

Abstract
Medullary thymic epithelial cells function as antigen-presenting cells in negative selection of self-reactive T cell clones, a process essential for the establishment of central self-tolerance. These cells mirror peripheral tissues through promiscuous expression of a diverse set of tissue-restricted self-antigens. The genes and signaling pathways that regulate the development of medullary thymic epithelial cells are not fully understood. Here we show that mice deficient in NF-κB2, a member of the NF-κB family, display a marked reduction in the number of mature medullary thymic epithelial cells that express CD80 and bind the lectin Ulex europaeus agglutinin-1, leading to a significant decrease in the extent of promiscuous gene expression in the thymus of NF-κB2−/− mice. Moreover, NF-κB2−/− mice manifest autoimmunity characterized by multiorgan infiltration of activated T cells and high levels of autoantibodies to multiple organs. A subpopulation of the mice also develops immune-complex glomerulonephritis. These findings identify a physiological function of NF-κB2 in the development of medullary thymic epithelial cells and, thus, the control of self-tolerance induction.
In the thymus, self-reactive T cells are eliminated through negative selection in which the T-cell receptor of a thymocyte engages a high affinity peptide-MHC ligand presented by an antigen-presenting cell, leading to the apoptotic death of the thymocyte (1). Although it has been known for many years that medullary thymic epithelial cells (mTECs)1 have a crucial role in negative selection by acting as antigen-presenting cells (2–4), only recently is the underlying mechanism beginning to emerge. mTECs express a broad spectrum of peripheral tissue-restricted self-antigens, termed promiscuous gene expression (5,6). Evidence for a crucial role of this promiscuous gene expression in self-tolerance induction comes from analysis of mice lacking Aire, a transcription factor that is mutated in the human disease autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) (7,8). mTECs from mice deficient in Aire have diminished expression of tissue-restricted self-antigens, and these mice develop a multiorgan autoimmune syndrome similar to APECED (9). The development of mTECs is accompanied by an increase in the expression levels of CD80 and a carbohydrate-epitope that binds the lectin UEA-1 (10–12). Both UEA-1− and UEA-1+ mTECs display promiscuous gene expression (5). However, a more recent study shows a close correlation between the expression levels of CD80 and the extent of promiscuous gene expression (6). The genes and signaling pathways that regulate the development of mTECs are not fully understood.
NF-κB2 is a member of the NF-κB family of transcription factors that also include p105/p50 (NF-κB1), RelA (p65), RelB, and c-Rel. The full-length NF-κB2 protein p100 is preferentially associated with RelB in the cytoplasm (13,14), which prevents RelB nuclear translocation and represses RelB-dependent transcription. Phosphorylation of the C-terminus of p100 by IKKα, which itself is activated by NIK, leads to proteolytic processing of p100 into p52 (15,16). The resulting p52-RelB heterodimers then translocate into the nucleus and activate the transcription of their target genes. This alternative NF-κB signaling pathway is activated by engagement of receptors for BAFF, LTβ, and CD40 ligand (14,17–19). Previous studies with NF-κB2−/− mice demonstrate a crucial role of NF-κB2 in B cell development and secondary lymphoid organogenesis. These mice present a marked decrease in the B cell population in peripheral lymphoid organs, and the absence of discrete perifollicular marginal and mantle zones, and of germinal centers in the spleen (20,21).
Recently, several studies provide convincing evidence for a critical role of the LTβR signaling pathway in regulation of mTEC development. Mice deficient in LTβR, IKKα, or carrying a loss-of-function mutant of NIK (NIKaly/aly) all display disorganized thymic medulla, reduced numbers of mTECs, and overt autoimmunity (22–24). As the LTβR signaling pathway is intimately involved in activation of NF-κB2 (25,26), these findings also implicate a role for NF-κB2 in the development of mTECs (12). However, defects in LTβR signaling not only impair processing of NF-κB2 p100 into p52, but also result in accumulation of p100, which may lead to repression of RelB-dependent transcription. In fact, it was recently suggested that it is the increase in the p100 levels, rather than the absence of p52, that might be responsible for the impaired mTEC development observed in IKKα-deficient and NIKaly/aly mice (24).
In this report, we describe an autoimmune phenotype for NF-κB2−/− mice that lack both p100 and p52, and present evidence for a physiological function of NF-κB2 in the development of mTECs. Our findings, in conjunction with studies of other mutant mouse strains, delineate an NF-κB2-activation signaling pathway that links thymic organogenesis to the establishment of self-tolerance.
EXPERIMENTAL PROCEDURES
Mice
NF-κB2−/− mice (20) were crossed to B6129SF1/J (Jackson Laboratory), and the heterozygous offspring were interbred to obtain NF-κB2−/−, heterozygous, and wild-type littermates. NOD.SCID/NCr mice were purchased from the National Cancer Institute at Frederick. All of the animals were maintained under specific pathogen-free conditions at the animal facility of the Medical University of Ohio, and all of the animal procedures were pre-approved by the Institutional Animal Care and Use Committee.
Histology and immunohistochemistry
Tissues were fixed in 10% neutral buffered formalin, embedded in paraffin blocks, sectioned at 5 μm, and stained with H&E. Histological examination of silver stained lung sections for possible Pneumocystis infection was conducted by the Research Animal Diagnostic Laboratory at the University of Missouri. For immunohistochemistry, the paraffin was removed and sections were rehydrated according to standard procedures. For retrieval of B220 and CD3 antigen, the sections were subjected to boiling in 10 mM citrate buffer (pH 6.0) or 1 mM EDTA (pH 8.0) for 10 min, respectively. Following quenching of endogenous peroxidase activity with H2O2 and blocking with normal serum, the sections were incubated for 1 h with rat mAb against CD45R/B220 (RA3-6B2, BD Pharmingen, 5 μg/ml) or CD3 (CD3-12, Serotec, 10 μg/ml). Isotype-matched rat mAb (BD Pharmingen, 10 μg/ml) was used as control. After washing, biotinylated rabbit anti-rat antibody (Vector Laboratories) was applied for 30 min. The sections were then incubated for 30 min with ABC reagent (Vector Laboratories), and the immunostaining was visualized with DAB, (Sigma). The tissue sections were counter-stained with Hematoxylin and examined under a light microscope.
Immunofluorescence
Thymi from 4- to 6-week-old NF-κB2−/− and wild type mice were embedded in OCT compound (Sakura) and snap frozen. Sections at 5 μm were cut from the frozen blocks, fixed in cold acetone, rehydrated in PBS plus 0.1% saponin (Sigma), and blocked for 1 h at room temperature with 5% goat serum in PBS. The sections were incubated with rabbit anti-mouse Aire polyclonal antibody (27) (1:1000), hamster anti-mouse CD11c mAb (HL3, 1:200), rat anti-mouse Ep-CAM mAb (G8.8, 1:100), biotinylated hamster anti-mouse CD80 mAb (16-10A1, 1:200), and biotinylated UEA-1 (2 μg/ml, Sigma) for 1 h at room temperature. After washing with PBS, the sections were incubated with FITC-goat anti-rabbit IgG (Molecular Probes, 1:500), FITC-mouse anti-hamster IgG (1:200), and biotin-mouse anti-rat IgG2a (1:100) for 1 h at room temperature. The biotin-conjugated antibodies were detected with PE-Streptavidin (Southern Biotechnology Associates). For direct immunofluorescence staining, sections were incubated with FITC-rat anti-mouse B220 mAb (RA3-6B2, 1:1000) or PE-rat anti-mouse IgM mAb (R6-60.2, 1:200). Unless indicated, all antibodies were obtained from BD Pharmingen. DAPI (Molecular Probes, 300 nM) was used for counterstaining of nuclei. For detection of immune complexes in renal glomeruli, cryostat sections of kidney were fixed in cold acetone for 15 min, rehydrated in PBS, and blocked with 10% goat serum/3% BSA in PBS for 2 h at room temperature. The sections were then incubated with FITC-goat anti-mouse IgG (Molecular Probes, 1:500) for 1 h at room temperature. To detect autoantibodies, cryostat sections of various organs from 8-weeks-old NOD.SCID/NCr mice were fixed in cold methanol for 5 min, blocked with 10% goat serum in PBS for 1 h at room temperature, and incubated with 1:40 dilutions of the serum from individual 1-year-old NF-κB2−/− and wild type littermates. The sections were then incubated with FITC-goat anti-mouse IgM (Southern Biotechnology Associates, 1:200) for 1 h. DAPI was used for counterstaining of nuclei as described above. Fluorescent images were taken on Nikon Eclipse E800 microscope.
Real-time PCR
Real-time PCR quantification was conducted with cDNA prepared from total RNA extracted from individual thymi of 4-week-old NF-κB2−/− and wild-type mice. The primers and probes for Aire, Spt1, FABP, GAD67, and glyceraldehyde phosphodehydrogenase (GAPDH) were as previously described (28,29). PCR reactions in triplicate were performed using the TaqMan Universal PCR Master Mix (Applied Biosystems) and run on an Applied Biosystems 7500 Real-time PCR system, according to the manufacturer’s instruction.
In vivo anti-CD3 antibody induced apoptosis
NF-κB2−/− and wild type control mice (4–6 weeks) were injected intraperitoneally with 20 μg of hamster anti-mouse CD3ε mAb (145-2C11, BD Pharmingen) or, as control, with PBS. Mice were sacrificed 40 h later. Thymocytes were collected and counted, and cell subset distribution was determined by flow cytometry (Epics Elite, Beckman-Coulter) after staining with FITC-rat anti-mouse CD4 (GK1.5) and PE-rat anti-mouse CD8 (53−6.7, both from BD Pharmingen).
Analysis of lung infiltrating cells
The infiltrating cells were isolated as described (28). Briefly, lung tissues were finely minced and digested for 30 min in 15 ml of RPMI 1640 containing 1 mg/ml collagenase VIII (Sigma) and 2% FBS at 37°C. Cell suspensions were passed through a 100-μm Nitex filter, and red blood cells were depleted with ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.3). The cells were stained with PE-rat anti-mouse CD4 (RM4-5), PE-rat anti-mouse CD8, FITC-rat anti-mouse CD44 (IM7), FITC-hamster anti-mouse CD69 (H1.2F3, all from BD Pharmingen), and then analyzed by flow cytometry.
Analysis of thymic stromal cells
Thymic stromal cells were prepared as described (30,31). Briefly, thymi of 4- to 6-week-old mice were minced and slowly stirred in the medium (RPMI 1640 plus 2% FBS) to release the majority of thymocytes. The tissue fragments were sequentially digested with 0.5 mg/ml collagenase D (Roche) and collagenase D/dispase I (Roche, 0.2 mg/ml each) in the presence of 25 μg/ml DNase I (Worthington) in the same medium to release epithelial cells. The digests were pooled, disrupted by 5 mM EDTA, and separated on a discontinuous Percoll gradient (Pharmacia) with densities of ρ= 1.115 g/ml, 1.06 g/ml, and 1.0 g/ml (from bottom to top) by centrifugation at 1,350 ×g for 30 min at 4°C. The thymic epithelial cell fraction was collected from the interphase between ρ= 1.06 and 1.0, and subjected to three-step staining with Mouse BD Fc Block (2.4G2), with rat anti-mouse Ep-CAM (G8.8) and Biotin-UEA-1 or Biotin-CD80 (16-10A1), and then with PE-Cy5-conjugated CD45 (30-F11), PE-conjugated Ly51 (BP-1), FITC-conjugated mouse anti-rat IgG2a (RG7/1.30), and PE-Texas Red-conjugated Streptavidin (all from BD Pharmingen). Cells were sorted on Epics Elite (Beckman-Coulter) with appropriate forward- and side-scatter setting to exclude thymocytes, and the data were analyzed with WinMDI 2.8 software.
Analysis of regulatory T cells
Single-cell suspensions were prepared from the spleen of 4- to 6-week-old mice according to standard procedures. Red blood cells were depleted as described above. Regulatory T cells were detected by staining for the expression of CD4, CD25, and Foxp3, using a mouse regulatory T cell staining kit (eBioscience) according to the manufacturer’s instructions.
RESULTS
Multiorgan lymphocytic infiltration in NF-κB2−/− mice
During a study of apoptosis regulation in thymocytes and T cells by NF-κB2 p100 and its processed product p52, we noticed that a significant number of NF-κB2−/− mice died prematurely when compared with their heterozygous and wild-type littermates (Figure 1A). Histological examination of various tissue samples revealed that, of the 20 deceased NF-κB2−/− mice, 5 had leukemia or lymphoma, indicated by complete effacement of normal bone marrow and spleen architecture by massive infiltration of medium-sized lymphocytes with pleomorphic nuclei and abundant cytoplasm (data not shown). However, the remaining 15 deceased NF-κB2−/− mice showed no obvious sign of leukemia in their bone marrow and spleen samples. Instead, these mice had extensive perivascular infiltration of morphologically normal lymphocytes in multiple organs (data not shown, see also Figure 1B). The cellular infiltrates might result in organ dysfunction, leading to premature death.
Fig. 1. Multiorgan infiltration of activated T cells in NF-κB2−/− mice.
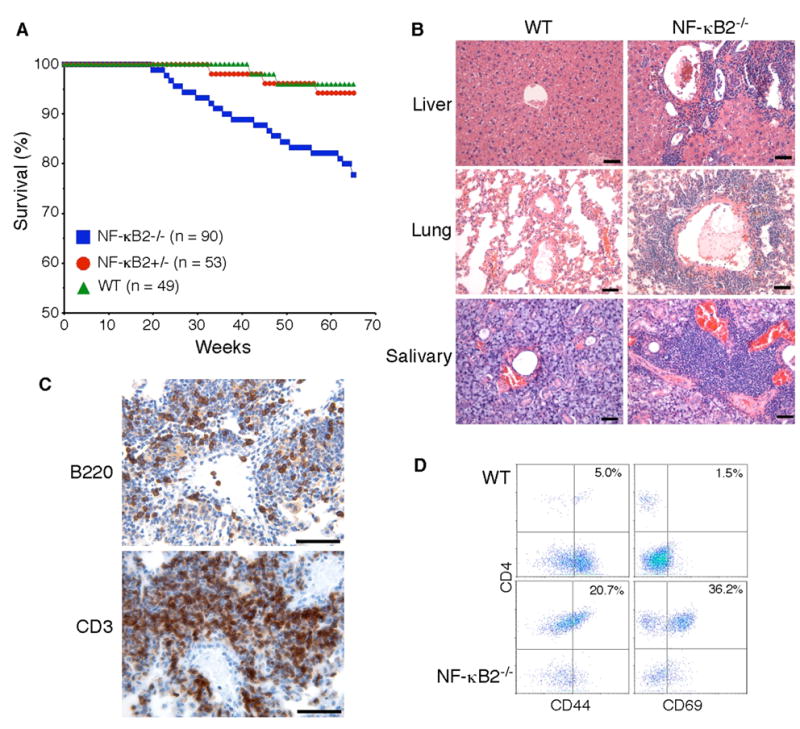
A, Survival curves of NF-κB2−/−, NF-κB2+/− and wild-type mice. Numbers of mice for each genotype are indicated. B, Infiltration of peripheral organs revealed by H&E staining of formalin-fixed sections of the liver, lung, and salivary gland from a 10-month-old NF-κB2−/− mouse, with an age-matched wild-type mouse as control. Scale bars, 100 μm. C, Immunohistochemical staining of formalin-fixed lung sections from a 1-year-old NF-κB2−/− mouse. Most of the infiltrating cells were stained strongly for CD3, a T-cell marker, and a significant number of the infiltrating cells stained positively for B220, a B-cell marker. Data are representatives of 4 mice for each genotype. Scale bars, 100 μm. D, CD4+ T cell infiltrates in the lung of a 1-year-old NF-κB2−/− mouse show increased expression of the memory marker CD44 and the activation marker CD69 in comparison with an age-matched wild-type mouse. Numbers indicate percentages of the gated populations. Data are representatives of 4 mice for each genotype.
We next examined 20 NF-κB2−/− mice at 10–12 months of age to determine whether the multiorgan lymphocytic infiltration observed in the deceased mice was a consistent feature in NF-κB2−/− mice. All of the NF-κB2−/− mice showed marked lymphocytic infiltration in the lung, liver and salivary gland (Figure 1B). Of note, many of the lung lesions appeared to have lymphocytic invasion of artery walls, consistent with vasculitis (Figure 1B). Immunohistochemical staining of lung sections revealed that most of the infiltrating cells were CD3+ T cells (Figure 1C). No significant lymphocytic infiltration was found in major organs of the 15 age-matched wild-type littermates examined (Figure 1B). This phenotype of multiorgan lymphocytic infiltration in NF-κB2−/− mice is very similar to that described for various mouse strains with autoimmunity, such as those lacking Aire, LTβR, IKKα, or with NIK mutation (9,22–24,28), suggesting that NF-κB2−/− mice may also develop autoimmunity.
Given the profound defect in B cell-mediated responses in NF-κB2−/− mice (20,21), we examined the possibility that the multiorgan infiltration might represent responses to chronic infection by opportunistic pathogens such as Pneumocystis carinii. Histological examination of silver stained sections of the lungs from 1-year-old wild-type (n=2) and NF-κB2−/− (n=3) mice revealed no indication of Pneumocystis infection (data not shown).
Compared to wild-type and heterozygous littermates, NF-κB2−/− mice have significantly fewer and smaller lymph nodes, probably due to a defect in lymph node development (Zhang and Ding, unpublished data). Thus, the observed multiorgan lymphocytic infiltration could be a result of homeostatic reorganization. To investigate this possibility, we isolated infiltrating cells from the lungs and performed flow cytometry analysis for expression of T-cell activation and memory phenotype markers (Figure 1D). The majority of the infiltrating cells in the lungs of NF-κB2−/− mice were CD4+ T cells. Staining for the activation marker CD69 revealed an average of 25-fold increase in the percentage of activated CD4+ cells in the lungs of NF-κB2−/− mice. The percentage of the CD4+ cells with the CD44+ memory phenotype was increased by an average of 4-fold. These data indicate that the lymphocytic infiltration in the organs of NF-κB2−/− mice was a result of an active ongoing immune response, rather than a passive homeostatic process.
Autoimmune diseases in NF-κB2−/− mice
Given the apparent autoimmune phenotype of NF-κB2−/− mice, we further examined these mice for signs of autoimmune disease. Of the deceased NF-κB2−/− mice (n = 20), 55% showed pathological evidence of glomerulopathy, including mesangial proliferation, crescent formation, and diffuse interstitial lymphocytic infiltrates (Figure 2A). Immunofluorescence staining of cryostat sectioned renal tissues using anti-IgG revealed deposits of immune complexes with a granular pattern in both the mesangial matrix and capillary loops (Figure 2B), suggesting that these mice had immune complex glomerulonephritis. We also examined 20 NF-κB2−/− mice at 10–12 months of age and found 30% of them had immune complex glomerulonephritis. None of the 15 age-matched or the two deceased wild-type littermates showed any pathological features of autoimmune renal disease.
Fig. 2. Autoimmune diseases in NF-κB2−/− mice.
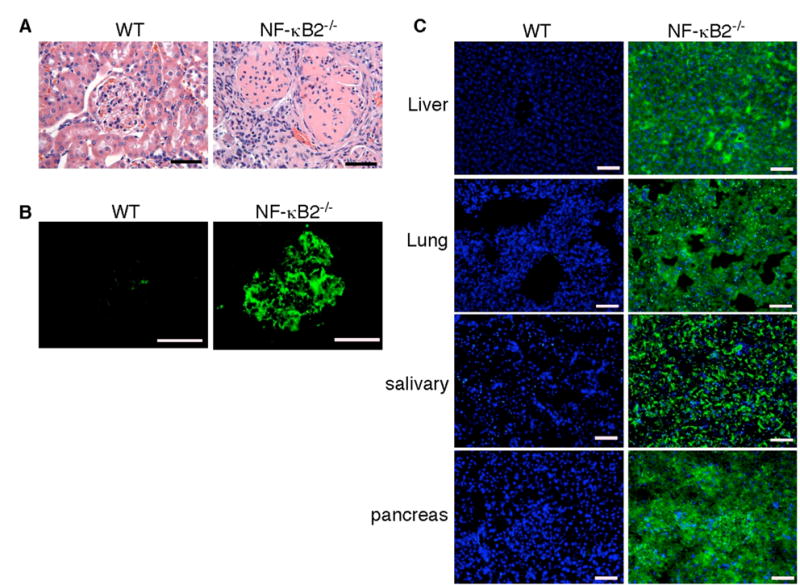
A, H&E staining of formalin-fixed renal sections of deceased NF-κB2−/− and wild-type mice. The renal cortex of the NF-κB2−/− mouse shows mesangial proliferation, crescent formation, and diffuse interstitial lymphocytic infiltrates. B, The presence of IgG-containing immune complexes in glomeruli of the same NF-κB2−/− mouse, revealed by staining cryostat renal sections with FITC-labeled anti-mouse IgG. C, NF-κB2−/− mice have autoantibodies to multiple organs, revealed by FITC-labeled anti-mouse IgM staining of NOD/SCID mouse tissue sections of liver, lung, salivary gland, and pancreas preincubated with serum from individual 1-year-old NF-κB2−/− or wild-type mice. Nuclei were stained with DAPI. Shown is representative staining of the tissue sections with sera from 6 mice for each genotype. Scale bars, 100 μm.
It has been shown previously that, despite of a marked reduction in the peripheral B-cell population and impaired B-cell mediated immune response, NF-κB2−/− mice had a significant increase (4.5-fold) in the levels of serum IgM in comparison with control wild-type littermates (20). Immunostaining of NOD/SCID mouse tissues with sera from 1-year-old NF-κB2−/− mice (n = 6) revealed the presence of high levels of IgM autoantibodies against multiple organs including the liver, lung, salivary gland, and pancreas (Figure 2C). We also detected high levels of antibodies against double-stranded DNA in 2 out of the 6 serum samples from NF-κB2−/− mice, but not in any of the 6 serum samples from age-matched wild-type littermates (data not shown). Together with the results of kidney histopathological analyses, these findings indicate that NF-κB2−/− mice developed systemic autoimmune disease, a phenotype consistent with a broad defect in self-tolerance induction.
NF-κB2 deficiency has no significant effect on activation-induced thymocyte apoptosis and the number of Aire-expressing cells in the thymus
Self-tolerance is maintained by multiple mechanisms, such as expression and presentation of self-antigens in the thymus, induction of apoptosis during negative selection, and production of regulatory T cells (32). Defects in any one of these mechanisms could lead to autoimmunity. As NF-κB2 regulates death receptor-mediated apoptosis (33), which has been shown to play a role in negative selection (34), we examined NF-κB2−/− mice for the ability of anti-CD3 antibody to induce apoptosis in CD4+CD8+ thymocytes, a widely used model of negative selection (35). No significant difference was observed in the levels of thymocyte death between NF-κB2−/− mice and their age-matched wild-type littermates after injection of anti-CD3 antibody (Figure 3A). We also performed in vitro assays of anti-CD3-induced apoptosis of thymocytes and obtained similar results (data not shown). Thus, NF-κB2 deficiency has no apparent effect on the ability of thymocytes to undergo apoptosis induced by T-cell receptor ligation, at least in the model systems examined.
Fig. 3. Effects of NF-κB2 deficiency on activation-induced thymocyte apoptosis and Aire expression.
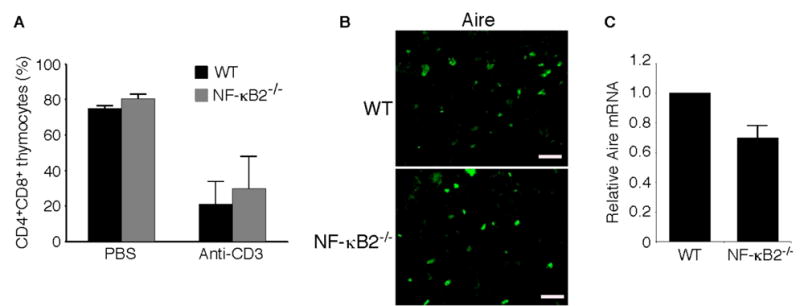
A, In vivo anti-CD3-induced apoptosis in CD4+CD8+ immature thymocytes in 4- to 6-week-old NF-κB2−/− and wild-type mice. Thymocytes were collected 40 h after intraperitoneal injection of 20 μg of anti-CD3ε (145-2C11) and analyzed by flow cytometry. Data represent means ± standard deviations in percentages of total thymocytes from 3 mice for each genotype. B, Immunofluorescence staining for Aire expression of cryostat thymic sections from 4-week-old NF-κB2−/− and wild-type mice. Shown is representative staining of thymic sections from 3 mice for each genotype. Scale bars, 100 μm. C, Real-time PCR analysis of relative abundance of thymic Aire mRNA in 4- to 6-week-old NF-κB2−/− and wild-type mice. Data represent means ± standard deviations from 4 mice for each genotype.
RelB has been shown to play a critical role in regulation of Aire expression or the survival of Aire-expressing cells in the thymus (27). As the NF-κB2 p52/RelB heterodimer is a major component of the κB-binding activity found in the thymus (36), we speculated that NF-κB2 might have a role similar to that of RelB in the thymus. Immunofluorescence staining of cryostat sectioned thymic tissues using an antibody against Aire revealed no apparent difference in the frequency of Aire positive cells between age-matched NF-κB2−/− and wild-type mice (Figure 3B), whereas quantitative real-time PCR analysis showed a modest (30%), but consistent, reduction in the Aire mRNA levels in NF-κB2−/− mice (Figure 3C). These findings suggest that NF-κB2 does not appear to play a significant role in regulation of Aire expression or the number of Aire-expressing cells in the thymus.
Impaired mTEC development in NF-κB2−/− mice
To search further for the mechanism underlying the autoimmune phenotype of NF-κB2-deficient mice, we investigated a possible role of NF-κB2 in the development of the thymic medulla and its cellular constituents. Histological examination of the thymi from 4- to 6-week-old NF-κB2−/− mice revealed no gross alterations in the thymic architecture (Figure 4A), as previously reported (20). We also visualized thymic dendritic cells (DCs) with an antibody against the DC marker CD11c and epithelial cells with the antibody G8.8 that recognizes the marker epithelial cell adhesion molecule (Ep-CAM) (5). Immunofluorescence staining showed no differences in the numbers and distribution patterns of these cells between age-matched NF-κB2- deficient and wild-type mice (Figure 4B). In contrast, NF-κB2−/− mice have a marked reduction in the number of mTECs that bind the lectin UEA-1 (Figure 4B).
Fig. 4. Impaired mTEC development in NF-κB2−/− mice.
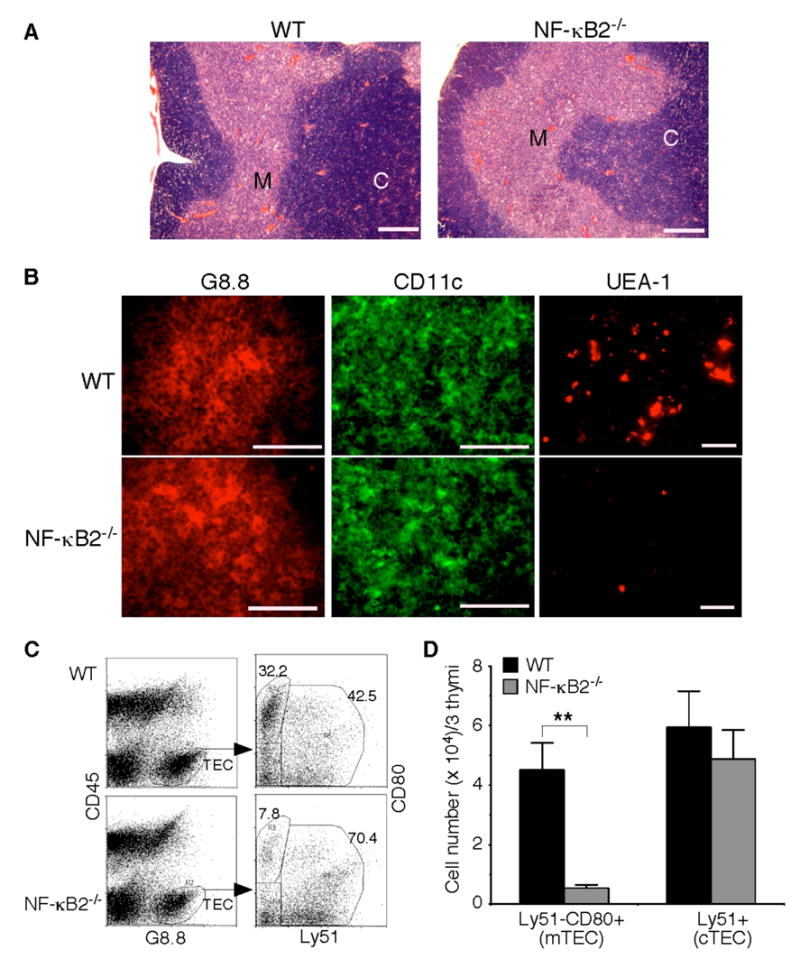
A, H&E staining of formalin-fixed thymic sections of 4- to 6-week-old NF-κB2−/− and wild-type mice. Shown is representative staining of thymic sections from 5 mice for each genotype. Scale bars, 500 μm. M, medulla; C, cortex. B, Immunofluorescence staining of thymic sections of 4- to 6-week-old NF-κB2−/− and wild-type mice for G8.8 (an epithelial cell marker), CD11c (a DC marker), or UEA-1 (a marker for mature mTECs). Shown is representative staining of thymic sections from 5 mice for each genotype. Scale bars, 100 μm. C–D, Flow cytometry analysis of thymic epithelial cell populations. Pooled thymic stromal cells from 3 NF-κB2−/− or wild-type mice of 4 to 6 weeks of age were stained with antibodies to CD45, G8.8, CD80, and Ly51. Mature mTECs are CD45−G8.8+Ly51−CD80+, and cTECs are CD45−G8.8+Ly51+. Data in D represent means ± standard deviations from three independent experiments with total 9 mice for each genotype. ** Student’s t-test, p<0.01.
To confirm these findings, we performed flow cytometry analysis of thymic epithelial cell populations (CD45−G8.8+) in 4- to 6-week-old NF-κB2−/− and wild-type mice (Figure 4C). No significant difference in the numbers of cortical TECs (cTECs, Ly51+) was observed between the knockout and wild-type mice (Figure 4D). However, the number of CD80+ cells was reduced by 88% in the thymus of NF-κB2−/− mice, in comparison with their wild-type littermates (Figure 4D). CD80 is a marker for mature mTECs (12) but also expressed on activated B cells (37,38). Dual immunofluorescence staining of thymic sections from both wild-type and NF-κB2−/− mice revealed that all of the CD80+ cells were negative for the B-cell markers B220 and surface IgM (data not shown). Taken together, these data indicate that NF-κB2−/− mice have a marked reduction in the number of CD80+ mTECs, suggesting that NF-κB2 has a specific and crucial role in the differentiation of mTECs and/or the survival of mature mTECs.
NF-κB2−/− mice show defects in promiscuous gene expression in the thymus
It was recently shown that CD80+ mTECs display the highest degree of promiscuous gene expression (6). As NF-κB2−/− mice show a marked reduction in the number of CD80+ mTECs, they may have defects in promiscuous expression of tissue-restricted self-antigens in the thymus. We investigated this possibility by examining the expression levels of three representative tissue-restricted self-antigens, Spt1, FABP, and GAD67 (9,22–24). Real-time PCR analysis revealed that the expression levels of Spt1 and FABP in the thymus of NF-κB2−/− mice were decreased by 65% and 85%, respectively, in comparison with their age-matched, wild-type littermates (Figure 5A). However, NF-κB2 deficiency had no apparent effect on the expression of GAD67 in the thymus (Figure 5A). These data suggest that NF-κB2−/− mice are defective in promiscuous expression of some, but not all, of tissue-restricted self-antigens, which may impair the process of self-tolerance induction, leading to the development of autoimmunity.
Fig. 5. NF-κB2−/− mice show defects in promiscuous gene expression in the thymus.
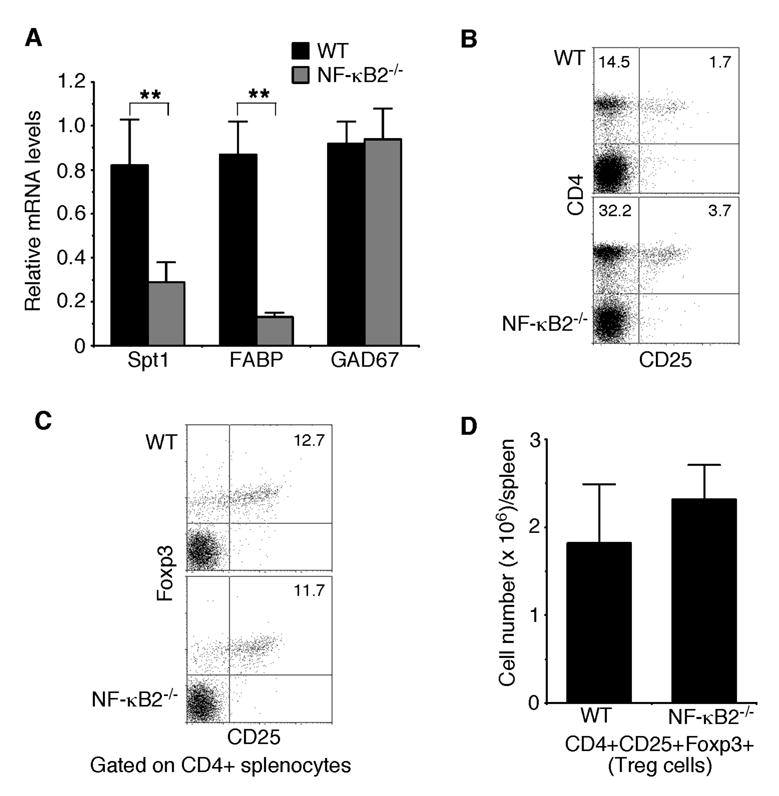
A, Real-time PCR analysis of the relative expression levels of three representative tissue-restricted self-antigens, Spt1, FABP, and GAD67. The highest expression levels of these antigens in wild-type mice are defined as 1.0. Data represent means ± standard deviations from 4 mice for each genotype. ** Student’s t-test, p<0.01. B, Flow cytometry analysis of CD4+CD25+ regulatory T cell populations in spleens of 4- to 6-week-old NF-κB2−/− and wild-type mice. Numbers indicate percentages of the CD4+ and CD4+CD25+ T cell populations. Data are representatives of 6 mice for each genotype. C–D, Flow cytometry analysis of CD4+CD25+Foxp3+ regulatory T cells in spleens of 4- to 6-week-old NF-κB2−/− and wild-type mice. The plots in C contain only the CD4+ cells in which percentages of the CD25+Foxp3+ T cells are shown. Data in D represent means ± standard deviations from 4 mice for each genotype.
We next examined the possibility that the reduced number of mTECs and the resulting defect in promiscuous gene expression in NF-κB2−/− mice may impair the production of CD4+CD25+ regulatory T cells (39,40), a population of T cells important for suppression of CD4+ T cell-mediated organ-specific autoimmune diseases (41,42). Flow cytometry analysis revealed that spleens from NF-κB2−/− mice actually have a 2.2-fold increase in the percentage of CD4+CD25+ T cells, compared to their wild-type littermates (Figure 5B). We wanted to point out that NF-κB2−/− mice also have more CD4+ T cells in their spleens (Figure 5B), and therefore, the ratio of CD4+CD25+ to CD4+ T cells is about same between NF-κB2−/− (10.5%) and wild-type (10.6%) mice. Given the activated phenotype of peripheral CD4+ T cells in NF-κB2−/− mice (Figure 1D), which may co-express CD25, we further analyzed the CD4+CD25+ T cells for the expression of Foxp3 (Figure 5C), a recently identified marker for regulatory T cells (43–45). The analysis revealed that NF-κB2−/− mice had a modest (27%) increase in the number of regulatory T cells in the spleen, compared to their wild-type littermates (Figure 5D). We also examined the frequency of regulatory T cells in the thymus and found no significant differences between NF-κB2-deficient and wild-type mice (data not shown). Together, these data indicate that NF-κB2 deficiency does not significantly affect the production of regulatory T cells. Thus, the autoimmune phenotype of NF-κB2−/− mice probably results from impaired elimination of auto-reactive T cells.
DISCUSSION
In this report we describe an autoimmune phenotype for NF-κB2−/− mice, manifesting as multiorgan infiltration of activated T cells, high levels of autoantibodies in the serum, and spontaneous development of immune complex glomerulonephritis in a subpopulation of the mice. We further show that NF-κB2−/− mice have a specific defect in the generation of UEA-1+ and CD80+ mature mTECs, leading to a marked reduction in promiscuous expression of some peripheral tissue-specific antigens critical for the induction of self-tolerance (5,6,46). The impaired development of mTECs and resulting breakdown in self-tolerance induction probably lead to autoimmunity in NF-κB2−/− mice. Thus, NF-κB2 not only is essential for B cell development and secondary lymphoid organogenesis, as reported before (20,21), but also has a physiological function in the development of mTECs and, thus, the control of self-tolerance induction.
Several recent studies demonstrate a crucial role for the LTβR signaling pathway in regulation of mTEC development. Mice deficient in LTβR, IKKα, or carrying NIKaly/aly mutation all display reduced numbers of mTECs and overt autoimmunity (22–24), a phenotype shared by NF-κB2−/− mice, as demonstrated in this study. Activation of the LTβR signaling pathway induces the processing of NF-κB2 p100 into p52 (17). As a result, defects in this signaling pathway lead to both p52 reduction and p100 accumulation. It was recently suggested that it is the increase in the p100 levels that might be responsible for the impaired mTEC development observed in IKKα −/− and NIKaly/aly mice (24). However, this model is not supported by our data, which, instead, suggest that the reduced p52 production is most likely the cause of the impaired mTEC development in mice deficient in LTβR signaling. Thus, our study provides the final evidence for an essential role of NF-κB2 activation by LTβR signaling in the differentiation, proliferation, and/or survival of mTECs.
We noticed that NIKaly/aly and IKKα −/− mice display more severe structural and cellular defects than do NF-κB2-deficient mice. Unlike NF-κB2−/− mice, NIKaly/aly and IKKα −/− mice show disorganization of thymic medulla and a marked reduction in the number of CD4+CD25+ regulatory T cells (23,24). Although the underlying mechanism remains to be defined, we speculate that it may be related to the control of RelB expression and activation. NF-κB2 p100 is an inhibitor of RelB transcriptional activity by forming a complex with RelB in the cytoplasm, which prevents RelB from associating with other NF-κB molecules and entering into the nucleus (13,14). Therefore, the absence of p100 in NF-κB2−/− mice is expected to result in activation of NF-κB complexes containing RelB. On the other hand, the expression of RelB is downregulated in NIKaly/aly and IKKα −/− mice (23,24). Thus, RelB may play a more general role in thymic organogenesis and regulatory T cell production. Consistent with the model, mice lacking TNF receptor-associated factor 6 (TRAF6), in which RelB expression is also downregulated, show a dramatic reduction in the size of the thymic medulla and in the number of CD4+CD25+ regulatory T cells (47). Also, the phenotype of RelB−/− mice more closely resembles that of NIKaly/aly, IKKα −/−, and TRAF6−/− mice, with complete disruption of the thymic medulla (48,49). A further examination of RelB-deficient mice will reveal whether they also have defects in the production of regulatory T cells.
The distinct phenotypes of knockout mice lacking individual NF-κB members suggest that they have non-redundant physiological functions. Since the NF-κB family members exert their biological functions as homo- or heterodimers, the functional specificity must be encoded in the particular NF-κB dimers. Thus, identification of particular NF-κB dimers that drive distinct biological processes is essential for a molecular understanding of NF-κB biology. Our study, coupled to the phenotypic analysis of RelB−/− mice (48,49), suggests a cell-type dependent specificity in RelB binding partners. The p52/RelB heterodimer appears to have an essential role in the control of mTEC differentiation and maturation, as both RelB−/− and NF-κB2−/− mice display a significant reduction in the number of UEA-1+ mTECs. However, in contrast to NF-κB2−/− mice, RelB−/− mice also show an absence of thymic DCs (48). Since the predominant κB-binding activity in thymic extracts is composed of p52/RelB and p50/RelB heterodimers (36), it is most likely that the p50/RelB dimer plays a critical role in the development of thymic DCs. This model is consistent with the reported defect in thymic DC function and development in NF-κB1 and NF-κB2 double knockout mice (50).
It was recently demonstrated that mTECs and cTECs share a common epithelial precursor (51–53). However, NF-κB2 deficiency has no apparent effect on the development of cTECs, despite of its essential role in the generation and/or maintenance of mTECs. A molecular understanding of this cell type-dependent activation of NF-κB2 will likely provide valuable insights into the process of thymic epithelial cell differentiation. Finally, the specific deficiency of mature mTECs in NF-κB2−/− mice provides an experimental system for identifying NF-κB2 target genes that regulate the development of mTECs.
The abbreviations used are:
- Aire
autoimmune regulator
- APECED
autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy
- BAFF
B-cell activating factor
- cTEC
cortical thymic epithelial cells
- DAB
3,3’-diaminobenzidine
- DAPI
4’,6’-diamidino-2-phenylindole
- DC
dendritic cell
- FABP
fatty acid-binding protein
- FITC
fluorescein isothiocyanate
- GAD67
glutamic acid decarboxylase
- H&E
Hematoxylin and Eosin; IKKα, IκB kinase α
- LTβ
lymphotoxin-β
- LTβR
lymphotoxin-β receptor
- mTEC
medullary thymic epithelial cells
- NIK
NF-κB-inducing kinase
- PE
phycoerythrin
- Spt1
salivary protein 1
- TRAF6
TNF receptor-associated factor 6
- UEA-1
Ulex europaeus agglutinin-1
Footnotes
We thank Tom Sawyer and Karen Domenico for help with flow cytometry analysis, Judy Meredith, Linda Bauman, and Connie Nowak for assistance in histological studies, and Bristol-Myers Squibb for providing NF-κB2 knockout mice. This work was supported by grants from the American Cancer Society (RSG-03-173-01-CCG) and National Cancer Institute (R01 CA106550) to H.-F.D.
References
- 1.Palmer E. Nat Rev Immunol. 2003;3:383–391. doi: 10.1038/nri1085. [DOI] [PubMed] [Google Scholar]
- 2.Hoffmann MW, Allison J, Miller JF. Proc Natl Acad Sci U S A. 1992;89:2526–2530. doi: 10.1073/pnas.89.7.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burkly LC, Degermann S, Longley J, Hagman J, Brinster RL, Lo D, Flavell RA. J Immunol. 1993;151:3954–3960. [PubMed] [Google Scholar]
- 4.Degermann S, Surh CD, Glimcher LH, Sprent J, Lo D. J Immunol. 1994;152:3254–3263. [PubMed] [Google Scholar]
- 5.Derbinski J, Schulte A, Kyewski B, Klein L. Nat Immunol. 2001;2:1032–1039. doi: 10.1038/ni723. [DOI] [PubMed] [Google Scholar]
- 6.Derbinski J, Gabler J, Brors B, Tierling S, Jonnakuty S, Hergenhahn M, Peltonen L, Walter J, Kyewski B. J Exp Med. 2005;202:33–45. doi: 10.1084/jem.20050471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Consortium TFGA. Nat Genet. 1997;17:399–403. [Google Scholar]
- 8.Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE, Kawasaki K, Asakawa S, Ito F, Shimizu N. Nat Genet. 1997;17:393–398. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- 9.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 10.Farr AG, Anderson SK. J Immunol. 1985;134:2971–2977. [PubMed] [Google Scholar]
- 11.Nelson AJ, Hosier S, Brady W, Linsley PS, Farr AG. J Immunol. 1993;151:2453–2461. [PubMed] [Google Scholar]
- 12.Derbinski J, Kyewski B. Trends Immunol. 2005;26:503–506. doi: 10.1016/j.it.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Solan NJ, Miyoshi H, Carmona EM, Bren GD, Paya CV. J Biol Chem. 2002;277:1405–1418. doi: 10.1074/jbc.M109619200. [DOI] [PubMed] [Google Scholar]
- 14.Coope HJ, Atkinson PG, Huhse B, Belich M, Janzen J, Holman MJ, Klaus GG, Johnston LH, Ley SC. Embo J. 2002;21:5375–5385. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao G, Harhaj EW, Sun SC. Mol Cell. 2001;7:401–419. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 16.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 17.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 18.Claudio E, Brown K, Park S, Wang H, Siebenlist U. Nat Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- 19.Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, Grewal IS, Cochran AG, Gordon NC, Yin J, Starovasnik MA, Dixit VM. Immunity. 2002;17:515–524. doi: 10.1016/s1074-7613(02)00425-9. [DOI] [PubMed] [Google Scholar]
- 20.Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, Bravo R. J Exp Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, Grinberg A, Tran T, Scharton-Kersten T, Anver M, Love P, Brown K, Siebenlist U. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boehm T, Scheu S, Pfeffer K, Bleul CC. J Exp Med. 2003;198:757–769. doi: 10.1084/jem.20030794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kajiura F, Sun S, Nomura T, Izumi K, Ueno T, Bando Y, Kuroda N, Han H, Li Y, Matsushima A, Takahama Y, Sakaguchi S, Mitani T, Matsumoto M. J Immunol. 2004;172:2067–2075. doi: 10.4049/jimmunol.172.4.2067. [DOI] [PubMed] [Google Scholar]
- 24.Kinoshita D, Hirota F, Kaisho T, Kasai M, Izumi K, Bando Y, Mouri Y, Matsushima A, Niki S, Han H, Oshikawa K, Kuroda N, Maegawa M, Irahara M, Takeda K, Akira S, Matsumoto M. J Immunol. 2006;176:3995–4002. doi: 10.4049/jimmunol.176.7.3995. [DOI] [PubMed] [Google Scholar]
- 25.Dixit V, Mak TW. Cell. 2002;111:615–619. doi: 10.1016/s0092-8674(02)01166-2. [DOI] [PubMed] [Google Scholar]
- 26.Pomerantz JL, Baltimore D. Mol Cell. 2002;10:693–695. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- 27.Heino M, Peterson P, Sillanpaa N, Guerin S, Wu L, Anderson G, Scott HS, Antonarakis SE, Kudoh J, Shimizu N, Jenkinson EJ, Naquet P, Krohn KJ. Eur J Immunol. 2000;30:1884–1893. doi: 10.1002/1521-4141(200007)30:7<1884::AID-IMMU1884>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 28.Chin RK, Lo JC, Kim O, Blink SE, Christiansen PA, Peterson P, Wang Y, Ware C, Fu YX. Nat Immunol. 2003;4:1121–1127. doi: 10.1038/ni982. [DOI] [PubMed] [Google Scholar]
- 29.Kuroda N, Mitani T, Takeda N, Ishimaru N, Arakaki R, Hayashi Y, Bando Y, Izumi K, Takahashi T, Nomura T, Sakaguchi S, Ueno T, Takahama Y, Uchida D, Sun S, Kajiura F, Mouri Y, Han H, Matsushima A, Yamada G, Matsumoto M. J Immunol. 2005;174:1862–1870. doi: 10.4049/jimmunol.174.4.1862. [DOI] [PubMed] [Google Scholar]
- 30.Klein L, Klugmann M, Nave KA, Tuohy VK, Kyewski B. Nat Med. 2000;6:56–61. doi: 10.1038/71540. [DOI] [PubMed] [Google Scholar]
- 31.Muller SM, Terszowski G, Blum C, Haller C, Anquez V, Kuschert S, Carmeliet P, Augustin HG, Rodewald HR. Proc Natl Acad Sci U S A. 2005;102:10587–10592. doi: 10.1073/pnas.0502752102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hogquist KA, Baldwin TA, Jameson SC. Nat Rev Immunol. 2005;5:772–782. doi: 10.1038/nri1707. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Cui H, Schroering A, Ding JL, Lane WS, McGill G, Fisher DE, Ding HF. Nat Cell Biol. 2002;4:888–893. doi: 10.1038/ncb872. [DOI] [PubMed] [Google Scholar]
- 34.Lamhamedi-Cherradi SE, Zheng SJ, Maguschak KA, Peschon J, Chen YH. Nat Immunol. 2003;4:255–260. doi: 10.1038/ni894. [DOI] [PubMed] [Google Scholar]
- 35.Shi YF, Bissonnette RP, Parfrey N, Szalay M, Kubo RT, Green DR. J Immunol. 1991;146:3340–3346. [PubMed] [Google Scholar]
- 36.Weih F, Carrasco D, Bravo R. Oncogene. 1994;9:3289–3297. [PubMed] [Google Scholar]
- 37.Hathcock KS, Laszlo G, Pucillo C, Linsley P, Hodes RJ. J Exp Med. 1994;180:631–640. doi: 10.1084/jem.180.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lenschow DJ, Walunas TL, Bluestone JA. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 39.Modigliani Y, Thomas-Vaslin V, Bandeira A, Coltey M, Le Douarin NM, Coutinho A, Salaun J. Proc Natl Acad Sci U S A. 1995;92:7555–7559. doi: 10.1073/pnas.92.16.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohki H, Martin C, Corbel C, Coltey M, Le Douarin NM. Science. 1987;237:1032–1035. doi: 10.1126/science.3616623. [DOI] [PubMed] [Google Scholar]
- 41.O’Garra A, Vieira P. Nat Med. 2004;10:801–805. doi: 10.1038/nm0804-801. [DOI] [PubMed] [Google Scholar]
- 42.Schwartz RH. Nat Immunol. 2005;6:327–330. doi: 10.1038/ni1184. [DOI] [PubMed] [Google Scholar]
- 43.Fontenot JD, Gavin MA, Rudensky AY. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 44.Hori S, Nomura T, Sakaguchi S. Science. 2003;299:1057–1061. [Google Scholar]
- 45.Khattri R, Cox T, Yasayko SA, Ramsdell F. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 46.Kyewski B, Derbinski J. Nat Rev Immunol. 2004;4:688–698. doi: 10.1038/nri1436. [DOI] [PubMed] [Google Scholar]
- 47.Akiyama T, Maeda S, Yamane S, Ogino K, Kasai M, Kajiura F, Matsumoto M, Inoue J. Science. 2005;308:248–251. doi: 10.1126/science.1105677. [DOI] [PubMed] [Google Scholar]
- 48.Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 49.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 50.Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bleul CC, Corbeaux T, Reuter A, Fisch P, Monting JS, Boehm T. Nature. 2006;441:992–996. doi: 10.1038/nature04850. [DOI] [PubMed] [Google Scholar]
- 52.Gordon J, Wilson VA, Blair NF, Sheridan J, Farley A, Wilson L, Manley NR, Blackburn CC. Nat Immunol. 2004;5:546–553. doi: 10.1038/ni1064. [DOI] [PubMed] [Google Scholar]
- 53.Rossi SW, Jenkinson WE, Anderson G, Jenkinson EJ. Nature. 2006;441:988–991. doi: 10.1038/nature04813. [DOI] [PubMed] [Google Scholar]