-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Krasilnikova, M. M., and S. M. Mirkin. 2004. Replication stalling at Friedreich's ataxia (GAA)n repeats in vivo. Mol. Cell. Biol. 24:2286-2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Krasilnikova, M. M., G. M. Samadashwily, A. S. Krasilnikov, and S. M. Mirkin. 1998. Transcription through a simple DNA repeat blocks replication elongation. EMBO J. 17:5095-5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Krasilnikova, M. M., E. V. Smirnova, A. S. Krasilnikov, and S. M. Mirkin. 2001. A new trick for an old dog: TraY binding to a homopurine- homopyrimidine run attenuates DNA replication. J. Mol. Biol. 313:271-282. [DOI] [PubMed] [Google Scholar]
- 143.Kremer, E. J., M. Pritchard, M. Lynch, S. Yu, K. Holman, E. Baker, S. T. Warren, D. Schlessinger, G. R. Sutherland, and R. I. Richards. 1991. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252:1711-1714. [DOI] [PubMed] [Google Scholar]
- 144.Krings, G., and D. Bastia. 2005. Sap1p binds to Ter1 at the ribosomal DNA of Schizosaccharomyces pombe and causes polar replication fork arrest. J. Biol. Chem. 280:39135-39142. [DOI] [PubMed] [Google Scholar]
- 145.Krings, G., and D. Bastia. 2004. swi1- and swi3-dependent and independent replication fork arrest at the ribosomal DNA of Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. USA 101:14085-14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kunkel, T. A. 1993. Slippery DNA and diseases. Nature 365:207-208. [DOI] [PubMed] [Google Scholar]
- 147.Kunst, C. B., and S. T. Warren. 1994. Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell 77:853-861. [DOI] [PubMed] [Google Scholar]
- 148.Lalioti, M. D., H. S. Scott, C. Buresi, C. Rossier, A. Bottani, M. A. Morris, A. Malafosse, and S. E. Antonarakis. 1997. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature 386:847-851. [DOI] [PubMed] [Google Scholar]
- 149.Lambert, S., and A. M. Carr. 2005. Checkpoint responses to replication fork barriers. Biochimie 87:591-602. [DOI] [PubMed] [Google Scholar]
- 150.Lambert, S., A. Watson, D. M. Sheedy, B. Martin, and A. M. Carr. 2005. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 121:689-702. [DOI] [PubMed] [Google Scholar]
- 151.Langley, D. B., M. T. Smith, P. J. Lewis, and R. G. Wake. 1993. Protein-nucleoside contacts in the interaction between the replication terminator protein of Bacillus subtilis and the DNA terminator. Mol. Microbiol. 10:771-779. [DOI] [PubMed] [Google Scholar]
- 152.Lanka, E., and B. M. Wilkins. 1995. DNA processing reactions in bacterial conjugation. Annu. Rev. Biochem. 64:141-169. [DOI] [PubMed] [Google Scholar]
- 153.Lapidot, A., N. Baran, and H. Manor. 1989. (dT-dC)n and (dG-dA)n tracts arrest single stranded DNA replication in vitro. Nucleic Acids Res. 17:883-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Leach, F. S., N. C. Nicolaides, N. Papadopoulos, B. Liu, J. Jen, R. Parsons, P. Peltomaki, P. Sistonen, L. A. Aaltonen, M. Nystrom-Lahti, X.-Y. Guan, J. Zhang, P. S. Meltzer, J.-W. Yu, F.-T. Kao, D. J. Chen, K. M. Cerosaletti, R. E. K. Fournier, S. Todd, T. Lewis, R. J. Leach, S. L. Naylor, J. Weissenbach, J.-P. Mecklin, H. Jarvinen, G. M. Petersen, S. R. Hamilton, J. Green, J. Jass, P. Watson, H. T. Lynch, J. M. Trent, A. de la Chapelle, K. W. Kinzler, and B. Vogelstein. 1993. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 75:1215-1225. [DOI] [PubMed] [Google Scholar]
- 155.Lee, E. H., A. Kornberg, M. Hidaka, T. Kobayashi, and T. Horiuchi. 1989. Escherichia coli replication termination protein impedes the action of helicases. Proc. Natl. Acad. Sci. USA 86:9104-9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Levine, A., S. Autret, and S. J. Sâeror. 1995. A checkpoint involving RTP, the replication terminator protein, arrests replication downstream of the origin during the stringent response in Bacillus subtilis. Mol. Microbiol. 15:287-925. [DOI] [PubMed] [Google Scholar]
- 157.Levine, A., F. Vannier, M. Dehbi, G. Henckes, and S. J. Seror. 1991. The stringent response blocks DNA replication outside the ori region in Bacillus subtilis and at the origin in Escherichia coli. J. Mol. Biol. 219:605-613. [DOI] [PubMed] [Google Scholar]
- 158.Lewis, P. J., M. T. Smith, and R. G. Wake. 1989. A protein involved in termination of chromosome replication in Bacillus subtilis binds specifically to the terC site. J. Bacteriol. 171:3564-3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lichten, M., and A. S. Goldman. 1995. Meiotic recombination hotspots. Annu. Rev. Genet. 29:423-444. [DOI] [PubMed] [Google Scholar]
- 160.Lilley, D. M. J. 1989. Structural isomerization in DNA: the formation of cruciform structures in supercoiled DNA molecules. Chem. Soc. Rev. 18:53-83. [Google Scholar]
- 161.Lindsay, H. D., D. J. Griffiths, R. J. Edwards, P. U. Christensen, J. M. Murray, F. Osman, N. Walworth, and A. M. Carr. 1998. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 12:382-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Linskens, M. H., and J. A. Huberman. 1988. Organization of replication of ribosomal DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 8:4927-4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liquori, C. L., K. Ricker, M. L. Moseley, J. F. Jacobsen, W. Kress, S. L. Naylor, J. W. Day, and L. P. Ranum. 2001. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293:864-867. [DOI] [PubMed] [Google Scholar]
- 164.Lisby, M., and R. Rothstein. 2005. Localization of checkpoint and repair proteins in eukaryotes. Biochimie 87:579-589. [DOI] [PubMed] [Google Scholar]
- 165.Little, R. D., T. H. Platt, and C. L. Schildkraut. 1993. Initiation and termination of DNA replication in human rRNA. Mol. Cell. Biol. 13:6600-6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liu, B., and B. M. Alberts. 1995. Head-on collision between a DNA replication apparatus and RNA. Science 267:1131-1137. [DOI] [PubMed] [Google Scholar]
- 167.Liu, B., M. L. Wong, R. L. Tinker, E. P. Geiduschek, and B. M. Alberts. 1993. The DNA replication fork can pass RNA polymerase without displacing the nascent transcript. Nature 366:33-39. [DOI] [PubMed] [Google Scholar]
- 168.Liu, L. F., and J. C. Wang. 1987. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 84:7024-7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lopes, M., C. Cotta-Ramusino, A. Pellicioli, G. Liberi, P. Plevani, M. Muzi-Falconi, C. S. Newlon, and M. Foiani. 2001. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412:557-561. [DOI] [PubMed] [Google Scholar]
- 170.Lopes, M., M. Foiani, and J. M. Sogo. 2006. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 21:15-27. [DOI] [PubMed] [Google Scholar]
- 171.Lopez-Estrano, C., J. B. Schvartzman, D. B. Krimer, and P. Hernandez. 1999. Characterization of the pea rDNA replication fork barrier: putative cis-acting and trans-acting factors. Plant Mol. Biol. 40:99-110. [DOI] [PubMed] [Google Scholar]
- 172.Lopez-Estrano, C., J. B. Schvartzman, D. B. Krimer, and P. Hernandez. 1998. Co-localization of polar replication fork barriers and rRNA transcription terminators in mouse rDNA. J. Mol. Biol. 277:249-256. [DOI] [PubMed] [Google Scholar]
- 173.Luo, Y., and R. C. Deonier. 1994. Mutational and physical analysis of F plasmid traY protein binding to oriT. Mol. Microbiol. 11:459-469. [DOI] [PubMed] [Google Scholar]
- 174.MacAlpine, D. M., Z. Zhang, and G. M. Kapler. 1997. Type I elements mediate replication fork pausing at conserved upstream sites in the Tetrahymena thermophila ribosomal DNA minichromosome. Mol. Cell. Biol. 17:4517-4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mahadevan, M., C. Tsilfidis, L. Sabourin, G. Shutler, C. Amemiya, G. Jansen, C. Neville, M. Narang, J. Barcelo, K. O'Hoy, et al. 1992. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 255:1253-1255. [DOI] [PubMed] [Google Scholar]
- 176.Maisnier-Patin, S., K. Nordstrèom, and S. Dasgupta. 2001. Replication arrests during a single round of replication of the Escherichia coli chromosome in the absence of DnaC activity. Mol. Microbiol. 42:1371-1382. [DOI] [PubMed] [Google Scholar]
- 177.Makovets, S., I. Herskowitz, and E. H. Blackburn. 2004. Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Mol. Cell. Biol. 24:4019-4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Manna, A. C., K. S. Pai, D. E. Bussiere, S. W. White, and D. Bastia. 1996. The dimer-dimer interaction surface of the replication terminator protein of Bacillus subtilis and termination of DNA replication. Proc. Natl. Acad. Sci. USA 93:3253-3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Marians, K. J. 2004. Mechanisms of replication fork restart in Escherichia coli. Philos. Trans. R. Soc. Lond. B 359:71-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mariappan, S. V., L. A. R. Silks, X. Chen, P. A. Springer, R. Wu, R. K. Moyzis, E. M. Bradbury, A. E. Garcia, and G. Gupta. 1998. Solution structures of the Huntington's disease DNA triplets, (CAG)n. Biomol. Struct. Dyn. 15:723-744. [DOI] [PubMed] [Google Scholar]
- 181.Maric, C., B. Levacher, and O. Hyrien. 1999. Developmental regulation of replication fork pausing in Xenopus laevis ribosomal RNA genes. J. Mol. Biol. 291:775-788. [DOI] [PubMed] [Google Scholar]
- 182.Massâe, E., and M. Drolet. 1999. R-loop-dependent hypernegative supercoiling in Escherichia coli topA mutants preferentially occurs at low temperatures and correlates with growth inhibition. J. Mol. Biol. 294:321-332. [DOI] [PubMed] [Google Scholar]
- 183.Matsuura, T., T. Yamagata, D. L. Burgess, A. Rasmussen, R. P. Grewal, K. Watase, M. Khajavi, A. E. McCall, C. F. Davis, L. Zu, M. Achari, S. M. Pulst, E. Alonso, J. L. Noebels, D. L. Nelson, H. Y. Zoghbi, and T. Ashizawa. 2000. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet. 26:191-194. [DOI] [PubMed] [Google Scholar]
- 184.McGlynn, P., and R. G. Lloyd. 2000. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell 101:35-45. [DOI] [PubMed] [Google Scholar]
- 185.McGlynn, P., and R. G. Lloyd. 2002. Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell. Biol. 3:859-870. [DOI] [PubMed] [Google Scholar]
- 186.McGowan, C. H., and P. Russell. 2004. The DNA damage response: sensing and signaling. Curr. Opin. Cell Biol. 16:629-633. [DOI] [PubMed] [Google Scholar]
- 187.McLean, M. J., K. H. Wolfe, and K. M. Devine. 1998. Base composition skews, replication orientation, and gene orientation in 12 prokaryote genomes. J. Mol. Evol. 47:691-696. [DOI] [PubMed] [Google Scholar]
- 188.McMurray, C. T. 1999. DNA secondary structure: a common and causative factor for expansion in human disease. Proc. Natl. Acad. Sci. USA 96:1823-1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.McMurray, C. T. 1995. Mechanisms of DNA expansion. Chromosoma 104:2-13. [DOI] [PubMed] [Google Scholar]
- 190.McMurry, M. T., and M. S. Krangel. 2000. A role for histone acetylation in the developmental regulation of VDJ recombination. Science 287:495-498. [DOI] [PubMed] [Google Scholar]
- 191.Meijer, W. J., M. Smith, R. G. Wake, A. L. de Boer, G. Venema, and S. Bron. 1996. Identification and characterization of a novel type of replication terminator with bidirectional activity on the Bacillus subtilis theta plasmid pLS20. Mol. Microbiol. 19:1295-1306. [DOI] [PubMed] [Google Scholar]
- 192.Mejâia-Ramâirez, E., A. Sâanchez-Gorostiaga, D. B. Krimer, J. B. Schvartzman, and P. Hernâandez. 2005. The mating type switch-activating protein Sap1 is required for replication fork arrest at the rRNA genes of fission yeast. Mol. Cell. Biol. 25:8755-8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mellon, I., G. Spivak, and P. C. Hanawalt. 1987. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 51:241-249. [DOI] [PubMed] [Google Scholar]
- 194.Michel, B. 2000. Replication fork arrest and DNA recombination. Trends Biochem. Sci. 25:173-178. [DOI] [PubMed] [Google Scholar]
- 195.Michel, B., G. Grompone, M. J. Floráes, and V. Bidnenko. 2004. Multiple pathways process stalled replication forks. Proc. Natl. Acad. Sci. USA 101:12783-12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mikhailov, V. S., and D. F. Bogenhagen. 1996. Termination within oligo(dT) tracts in template DNA by DNA polymerase gamma occurs with formation of a DNA triplex structure and is relieved by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 271:30774-30780. [DOI] [PubMed] [Google Scholar]
- 197.Miret, J. J., L. Pessoa-Brandao, and R. S. Lahue. 1998. Orientation-dependent and sequence-specific expansions of CTG/CAG trinucleotide repeats in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 95:12438-12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mirkin, E. V., D. Castro Roa, E. Nudler, and S. M. Mirkin. 2006. Transcription regulatory elements are punctuation marks for DNA replication. Proc. Natl. Acad. Sci. USA 103:7276-7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mirkin, E. V., and S. M. Mirkin. 2005. Mechanisms of transcription-replication collisions in bacteria. Mol. Cell. Biol. 25:888-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mirkin, S. M. 2006. DNA structures, repeat expansions and human hereditary disorders. Curr. Opin. Struct. Biol. 16:1-8. [DOI] [PubMed] [Google Scholar]
- 201.Mirkin, S. M. 2004. Molecular models for repeat expansions. Chemtracts Biochem. Mol. Biol. 17:639-662. [Google Scholar]
- 202.Mirkin, S. M. 2006. Replication of expandable repeats, p. 637-644. In R. D. Wells and T. Ashizawa (ed.), Genetic instabilities and neurological diseases, 2nd ed. Academic Press, New York, NY.
- 203.Mirkin, S. M. 1999. Structure and biology of H DNA, p. 193-222. In C. Malvy, A. Harel-Bellan, and L. L. Pritchard (ed.), Triple helix forming oligonucleotides. Kluwer Academic Publishers, Boston, MA.
- 204.Mirkin, S. M., and E. V. Smirnova. 2002. Positioned to expand. Nat. Genet. 31:5-6. [DOI] [PubMed] [Google Scholar]
- 205.Mitchell, J. E., S. F. Newbury, and J. A. McClellan. 1995. Compact structures of d(CNG)n oligonucleotides in solution and their possible relevance to fragile X and related human genetic diseases. Nucleic Acids Res. 23:1876-1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Mizuta, R., K. Iwai, M. Shigeno, M. Mizuta, T. Uemura, T. Ushiki, and D. Kitamura. 2003. Molecular visualization of immunoglobulin switch region RNA/DNA complex by atomic force microscope. J. Biol. Chem. 278:4431-4434. [DOI] [PubMed] [Google Scholar]
- 207.Modrich, P. 1994. Mismatch repair, genetic stability, and cancer. Science 266:1959-1960. [DOI] [PubMed] [Google Scholar]
- 208.Mohanty, B. K., and D. Bastia. 2004. Binding of the replication terminator protein Fob1p to the Ter sites of yeast causes polar fork arrest. J. Biol. Chem. 279:1932-1941. [DOI] [PubMed] [Google Scholar]
- 209.Mulcair, M. D., P. M. Schaeffer, A. J. Oakley, H. F. Cross, C. Neylon, T. M. Hill, and N. E. Dixon. 2006. A molecular mousetrap determines polarity of termination of DNA replication in E. coli. Cell 125:1309-1319. [DOI] [PubMed] [Google Scholar]
- 210.Mulugu, S., A. Potnis, Shamsuzzaman, J. Taylor, K. Alexander, and D. Bastia. 2001. Mechanism of termination of DNA replication of Escherichia coli involves helicase-contrahelicase interaction. Proc. Natl. Acad. Sci. USA 98:9569-9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Myung, K., A. Datta, and R. D. Kolodner. 2001. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 104:397-408. [DOI] [PubMed] [Google Scholar]
- 212.Nancarrow, J. K., E. Kremer, K. Holman, H. Eyre, N. A. Doggett, D. Le Paslier, D. F. Callen, G. R. Sutherland, and R. I. Richards. 1994. Implications of FRA16A structure for the mechanism of chromosomal fragile site genesis. Science 264:1938-1941. [DOI] [PubMed] [Google Scholar]
- 213.Nedelcheva, M. N., A. Roguev, L. B. Dolapchiev, A. Shevchenko, H. B. Taskov, A. F. Stewart, and S. S. Stoynov. 2005. Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J. Mol. Biol. 347:509-521. [DOI] [PubMed] [Google Scholar]
- 214.Neylon, C., A. V. Kralicek, T. M. Hill, and N. E. Dixon. 2005. Replication termination in Escherichia coli: structure and antihelicase activity of the Tus-Ter complex. Microbiol. Mol. Biol. Rev. 69:501-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nicolaides, N. C., N. Papadopoulos, B. Liu, Y.-F. Wei, K. C. Carter, S. M. Ruben, C. A. Rosen, W. A. Haseltine, R. D. Fleischmann, C. M. Fraser, M. D. Adams, J. C. Venter, M. G. Dunlop, S. R. Hamilton, G. M. Petersen, A. de la Chapelle, B. Vogelstein, and K. W. Kinzler. 1994. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 371:75-80. [DOI] [PubMed] [Google Scholar]
- 216.Niki, H., Y. Yamaichi, and S. Hiraga. 2000. Dynamic organization of chromosomal DNA in Escherichia coli. Genes Dev. 14:212-223. [PMC free article] [PubMed] [Google Scholar]
- 217.Noguchi, E., C. Noguchi, L. L. Du, and P. Russell. 2003. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol. Cell. Biol. 23:7861-7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Nomura, M., and E. A. Morgan. 1977. Genetics of bacterial ribosomes. Annu. Rev. Genet. 11:297-347. [DOI] [PubMed] [Google Scholar]
- 219.Norio, P., and C. L. Schildkraut. 2001. Visualization of DNA replication on individual Epstein-Barr virus episomes. Science 294:2361-2364. [DOI] [PubMed] [Google Scholar]
- 220.O'Donnell, M. 2006. Replisome architecture and dynamics in Escherichia coli. J. Biol. Chem. 281:10653-10656. [DOI] [PubMed] [Google Scholar]
- 221.Ohshima, K., L. Montermini, R. D. Wells, and M. Pandolfo. 1998. Inhibitory effects of expanded GAA · TTC triplet repeats from intron I of the Freidreich's ataxia gene on transcription and replication in vivo. J. Biol. Chem. 273:14588-14595. [DOI] [PubMed] [Google Scholar]
- 222.Olavarrieta, L., P. Hernandez, D. B. Krimer, and J. B. Schvartzman. 2002. DNA knotting caused by head-on collision of transcription and replication. J. Mol. Biol. 322:1-6. [DOI] [PubMed] [Google Scholar]
- 223.Osinga, K. A., M. De Haan, T. Christianson, and H. F. Tabak. 1982. A nonanucleotide sequence involved in promotion of ribosomal RNA synthesis and RNA priming of DNA replication in yeast mitochondria. Nucleic Acids Res. 10:7993-8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Panigrahi, G. B., J. D. Cleary, and C. E. Pearson. 2002. In vitro (CTG)*(CAG) expansions and deletions by human cell extracts. J. Biol. Chem. 277:13926-13934. [DOI] [PubMed] [Google Scholar]
- 225.Papadopoulos, N., N. C. Nicolaides, Y.-F. Wei, S. M. Ruben, K. C. Carter, C. A. Rosen, W. A. Haseltine, R. D. Fleischmann, C. M. Fraser, M. D. Adams, J. C. Venter, S. R. Hamilton, G. M. Petersen, P. Watson, H. T. Lynch, P. Peltomaki, J.-P. Mecklin, A. de la Chapelle, K. W. Kinzler, and B. Vogelstein. 1994. Mutation of a mutL homolog in hereditary colon cancer. Science 263:1625-1629. [DOI] [PubMed] [Google Scholar]
- 226.Paques, F., and J. E. Haber. 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63:349-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Parsons, R., G.-M. Li, M. J. Longley, W.-H. Fang, N. Papadopoulos, J. Jen, A. de la Chapelle, K. W. Kinzler, B. Vogelstein, and P. Modrich. 1993. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 75:1227-1236. [DOI] [PubMed] [Google Scholar]
- 228.Patel, H. P., L. Lu, R. T. Blaszak, and J. J. Bissler. 2004. PKD1 intron 21: triplex DNA formation and effect on replication. Nucleic Acids Res. 32:1460-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pearson, C. E., K. N. Edamura, and J. D. Cleary. 2005. Repeat instability: mechanisms of dynamic mutations. Nat. Rev. Genet. 6:729-742. [DOI] [PubMed] [Google Scholar]
- 230.Pearson, C. E., and R. R. Sinden. 1996. Alternative structures in duplex DNA formed within the trinucleotide repeats of the myotonic dystrophy and fragile X loci. Biochemistry 35:5041-5053. [DOI] [PubMed] [Google Scholar]
- 231.Pearson, C. E., and R. R. Sinden. 1998. Trinucleotide repeat DNA structures: dynamic mutations from dynamic DNA. Curr. Opin. Struct. Biol. 8:321-330. [DOI] [PubMed] [Google Scholar]
- 232.Pease, P. J., O. Levy, G. J. Cost, J. Gore, J. L. Ptacin, D. Sherratt, C. Bustamante, and N. R. Cozzarelli. 2005. Sequence-directed DNA translocation by purified FtsK. Science 307:586-590. [DOI] [PubMed] [Google Scholar]
- 233.Pelletier, A. J., T. M. Hill, and P. L. Kuempel. 1989. Termination sites T1 and T2 from the Escherichia coli chromosome inhibit DNA replication in ColE1-derived plasmids. J. Bacteriol. 171:1739-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Pelletier, R., M. M. Krasilnikova, G. M. Samadashwily, R. S. Lahue, and S. M. Mirkin. 2003. Replication and expansion of trinucleotide repeats in yeast. Mol. Cell. Biol. 23:1349-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Peng, X., R. K. Karuturi, L. D. Miller, K. Lin, Y. Jia, P. Kondu, L. Wang, L. S. Wong, E. T. Liu, M. K. Balasubramanian, and J. Liu. 2005. Identification of cell cycle-regulated genes in fission yeast. Mol. Biol. Cell 16:1026-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Perucho, M., M. A. Peinado, Y. Ionov, S. Casares, S. Malkhosyan, and E. Stanbridge. 1994. Defects in replication fidelity of simple repeated sequences reveal a new mutator mechanism for oncogenesis. Cold Spring Harbor Symp. Quant. Biol. 59:339-348. [DOI] [PubMed] [Google Scholar]
- 237.Peter, B. J., C. Ullsperger, H. Hiasa, K. J. Marians, and N. R. Cozzarelli. 1998. The structure of supercoiled intermediates in DNA replication. Cell 94:819-827. [DOI] [PubMed] [Google Scholar]
- 238.Petruska, J., N. Arnheim, and M. F. Goodman. 1996. Stability of intrastrand hairpin structures formed by the CAG/CTG class of DNA triplet repeats associated with neurological diseases. Nucleic Acids Res. 24:1992-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Possoz, C., S. R. Filipe, I. Grainge, and D. J. Sherratt. 2006. Tracking of controlled Escherichia coli replication fork stalling and restart at repressor-bound DNA in vivo. EMBO J. 25:2596-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Postow, L., C. Ullsperger, R. W. Keller, C. Bustamante, A. V. Vologodskii, and N. R. Cozzarelli. 2001. Positive torsional strain causes the formation of a four-way junction at replication forks. J. Biol. Chem. 276:2790-2796. [DOI] [PubMed] [Google Scholar]
- 241.Potaman, V. N., J. J. Bissler, V. I. Hashem, E. A. Oussatcheva, L. Lu, L. S. Shlyakhtenko, Y. L. Lyubchenko, T. Matsuura, T. Ashizawa, M. Leffak, C. J. Benham, and R. R. Sinden. 2003. Unpaired structures in SCA10 (ATTCT)n · (AGAAT)n repeats. J. Mol. Biol. 326:1095-1111. [DOI] [PubMed] [Google Scholar]
- 242.Prado, F., and A. Aguilera. 2005. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 24:1267-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Putter, V., and F. Grummt. 2002. Transcription termination factor TTF-I exhibits contrahelicase activity during DNA replication. EMBO Rep. 3:147-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Rao, B. S. 1994. Pausing of simian virus 40 DNA replication fork movement in vivo by (dG-dA)n · (dT-dC)n tracts. Gene 140:233-237. [DOI] [PubMed] [Google Scholar]
- 245.Rao, S., H. Manor, and R. G. Martin. 1988. Pausing in simian virus 40 DNA replication by a sequence containing (dG-dA)27 · (dT-dC)27. Nucleic Acids Res. 16:8077-8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Raumann, B. E., B. M. Brown, and R. Y. Sauer. 1994. Major groove DNA recognition by β-sheets: the ribbon-helix-helix family of gene regulatory proteins. Curr. Opin. Struct. Biol. 4:36-43. [Google Scholar]
- 247.Reaban, M. E., and J. A. Griffin. 1990. Induction of RNA-stabilized DNA conformers by transcription of an immunoglobulin switch region. Nature 348:342-344. [DOI] [PubMed] [Google Scholar]
- 248.Reaban, M. E., J. Lebowitz, and J. A. Griffin. 1994. Transciption induces the formation of a stable RNA · DNA hybrid in the immunoglobulin a switch region. J. Biol. Chem. 269:21850-21857. [PubMed] [Google Scholar]
- 249.Rich, A., A. Nordheim, and A. H.-J. Wang. 1984. The chemistry and biology of left-handed Z-DNA. Annu. Rev. Biochem. 53:791-846. [DOI] [PubMed] [Google Scholar]
- 250.Rocha, E. P., and A. Danchin. 2003. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat. Genet. 34:377-378. [DOI] [PubMed] [Google Scholar]
- 251.Rothstein, R., B. Michel, and S. Gangloff. 2000. Replication fork pausing and recombination or “gimme a break.” Genes Dev. 14:1-10. [PubMed] [Google Scholar]
- 252.Sahoo, T., B. K. Mohanty, I. Patel, and D. Bastia. 1995. Termination of DNA replication in vitro: requirement for stereospecific interaction between two dimers of the replication terminator protein of Bacillus subtilis with the terminator site to elicit polar contrahelicase and fork impedance. EMBO J. 14:619-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sakamoto, N., P. D. Chastain, P. Parniewski, K. Oshima, M. Pandolfo, J. D. Griffith, and R. D. Wells. 1999. Sticky DNA: self association properties of long GAA · TTC repeats in R · R · Y triplex structures from Friedreich's ataxia. Mol. Cell 3:465-475. [DOI] [PubMed] [Google Scholar]
- 254.Samadashwily, G. M., A. Dayn, and S. M. Mirkin. 1993. Suicidal nucleotide sequences for DNA polymerization. EMBO J. 12:4975-4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Samadashwily, G. M., G. Raca, and S. M. Mirkin. 1997. Trinucleotide repeats affect DNA replication in vivo. Nat. Genet. 17:298-304. [DOI] [PubMed] [Google Scholar]
- 256.Sanchez-Gorostiaga, A., C. Lopez-Estrano, D. B. Krimer, J. B. Schvartzman, and P. Hernandez. 2004. Transcription termination factor reb1p causes two replication fork barriers at its cognate sites in fission yeast ribosomal DNA in vivo. Mol. Cell. Biol. 24:398-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Sandler, S. J. 2000. Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics 155:487-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Sandler, S. J., and K. J. Marians. 2000. Role of PriA in replication fork reactivation in Escherichia coli. J. Bacteriol. 182:9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Santamaria, D., G. de la Cueva, M. L. Martinez-Robles, D. B. Krimer, P. Hernandez, and J. B. Schvartzman. 1998. DnaB helicase is unable to dissociate RNA-DNA hybrids. Its implication in the polar pausing of replication forks at ColE1 origins. J. Biol. Chem. 273:33386-33396. [DOI] [PubMed] [Google Scholar]
- 260.Santamaria, D., P. Hernâandez, M. L. Martinez-Robles, D. B. Krimer, and J. B. Schvartzman. 2000. Premature termination of DNA replication in plasmids carrying two inversely oriented ColE1 origins. J. Mol. Biol. 300:75-82. [DOI] [PubMed] [Google Scholar]
- 261.Schlissel, M. S. 2000. Perspectives: transcription. A tail of histone acetylation and DNA recombination. Science 287:438-440. [DOI] [PubMed] [Google Scholar]
- 262.Schroth, G. P., and P. S. Ho. 1995. Occurrence of potential cruciform and H-DNA forming sequences in genomic DNA. Nucleic Acids Res. 23:1977-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Schwartz, M., E. Zlotorynski, and B. Kerem. 2006. The molecular basis of common and rare fragile sites. Cancer Lett. 232:13-26. [DOI] [PubMed] [Google Scholar]
- 264.Schweitzer, J. K., and D. M. Livingston. 1999. The effect of DNA replication mutations on CAG tract stability in yeast. Genetics 152:953-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Schweitzer, J. K., and D. M. Livingston. 1998. Expansions of CAG repeat tracts are frequent in a yeast mutant defective in Okazaki fragment maturation. Hum. Mol. Genet. 7:69-74. [DOI] [PubMed] [Google Scholar]
- 266.Seigneur, M., V. Bidnenko, S. D. Ehrlich, and B. Michel. 1998. RuvAB acts at arrested replication forks. Cell 95:419-430. [DOI] [PubMed] [Google Scholar]
- 267.Sherman, L. A., and M. L. Gefter. 1976. Studies of the mechanism of enzymatic DNA elongation of by Escherichia coli DNA polymerase II. J. Mol. Biol. 103:61-76. [DOI] [PubMed] [Google Scholar]
- 268.Shimizu, M., R. Gellibolian, B. A. Oostra, and R. D. Wells. 1996. Cloning, characterization and properties of plasmids containing CGG triplet repeats from the FMR-1 gene. J. Mol. Biol. 258:614-626. [DOI] [PubMed] [Google Scholar]
- 269.Sinden, R. R. 1994. DNA structure and function. Academic Press, Inc., San Diego, CA.
- 270.Sista, P. R., S. Mukherjee, P. Patel, G. S. Khatri, and D. Bastia. 1989. A host-encoded DNA-binding protein promotes termination of plasmid replication at a sequence-specific replication terminus. Proc. Natl. Acad. Sci. USA 86:3026-3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Smith, M. T., and R. G. Wake. 1992. Definition and polarity of action of DNA replication terminators in Bacillus subtilis. J. Mol. Biol. 227:648-657. [DOI] [PubMed] [Google Scholar]
- 272.Sogo, J. M., M. Lopes, and M. Foiani. 2002. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297:599-602. [DOI] [PubMed] [Google Scholar]
- 273.Spellman, P. T., G. Sherlock, M. Q. Zhang, V. R. Iyer, K. Anders, M. B. Eisen, P. O. Brown, D. Botstein, and B. Futcher. 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9:3273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Spiro, C., R. Pelletier, M. L. Rolfsmeier, M. J. Dixon, R. S. Lahue, G. Gupta, M. S. Park, X. Chen, S. V. S. Mariappan, and C. T. McMurray. 1999. Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol. Cell 4:1079-1085. [DOI] [PubMed] [Google Scholar]
- 275.Stewart, S. E., and G. S. Roeder. 1989. Transcription by RNA polymerase I stimulates mitotic recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 9:3464-3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Strauss, B. S. 2005. Excision repair and bypass, p. 431-447. In N. P. Higgins (ed.), The bacterial chromosome. ASM Press, Washington, DC.
- 277.Subramanian, P. S., D. L. Nelson, and A. C. Chinault. 1996. Large domains of apparent delayed replication timing associated with triplet repeat expansion at FRAXA and FRAXE. Am. J. Hum. Genet. 59:407-416. [PMC free article] [PubMed] [Google Scholar]
- 278.Sutherland, G. R., E. Baker, and R. I. Richards. 1998. Fragile sites still breaking. Trends Genet. 14:501-506. [DOI] [PubMed] [Google Scholar]
- 279.Takeuchi, Y., T. Horiuchi, and T. Kobayashi. 2003. Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev. 17:1497-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Tercero, J. A., M. P. Longhese, and J. F. Diffley. 2003. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 11:1323-1336. [DOI] [PubMed] [Google Scholar]
- 281.Thomas, B. J., and R. Rothstein. 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56:619-630. [DOI] [PubMed] [Google Scholar]
- 282.Trinh, T. Q., and R. R. Sinden. 1991. Preferential DNA secondary structure mutagenesis in the lagging strand of replication in E. coli. Nature 352:544-547. [DOI] [PubMed] [Google Scholar]
- 283.Usdin, K. 1998. NGG-triplet repeats form similar intrastrand structures: implications for the triplet expansion diseases. Nucleic Acids Res. 26:4078-4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Usdin, K., and E. Grabczyk. 2000. DNA repeat expansions and human disease. Cell. Mol. Life Sci. 57:914-931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Usdin, K., and K. J. Woodford. 1995. CGG repeats associated with DNA instability and chromosome fragility from structures that block DNA synthesis in vitro. Nucleic Acids Res. 23:4202-4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Verkerk, A. J. M. H., M. Pieretti, J. S. Sutcliffe, Y. H. Fu, D. P. A. Kuhl, A. Pizzuti, O. Reiner, S. Richards, M. F. Victoria, F. Zhang, B. W. Eussen, G.-J. B. van Ommen, L. A. J. Blonden, G. J. Riggins, J. L. Chastain, C. B. Kunst, H. Galjaard, C. T. Caskey, D. R. Nelson, B. A. Oostra, and S. T. Warren. 1991. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65:905-914. [DOI] [PubMed] [Google Scholar]
- 287.Vetcher, A. A., M. Napierala, R. R. Iyer, P. D. Chastain, J. D. Griffith, and R. D. Wells. 2002. Sticky DNA, a long GAA · GAA · TTC triplex that is formed intramolecularly, in the sequence of intron 1 of the frataxin gene. J. Biol. Chem. 277:39217-39227. [DOI] [PubMed] [Google Scholar]
- 288.Viguera, E., P. Hernandez, D. B. Krimer, A. S. Boistov, R. Lurz, J. C. Alonso, and J. B. Schvartzman. 1996. The ColE1 unidirectional origin acts as a polar replication fork pausing site. J. Biol. Chem. 271:22414-22421. [DOI] [PubMed] [Google Scholar]
- 289.Vilette, D., S. D. Ehrlich, and B. Michel. 1995. Transcription-induced deletions in Escherichia coli plasmids. Mol. Microbiol. 17:493-504. [DOI] [PubMed] [Google Scholar]
- 290.Vincent, S. D., A. A. Mahdi, and R. G. Lloyd. 1996. The RecG branch migration protein of Escherichia coli dissociates R-loops. J. Mol. Biol. 264:713-721. [DOI] [PubMed] [Google Scholar]
- 291.Waga, S., and B. Stillman. 1998. The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem. 67:721-751. [DOI] [PubMed] [Google Scholar]
- 292.Wang, J., and B. Sugden. 2005. Origins of bidirectional replication of Epstein-Barr virus: models for understanding mammalian origins of DNA synthesis. J. Cell Biochem. 94:247-256. [DOI] [PubMed] [Google Scholar]
- 293.Wang, Y., M. Vujcic, and D. Kowalski. 2001. DNA replication forks pause at silent origins near the HML locus in budding yeast. Mol. Cell. Biol. 21:4938-4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Ward, T. R., M. L. Hoang, R. Prusty, C. K. Lau, R. L. Keil, W. L. Fangman, and B. J. Brewer. 2000. Ribosomal DNA replication fork barrier and HOT1 recombination hot spot: shared sequences but independent activities. Mol. Cell. Biol. 20:4948-4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Weaver, D. T., and M. L. DePamphilis. 1982. Specific sequences in native DNA that arrest synthesis by DNA polymerase alpha. J. Biol. Chem. 257:2075-2086. [PubMed] [Google Scholar]
- 296.Weitzmann, M. N., K. J. Woodford, and K. Usdin. 1997. DNA secondary structures and the evolution of hypervariable tandem arrays. J. Biol. Chem. 272:9517-9523. [DOI] [PubMed] [Google Scholar]
- 297.Wells, R. D. 1996. Molecular basis of genetic instability of triplet repeats. J. Biol. Chem. 271:2875-2878. [DOI] [PubMed] [Google Scholar]
- 298.Wells, R. D., and S. T. Warren (ed.). 1998. Genetic instabilities and hereditary neurological disorders. Academic Press, San Diego, CA.
- 299.White, P. J., R. H. Borts, and M. C. Hirst. 1999. Stability of the human fragile X (CGG)n triplet repeat array in Saccharomyces cerevisiae deficient in aspects of DNA metabolism. Mol. Cell. Biol. 19:5675-5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Wiesendanger, B., R. Lucchini, T. Koller, and J. M. Sogo. 1994. Replication fork barriers in the Xenopus rDNA. Nucleic Acids Res. 22:5038-5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Williamson, J. R., M. K. Raghuraman, and T. R. Cech. 1989. Monovalent cation-induced structure of telomeric DNA: the G-quartet model. Cell 59:871-880. [DOI] [PubMed] [Google Scholar]
- 302.Woodford, K. J., R. M. Howell, and K. Usdin. 1994. A novel K(+)-dependent DNA synthesis arrest site in a commonly occuring sequence motif in eukaryotes. J. Biol. Chem. 269:27029-27035. [PubMed] [Google Scholar]
- 303.Wu, H.-Y., S. Shyy, J. C. Wang, and L. F. Liu. 1988. Transcription generates positively and negatively supercoiled domains in the template. Cell 53:433-440. [DOI] [PubMed] [Google Scholar]
- 304.Wu, T. C., and M. Lichten. 1994. Meiosis-induced double-strand break sites determined by yeast chromatin structure. Science 263:515-518. [DOI] [PubMed] [Google Scholar]
- 305.Xu, L., and N. Kleckner. 1995. Sequence non-specific double-strand breaks and interhomolog interactions prior to double-strand break formation at a meiotic recombination hot spot in yeast. EMBO J. 14:5115-5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Yancopoulos, G. D., and F. W. Alt. 1985. Developmentally controlled and tissue-specific expression of unrearranged VH gene segments. Cell 40:271-281. [DOI] [PubMed] [Google Scholar]
- 307.Yu, A., M. D. Barron, R. M. Romero, M. Christy, B. Gold, J. Dai, D. M. Gray, I. S. Haworth, and M. Mitas. 1997. At physiological pH, d(CCG)15 forms a hairpin containing protonated cytosines and a distorted helix. Biochemistry 36:3687-3699. [DOI] [PubMed] [Google Scholar]
- 308.Yu, A., J. Dill, S. S. Wirth, G. Huang, V. H. Lee, I. S. Haworth, and M. Mitas. 1995. The trinucleotide repeat sequence d(GTC)15 adopts a hairpin conformation. Nucleic Acids Res. 23:2706-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Yu, S., M. Mangelsdorf, D. Hewett, L. Hobson, E. Baker, H. J. Eyre, N. Lapsys, D. Le Paslier, N. A. Doggett, G. R. Sutherland, and R. I. Richards. 1997. Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell 88:367-374. [DOI] [PubMed] [Google Scholar]
- 310.Zhang, Z., D. M. Macalpine, and G. M. Kapler. 1997. Developmental regulation of DNA replication: replication fork barriers and programmed gene amplification in Tetrahymena thermophila. Mol. Cell. Biol. 17:6147-6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Zheng, M., X. Huang, G. K. Smith, X. Yang, and X. Gao. 1996. Genetically unstable CXG repeats are structurally dynamic and have a high propensity for folding. An NMR and UV spectroscopic study. J. Mol. Biol. 264:323-336. [DOI] [PubMed] [Google Scholar]