

Abstract
ApoB mRNA editing involves site-specific deamination of cytidine 6666 producing an in-frame translation stop codon. Editing minimally requires APOBEC-1 and APOBEC-1 complementation factor (ACF). Metabolic stimulation of apoB mRNA editing in hepatocytes is associated with serine phosphorylation of ACF localized to editing competent, nuclear 27S editosomes. We demonstrate that activation of protein kinase C (PKC) stimulated editing and enhanced ACF phosphorylation in rat primary hepatocytes. Conversely, activation of protein kinase A (PKA) had no effect on editing. Recombinant PKC efficiently phosphorylated purified ACF64 protein in vitro, whereas PKA did not. Mutagenesis of predicted PKC phosphorylation sites S154 and S368 to alanine inhibited ethanol-stimulated induction of editing suggesting that these sites function in the metabolic regulation of editing. Consistent with this interpretation, substitution of S154 and S368 with aspartic acid stimulated editing to levels comparable to ethanol treatment in control McArdle RH7777 cells. These data suggest that phosphorylation of ACF by PKC may be a key regulatory mechanism of apoB mRNA editing in rat hepatocytes.
Keywords: apoB, RNA editing, APOBEC-1 Complementation Factor, phosphorylation, regulation
Introduction
ApoB mRNA editing occurs post-transcriptionally and causes the site-specific deamination of cytidine 6666 to uridine thereby creating an in-frame translation stop codon [1, 2]. Consequently, two different apoB proteins are expressed, full-length apoB100 and the truncated protein apoB48, each with distinct physiological functions [3]. Minimally, apoB mRNA editing requires the cytidine deaminase APOBEC-1 as a homodimer [4, 5], APOBEC-1 complementation factor (ACF) [6], and the RNA substrate. Limited tissue expression of APOBEC-1 restricts editing in humans to the small intestine (≥ 85% editing), whereas in some rodents apoB mRNA editing also occurs in the liver where it is subject to metabolic regulation [2, 3, 7, 8].
Under normal, physiological conditions, apoB mRNA editing is a nuclear event occurring on spliced and polyadenylated RNA [9, 10]. However, in vitro editing activity could be detected in both cytoplasmic and nuclear S100 extracts [11, 12]. In vitro and in vivo data demonstrate that the proteins involved in editing exist in two distinct complexes; active nuclear 27S editosomes and inactive, 60S cytoplasmic complexes that can be re-organized into active 27S complexes in vitro [11, 13, 14]. Proteins that form these complexes traffick between the cytoplasm and nucleus and their subcellular compartmentalization is regulated to control editing activity [14–17].
ApoB mRNA editing is regulated by developmental, hormonal, and dietary factors as well as being under tissue-specific control [7, 18–23]. Editing can be stimulated primarily through two mechanisms: increased APOBEC-1 expression [24] and/or through modulation of pre-existing editing factors [25]. The fasting/refeeding and insulin induced models of editing are associated with increased APOBEC-1 abundance [24]. Conversely, induction of editing with ethanol was not dependent on de novo protein or RNA synthesis [25], suggesting modification or redistribution of pre-existing auxiliary factors were sufficient to induce editing. In both cases, the mechanism of induction involved the redistribution of ACF from the cytoplasm to the nucleus [14].
Recently, ACF was shown to be a phosphoprotein in rat primary hepatocytes [26]. The phosphorylated form was strictly localized to nuclear 27S editing complexes, the biological site of editing [26]. Dephosphorylation of ACF diminished in vitro editing activity and the interaction between ACF and APOBEC-1, but did not significantly affect RNA-binding activity. Induction of editing by ethanol and insulin resulted in nuclear accumulation of phosphorylated ACF [14, 26] suggesting that phosphorylation is a common mechanism to modulate editing in response to metabolic stimuli. Inhibition of protein phosphatase 1 (PP1) activity was associated with nuclear accumulation of ACF, increased recovery of phosphorylated ACF and robust editing levels [26]. However, the protein kinase(s) responsible for phosphorylating ACF remain an outstanding question.
We identified serine 154 (S154) and serine 368 (S368) as two candidate phosphorylation sites. McArdle RH7777 cell lines expressing S154 and S368 mutated to alanine maintained normal basal editing levels, but these levels were refractory to ethanol induction. Conversely, substitution of aspartic acid at these sites significantly increased editing in the absence of ethanol to levels comparable with those achieved by ethanol treated cells expressing wild type ACF. The data support the possibility that these sites are necessary for ethanol-induced editing. Residues S154 and S368, are predicted PKC phosphorylation sites, and are conserved between rat and human ACF. Studies with PKC and PKA activators demonstrated that modulation of PKC activity enhanced editing levels in rat primary hepatocytes, whereas PKA did not. Similarly, purified ACF protein was efficiently labeled by PKC isozymes, but not by PKA. These findings are discussed in terms of structural and functional predictions of ACF phosphorylation.
Materials and methods
Isolation of rat primary hepatocytes and protein kinase activator studies
Rat primary hepatocytes were isolated from normal, fed male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) (250–275 g BW) and cultured on BIOCOAT type I collagen coated, 60 mm plastic dishes (Becton Dickinson Labware, Franklin Lakes, NJ) in Waymouth’s media (Sigma Chemical Co., St. Louis, MO) containing 0.1 nM porcine insulin (Sigma) as described previously [14]. Primary hepatocytes were treated for 6 hours with protein kinase activators indolactam V, 8-cpt cAMP, and forskolin at the concentrations encompassing their respective in vivo EC50 values, as described by the manufacturer (Calbiochem, La Jolla, CA). Total cellular RNA was harvested in TriReagent (MRC, Cincinnati, OH) RNA was harvested and processed through the poisoned-primer extension assay to quantify the effect on apoB mRNA editing [14].
To evaluate the effect of protein kinase activation on ACF phosphorylation, cultures were pre-incubated for 2 hours with 12 or 120 μM indolactam V in phosphate-free Waymouth’s media and subsequently supplemented with 0.5 mCi 32PO4 and incubated for an additional 4 hours. Cultures were harvested and subcellular extracts prepared. ACF was immunoprecipitated, resolved by SDS-PAGE, and specific activity was quantified as described previously [26].
Recombinant ACF purification and in vitro ACF phosphorylation
Recombinant ACF64 was expressed in E. coli and purified as described [27]. In vitro ACF labeling reactions were carried out using 2 μg of purified recombinant ACF64 in 5 mM MgCl2, 100 μM CaCl2, 1 mM DTT, 5 μCi 32P-γ ATP, 25 mM Tris-HCl pH 7.5 and 1X Lipid Activator (Invitrogen, Carlsbad, CA) with 25 mU of each PKC isozyme (Calbiochem) or 40 mM Tris-HCl pH 7.4, 20 mM Mg Acetate, 5 μCi 32P-γ ATP and 25 mU of bovine PKA catalytic subunit (Promega, Madison, WI). Reactions were incubated at 30 °C for 10 minutes, terminated by acetone precipitation, resolved by 10.5% SDS-PAGE, and transferred to nitrocellulose (Bio-Rad, Hercules, CA). Western transfers were probed with anti-HA monoclonal antibody (Covance Research Products, Berkeley, CA) and developed using Western Lightning reagent (NEN, Boston, MA). 32P incorporation was monitored by phosphorimager scanning densitometry. PKA enzymatic activity was confirmed via labeling of Kemptide peptide (Promega).
ACF site-directed mutagenesis and functional analysis
Rat ACF64 with a C-terminal V5 epitope tag was cloned into a modified pcDNAIII vector (Invitrogen) [14]. ACF64 cDNA was mutated at specific bases to convert specific serine or threonine codons to alanine or aspartic acid codons using the QuikChange® Multi System (Stratagene, La Jolla, CA). Mutagenic primers (T49AT50A;
| CCAGGCTGGGATGCTGCACCTCCTGAAAGGGGCTGC, | T49DT50D; |
| CCAGGCTGGGATGACGATCCTCCTGAAAGGGGCTGC, | S132A; |
| GGGCGTCTGTGCTGCTGTGGACAACTGCCGG, | S132D; |
| GGGCGTCTGTGCTGATGTGGACAACTGCCGG, | S154A; |
| GAGAGAAGAAATCTTGGCAGAGATGAAAAAGGTC, | S154D; |
| GAGAGAAGAAATCTTGGACGAGATGAAAAAGGTC, | S171A; |
| GTCATTGTCTACCCAGCCGCTGCCGATAAAACC, | S171D; |
| GTCATTGTCTACCCAGACGCTGCCGATAAAACC, | S176A; |
| GTCTACCCAAGCGCTGCTGATAAAGCCAAAAACCGGGGG, | S176D; |
| GTCTACCCAAGCGCTGCTGATAAAGACAAAAACCGGGGG, | S188A; |
| GCCTTTGTGGAATATGAAGCTCACCGCGCAGCCG, | S188D; |
| GCCTTTGTGGAATATGAAGATCACCGCGCAGCCG, | S368; |
| CTACCAAAGGACATCTCGCCAACAGAGCTCTCATCCG, | S368D; |
| CTACCAAAGGACATCTCGACAACAGAGCTCTCATCCG), | were extended using PfuTurbo® |
DNA polymerase according to the manufacture’s recommendations. The accuracy of the mutations was verified by sequencing the entire cDNA using T7 (Promega, Madison, WI), V5 (Sigma Genosys), and Seq481 (Sigma Genosys, The Woodlands, TX) sequencing primers (T7, TAATACGACTCTATAGGG, V5; CTAGAAGGCACGTCGAGGC, Seq481; GAACGAGTTGTTGATGTCATTG) and using Big Dye sequencing system (Applied Biosystems, Foster City, CA).
McArdle RH 7777 (McArdle) cells maintained in DMEM (Gibco-BRL, Carlsbad, CA; supplemented with 10% FBS, 10% horse serum,_1% Penicillin, Streptomycin, Fungizone (Invitrogen)) were transfected with 2 μg plasmid containing each ACF64 mutant using Fugene 6 (Roche, Mannheim, Germany). Stable cell lines were created by G418 selection. When applicable, stable cell lines were treated with 0.9% ethanol for 4 hours. RNA editing of endogenous apoB mRNA was assessed by poisoned-primer extension analysis as previously described [14].
Expression of ACF mutants was assessed by western blotting of whole-cell extracts prepared by lysing cells for 30 minutes in RIPA buffer (50 mM Tris pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 0.2 mM DTT) supplemented with 50 mM NaF and protease inhibitors. Equal amounts of cell extract protein were resolved by 10.5% SDS-PAGE, transferred to nitrocellulose, and probed with anti-V5 antibody (KPL; Gaithersburg, MD).
McArdle RH7777 cells were transfected with pcDNA3 (Invitrogen) containing rat ACF cDNA mutated at suspected phosphorylation sites using FuGene6 (Roche; Basel, Switzerland). Cells were harvested 48 hours post-transfection and fractionated into nuclear and cytoplasmic fractions using the NE-PER kit (Pierce; Rockford, IL). Lysates, 40 μg of nuclear and 60 μg of cytoplasmic, were resolved using 8% or 10% SDS-PAGE and transferred to nitrocellulose (BioRad, Hercules, CA). Blots were probed with anti-histone H1 (Santa Cruz Biotech; Santa Cruz, CA) and anti-actin (Sigma) to ensure proper fractionation. Exogenous ACF was detected using anti-V5 (Invitrogen) antibody, while both ACF populations were observed when probing with an anti C-terminal (CT) ACF antibody [26]. To evaluate total cellular ACF expression 60 μg of total cell lysate was resolved and probed with anti-ACF CT antibody or actin to ensure equal loading.
Immunological techniques
Affinity purified, peptide-specific polyclonal antibodies were raised against ACF N-terminal (NT) sequence (NHKSGDGLSGTQKE) and C-terminal (CT) sequence (HTLQTLGIPTEGGD) in rabbits ([14]); Bethyl Laboratories, Inc., Montgomery, TX). ACF CT antibody was conjugated to Sepharose (Bethyl Laboratories, Inc.) and ACF immunoprecipitated as follows. Whole cell extracts were incubated with the antibody-Sepharose bead column overnight at 4 °C. The resin was washed 3X with PBS, 1X with 10 mM phosphate buffer pH 6.8, and eluted using 100 mM glycine pH 2.5. The eluates were neutralized with 1/10th volume 1 M Tris pH 7.5 and precipitated with 5 volumes acetone at −80 °C. Samples were resolved by 10.5% SDS-PAGE, probed with anti-phosphoserine, threonine, or tyrosine antibodies (Zymed Laboratories, San Francisco, CA), stripped and re-probed with anti-ACF NT antibody.
Comparative modeling of ACF
RNA recognition motifs 1 and 2 of rat ACF64 (residues 54–223) were aligned with RRMs 1 and 2 (residues 37–203) of human HuD protein using ClustalX version 1.83 [28]. ACF RRM boundaries were predicted according to alignments with existing RRM crystal structures from HuD [29], polyA binding protein [31], and sex lethal [32]. ACF RRMs 1 and 2 were then comparatively modeled onto the 1.8 Å crystal structure of RRMs 1 and 2 of HuD [29] using MODELLER 6v2 [30]. Details of template assembly and utilization were as described in the MODELLER manual. HuD and ACF ribbon diagrams were constructed with PyMol [33]. Model quality was assessed by Ramachandran plots using RAMPAGE [34].
Results
Evaluation of ACF phosphorylation
Previous studies of in vivo 32P labeling of rat primary hepatocytes demonstrated that ACF possesses metabolically regulated, high turnover serine phosphorylation sites [26]. However, the possibility of threonine and tyrosine phosphorylation at lower turnover or constitutive sites was not excluded and thus remains a formal possibility. Rat ACF64 protein sequence was analyzed by NetPhosK (http://www.cbs.dtu.dk/services/NetPhosK/), ProSite (http://expasy.org/tools/scanprosite), ProteinScan (http://nci.nih.gov/MPR/ScanProteinForPKCSitesPageaspx), and MotifScan (http://scansite.mit.edu/motifscan_seq.phtml) algorithms for prediction of potential sites of phosphorylation. The algorithms detected several potential serine, threonine, and tyrosine residues with varying probabilities of being phosphorylated. To examine the types of residues phosphorylated in ACF we initially used an immunological approach.
ACF was immunoprecipitated from extracts of primary hepatocyte cultured in basal insulin using immobilized ACF CT antibody, resolved by SDS-PAGE and probed with anti-phosphoserine, phosphothreonine, and phosphotyrosine antibodies. ACF reacted with anti-phosphoserine and anti-phosphothreonine, but not anti-phosphotyrosine antibodies (Figure 1, right hand column). To confirm that phospho-ACF was responsible for the observed immunoreactivity, western transfers were stripped and re-probed with anti-ACF NT antibody and the images overlaid (Figure 1, left hand column). ACF and anti-phosphoserine/threonine immunoreactivities were superimposed demonstrating that ACF was responsible for the observed anti-phosphoserine/threonine reactivity.
Figure 1. Rat hepatic ACF immunoreactivity with anti-phosphoserine and threonine antibodies.
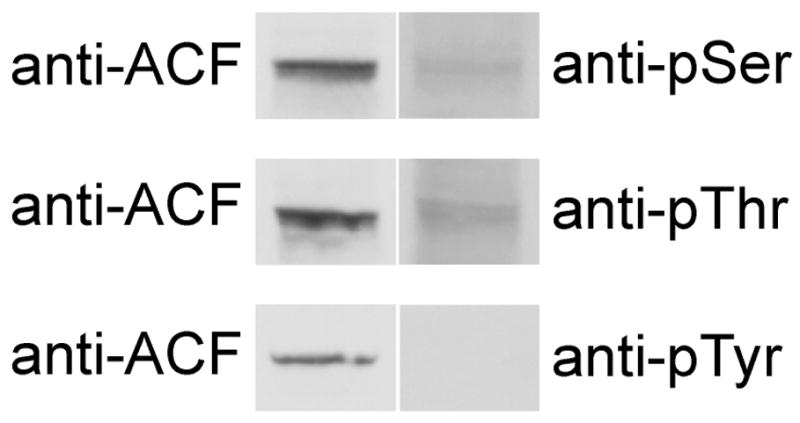
ACF was immunoprecipitated from liver nuclear extracts with anti-ACF CT antibody conjugated to Sepharose and western blotted with anti-phosphoserine, threonine, or tyrosine antibodies (right column). The blots were stripped and re-probed with anti-ACF NT antibody. ACF, position of ACF; anti-pSer, anti-pThr, anti-pTyr denotes position of phosphoserine, threonine, or tyrosine immunoreactivity.
Protein kinase activator studies
The majority of serine and threonine residues predicted to be phosphorylated fit consensus PKC or PKA target sequences (Table 1). Studies were therefore initiated to evaluate the effect of PKC and PKA activation on apoB mRNA editing in rat primary hepatocytes. Since inhibition of PP1 stimulated editing activity [26], we reasoned that activation of appropriate protein kinases would also induce editing. Commercially available activators of PKC (indolactam V, [28]) and PKA (8-cpt-cAMP [29] and forskolin [30]) were selected for evaluation.
Table 1. Sites selected for site-directed mutagenesis.
ACF64 protein sequence was analyzed using ProteinScan (http://nci.nih.gov/MPR/ScanProteinForPKCSitesPageaspx) algorithm to predict potential PKC phosphorylation sites. As an example of the range of site scoring, the average of PKC δ and ζ percentiles are shown. The specific serine (S) or threonine (T) predicted to be phosphorylated are shown in bold.
| Site | Sequence | Average Prediction Percentile |
|---|---|---|
| T49 | GGPPPGWD-T-TPPERGCE | 92 |
| T50 | GPPPGWDT-T-PPERGCE | 97 |
| S132 | GRLLGVCA-S-VDNCRLFV | 36 |
| S154 | TKKREEIL-S-EMKKVTEG | 52 |
| S171 | WDVIVYP-S-AADKTKN R | 66 |
| T176 | WYSAADK-T-KNRGFAFV | 7 |
| S188 | GFAFVETE-S-HRAAAMAR | 39 |
| S368 | FPATKGHL-S-NRALIRTP | 7 |
As anticipated, control primary hepatocytes edited 60% of apoB mRNA and DMSO treatment did not significantly increase editing (Table 2). Activation of PKC with 12 μM and 120 μM indolactam V significantly stimulated editing to 92% (P ≤ 0.01). Activation of PKA with 2 μM, 20 μM, or 200 μM 8-cpt-cAMP (EC50 2 μM) had no effect on editing (P > 0.05, Table 2) suggesting that PKA activation does not alter editing. To exclude the possibility that 8-cpt-cAMP was not active under our experimental conditions, we tested a second PKA activator, forskolin. Treatment with 4 μM, 40 μM, or 120 μM forskolin had also no effect on editing (Table 2). Furthermore, additional adenylate cyclase activators, glucagon and isoproterenol also failed to induce editing (data not shown). Taken together, these data suggest that PKC plays a role in editing regulation, while PKA does not.
Table 2. Activation of PKC modulates apoB mRNA editing.
Hepatocytes were treated with the indicated concentrations of protein kinase activators for 6 hours. ApoB mRNA editing activity was determined using apoB-specific RT-PCR and poisoned-primer extension analysis [14]. The mean, standard deviation (STD) and number of replicates (n) are listed below for each concentration tested.
| Indolactam V | Mean | STD | n |
|---|---|---|---|
| Primary hepatocytes | 63.3 | 3.9 | 3 |
| DMSO | 62.2 | 4.8 | 3 |
| 12μM | *92.5 | 0.8 | 3 |
| 120μM | *91.6 | 1.1 | 3 |
| PKC EC50 = μM | |||
|
| |||
| 8-cpt CAMP | Mean | STD | n |
|
| |||
| Primary hepatocytes | 58.8 | 4.8 | 4 |
| DMSO | 58.2 | 4.6 | 4 |
| 2μM | 61.2 | 4.3 | 4 |
| 20μM | 59.3 | (-) | 2 |
| 200μM | 59.6 | 3.4 | 4 |
| PKA EC50 = 2μM | |||
|
| |||
| Forskolin | Mean | STD | n |
|
| |||
| Primary hepatocytes | 57.6 | 3.4 | 7 |
| DMSO | 61.8 | 4.1 | 6 |
| 4μM | 70.6 | 5.2 | 6 |
| 40μM | 68.5 | 7.5 | 4 |
| 120μM | 69.0 | 0.8 | 4 |
| AC EC50 = 4μM | |||
Statistical significance was determined to be P < 0.05 by unpaired t-test relative to DMSO control when n ≥ 3.
In order to correlate the observed effects on editing with changes in ACF phosphorylation, ACF was immunoprecipitated from extracts isolated from hepatocytes treated with increasing concentrations of indolactam V (Figure 2) in the presence of 32P. ACF phosphorylation was increased in hepatocytes treated with 12 μM and 120 μM indolactam V. Taken together, these data demonstrate that the cellular effects of indolactam V that lead to enhanced editing activity are associated with increased ACF phosphorylation.
Figure 2.
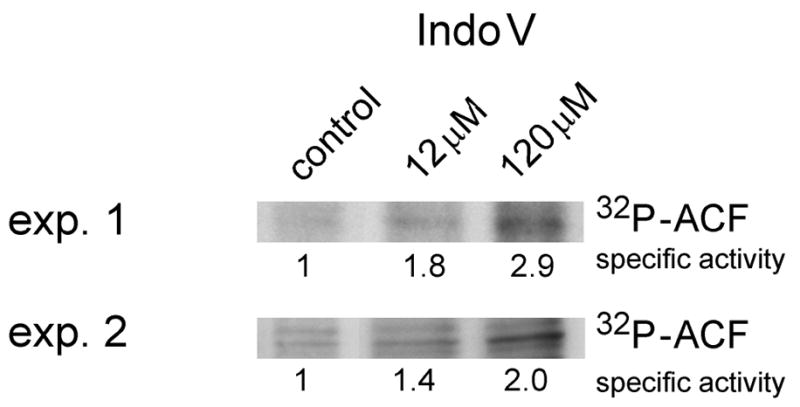
ACF phosphorylation is modulated by protein kinase C activation. In vivo ACF 32P incorporation was determined by Phosphorimager scanning of ACF immunoprecipitates prepared from rat hepatocytes treated with 12 or 120 μM indolactam v (Indo V). ACF specific activity (relative to control hepatocytes) was calculated as the ACF 32P density (Phosphorimager) divided by the recovery of ACF determined from densitometric scanning of ACF western blots (Image J). ACF immunopurified from control hepatocytes was arbitrarily assigned a value of 1 (n = 3). Exp. 1 and Exp. 2 denote independent experiments 1 and 2.
PKC phosphorylates ACF64
To determine if PKC can phosphorylate ACF, recombinant rat ACF64 was expressed and purified [27] and then incubated with equal units of various PKC isozymes or PKA catalytic subunit. Reactions were resolved by SDS-PAGE, transferred to nitrocellulose, and evaluated by autoradiography. Liver-expressed PKC isozymes (including α, βII, δ, ε, and ζ; reviewed in [31]) phosphorylated ACF64 in vitro (Figure 3, upper panel). To verify that the observed 32P incorporation was specific to ACF, western transfers were probed with anti-HA and the images overlaid (Figure 3, lower panel). PKC isozymesα, βII and ζ demonstrated the highest specific-activity with ACF64. No 32P labeling was detected in the absence of protein kinase demonstrating that the signal is PKC-dependent. These data are consistent with the PKC activator studies (Table 2) and suggest that liver-specific PKC isozymes are capable of phosphorylating ACF64.
Figure 3. PKC phosphorylates ACF64 in vitro.
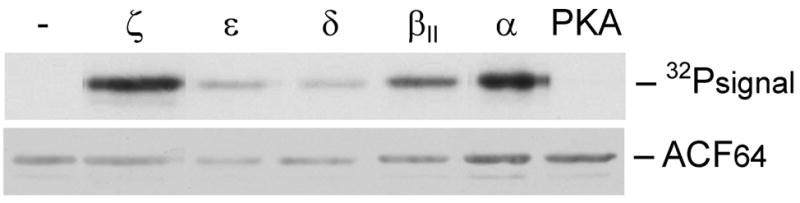
Recombinant ACF64 was reacted with PKC isozymes (α βII, δ, ε and ζ) or PKA, transferred to nitrocellulose and analyzed by autoradiography. Western transfers were subsequently probed with anti-HA antibody. (−) denotes kinase-free control reaction only containing buffer and ACF.
Given that ACF was predicted to contain potential PKA phosphorylation sites, the ability of PKA catalytic subunit to phosphorylate ACF was also evaluated. PKA catalytic subunit was unable to phosphorylate ACF64 in vitro using the same number of enzyme units as PKC. PKA-dependent phosphorylation of Kemptide peptide (Promega) demonstrated that the enzyme was active under our assay conditions (data not shown). However, 10-times greater number of units of PKA resulted in 32P incorporation into ACF (data not shown) and may represent non-selective phosphorylation due to excess enzyme. Although our data are consistent with phosphorylation of ACF by PKC, we cannot rule out PKA phosphorylation since in vivo phosphorylation of ACF may require other regulatory factors (e.g. PKA may require prior posttranslational modification of ACF by PKC).
Site-directed mutagenesis and apoB mRNA editing
To identify candidate ACF phosphorylation sites, ACF cDNA was selectively mutated to generate alanine or aspartic acid at predicted PKC phosphorylation sites (Table 1). Sites were selected for mutagenesis based on the following three criteria: (1) ACF contains serine and threonine phosphorylation sites (Figure 1 and [26]) predicted to be phosphorylated by PKC (Table 1), (2) the amino terminal 380 amino acids of ACF contain all domains necessary to complement editing including; APOBEC-1 interaction [32], RNA-binding, [16], and a nuclear localization sequence [33], and (3) only sites conserved in both rat and human ACF64 were considered for our studies (see sequence alignment Figure 6A). We also selected sites for mutagenesis with low and medium probabilities of being phosphorylated (Table 1) as there are examples of empirically determined and validated sites of phosphorylation that were not initially predicted by consensus sequence algorithms alignments [34].
Figure 6. Comparative modeling of ACF.

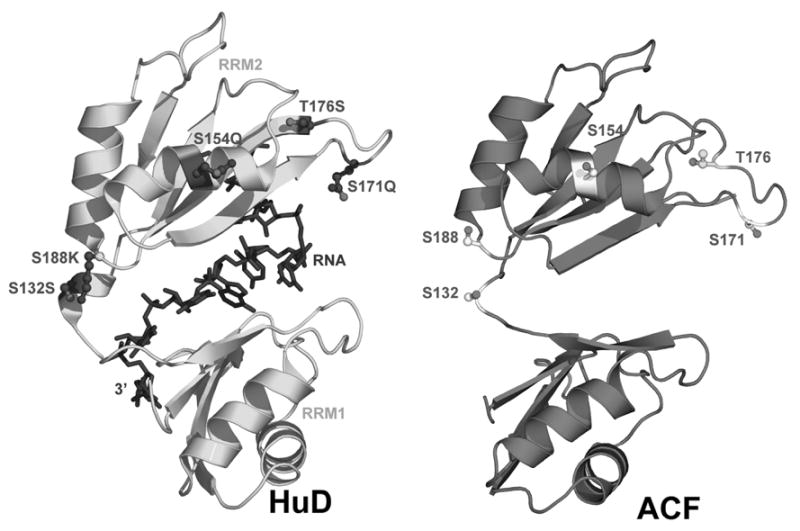
(A) Rat and human ACF sequence alignment and secondary structure prediction. The human ACF amino acid sequence is shown above that of rat. Black residue pairs are conserved; unique regions between ACF variants are blue and unique HuD residues are green. Red letters indicate contacts to HuD RNA. Arrows mark mutagenized residues. (B) Predicted tertiary structure of ACF and potential phosphorylation sites, provided alongside original HuD structure bound to substrate RNA. Figure 6B was made using PyMol [59].
Eight of the predicted sites of ACF phosphorylation were separately mutated to either alanine or aspartic acid (Figure 4 and Figure 5). ApoB mRNA editing activity was evaluated in stable McArdle cell lines that expressed similar levels of each phosphorylation site mutants to ensure that modulation of editing activity was not due to large differences in ACF expression. Expression of each mutant was assessed by western blot of whole cell extracts using an anti-V5 epitope antibody (Figure 4A). Cell lines were treated without or with 0.9% ethanol for 4 hours to test the ability of each mutant in ACF to support editing activity under basal and stimulated conditions.
Figure 4. Expression of ACF64 site-directed mutants in McArdle cells.
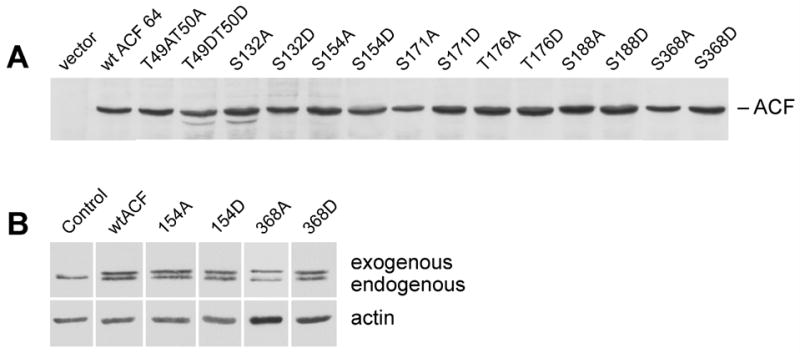
A) Rat ACF64 was previously cloned [14] into modified pcDNAIII vector containing 3′ V5 epitope tag. Equivalent number of cells and amount of protein from McArdle cell lines overexpressing V5-tagged ACF64 constructs (as labeled) were resolved by 10.5% SDS-PAGE, transferred to nitrocellulose and probed with anti-V5 antibody. B) Abundance endogenous ACF is unaffected by ectopic expression of ACF64 site-directed mutants. Whole cell extracts were generated from McArdle cells expressing ACF mutants. Equal micrograms of protein were resolved by SDS-PAGE, transferred to nitrocellulose, and blotted with anti-ACF CT antibody (upper panel), anti-actin (lower panel). Data shown is representative of 3 independent experiments.
Figure 5. Mutagenesis of predicted ACF64 phosphorylation sites modulates apoB mRNA editing in McArdle cells.
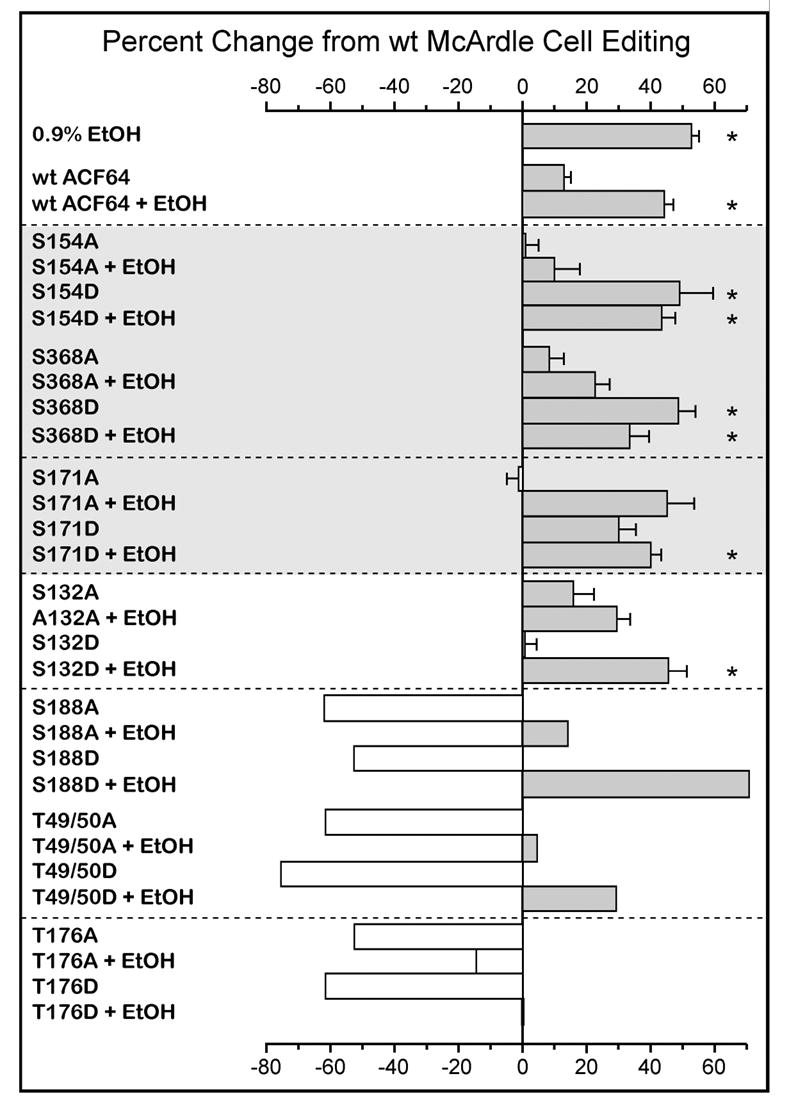
McArdle cells stably transfected with various ACF site-directed mutants were treated with 0.9% EtOH or as control for 4 hours. RNA was isolated, apoB RNA was amplified and evaluated for editing using the poisoned-primer extension assay. The central vertical line represents the level of wild type McArdle editing. Percent change from wild type (wt) editing was calculated by [(ACF mutant % editing − wt ACF % editing)/(wt ACF % editing)] × 100 and is shown as histograms to either side of the control value vertical line. *Statistical significance was defined as P ≤ 0.05 compared to control wtACF expressing cells when n ≥ 3. Error bars represent the calculated standard deviation (n ≥3).
Editing in vector only transfected cells was indistinguishable from control, parental McArdle cells without ethanol treatment (data not shown). ApoB mRNA editing efficiency in wild type McArdle cells was 14% (+/−4.4) (Figure 5, shown as a vertical line for comparison to other treatments). As anticipated [25], the addition of ethanol significantly stimulated editing by 50% (P ≤ 0.05) in wild type McArdle cells (Figure 5). A 13% (+/− 4.4%) increase in editing was observed in McArdle cells stably transfected with wild type ACF64. Editing activity in ACF64 transfected McArdle cells retained the ability to respond to ethanol stimulation (45% increase +/− 3.5%, P<0.05). These data demonstrated that over expression of ACF does not adversely affect editing activity and that ACF64 transfected McArdle cells responded robustly to ethanol stimulation.
There is a vast literature on protein phosphorylation supporting the use of mutagenesis to identify sites of phosphorylation. In this approach, mimicking a phosphorylation site by mutation to aspartic acid should induce editing in control cells, whereas ablation of a phosphorylation site by mutation to alanine should inhibit ethanol-induced editing. Cells transfected with an ACF64 S154 mutated to alanine (S154A) had basal editing values comparable to control untransfected cells. McArdle cells expressing ACF64 S368 mutated to alanine (S368A) demonstrated editing values only 8% (+/− 5.0) greater than control. In contrast, editing activity in cells transfected with S154A or S368A had a severely blunted the response to ethanol treatment with editing values only 10% (+/− 7.1) and 22% (+/− 4.4) greater than control untreated McArdle cells, respectively (Figure 5). These data suggested these mutants had a dominant negative effect on the ability of the endogenous McArdle cell machinery to respond regulated activity in response to ethanol treatment.
Mutation of ACF64 S154 to aspartic acid (S154D) stimulated editing 48% greater than control in the absence of ethanol (P ≤ 0.05). Mutation of ACF64 S368 to aspartic acid (S368D) also enhanced editing by 48% greater than control (P ≤ 0.05). Treatment of McArdle cells expressing either S154D or S368D with ethanol resulted in editing 43% (+/− 11.0) and 33% (+/− 4.8) greater than control, respectively (Figure 5). Taken together, the data suggested the phenotype of ala and asp mutation at S154 and S368 was consistent with regulation of editing through these sites by phosphorylation.
Several other serine and threonine residues were mutated to alanine or aspartic acid, but failed to behave in a manner consistent with regulated sites of phosphorylation. To facilitate discussion, the remaining mutants have been categorized as: (1) those for which mutation to alanine was inhibitory (e.g. S171), (2) those for which mutation to aspartic acid inhibited editing (e.g. S132), (3) those for which ethanol treatment rescued editing of alanine and aspartic acid mutations (e.g. S188 and T49/T50), (4) those whose editing activity never reached wild type McArdle cell editing levels (e.g. T176).
In the first case, cells transfected with S171 mutated to alanine (S171A) could not maintain editing levels comparable to cells transfected with wtACF. While these data suggest that this site is not essential to ACF64 function and S171 phosphorylation is not critical for ethanol stimulation of editing activity mutation of S171 to aspartic acid (S171D) stimulated editing compared to control cells and cells transfected with wtACF. The addition of ethanol further stimulated editing in cells expressing S171D to 40% greater than control (P ≤ 0.05). The data suggested that the ability of S171D to stimulate editing activity is likely due to the change in local charge that lies close to S154 (see structural model, Figure 6B).
S132 was also in its own category where mutation to alanine (S132A) displayed a phenotype similar to transfection with wild type ACF (stimulated editing by 17% (+/− 6.4) and 29% (+/− 4.3) in control and ethanol treated McArdle cells, respectively (Figure 5). However, mutation to aspartic acid (S132D) did not stimulate editing activity on its own, but cells retained the ability to respond to ethanol stimulation. The data suggest a preference for nonpolar or uncharged amino acids at position 132 and that ethanol treatment can activate ADF (presumably through phosphorylation at S154 and S368 regardless of the mutations at S132 studied here.
Mutation of S188 or T49/T50 to either alanine (S188A and T49A/T50A) or aspartic acid (S188D and T49D/T50D) markedly inhibited editing activity (Figure 5). Treatment with ethanol increased editing activity to varying levels greater than control cell levels, but not to the extent seen in ethanol-treated parental McArdle cells. The data suggest that S188 and T49/T50 are inherently important for ACF64 structure and function and that it is unlikely that they are phosphorylation sites. However, consistent with phosphorylation of S154 and S368, cell lines expressing S188D or T49/T50D mutants resulted in increased editing. Similar results were obtained when ACF64 alanine and aspartic acid substitution mutagenesis was performed at S253, S274, T160 and T316 (data not shown).
Mutation of T176 to either alanine (T176A) or aspartic acid (T176D) resulted in the most marked dominant negative editing phenotype (Figure 5). With this mutation ethanol treatment was only able to marginally restore editing to the levels, suggesting that this site is essential as a threonine. Similar results were obtained when ACF64 alanine and aspartic acid substitutions were evaluated at S241, S242, S243, S377, S308, T14 and T146 (data not shown).
Our findings suggest that mutagenesis of phosphorylation sites had an effect on ACF’s ability to complement APOBEC-1 and form functional editosomes. To rule out the possibility that ectopically expressed ACF mutants altered editing by changing the expression of the endogenous ACF we evaluated the abundance of endogenous ACF in the presence of our mutants. Irrespective of ectopic ACF64 expression, endogenous ACF abundance (normalized to cellular actin) was not changed (Figure 4B). The subcellular distribution of endogenous ACF also was not significantly changed through ectopic expression of wtACF or mutant ACF (data not shown). These findings demonstrate that the observed changes in editing activity are due to the properties of individual ACF mutants.
While not exhaustive, the mutagenesis data suggested that phosphorylation of 154, 171 and 368 would stimulate editing activity, whereas phosphorylation at other serines or threonines would markedly inhibited ACF function and editing activity.
ACF structure-based prediction of phosphorylation site accessibility
The positions of ACF phosphorylation-site mutants were evaluated based on the comparative model to HuD. This model served to demonstrate that the predicted sites of ACF phosphorylation might be in accessible regions of the protein. Alignment of rat [14] and human [3] ACF64 protein sequences demonstrated 93.5% identity (Figure 6A). All predicted sites of ACF phosphorylation selected for mutagenesis were conserved between both species (Figure 6A). HuD and ACF both contain three RRMs and regulate mRNA stability through binding AU-rich 3′-UTRs [42]. Alignment of the first two RRMs of ACF (residues 55 – 223) and HuD (37 – 203) maintained RNP consensus alignment and yielded a primary sequence identity of 23%, with 57% homology. A structural model of ACF was generated from this alignment using the 1.8 Å structure of HuD RRMs 1 and 2 bound to a short AU-rich RNA oligomer (PDB 1FXL) [29]. These RRMs exhibited the canonical four-stranded antiparallel β-sheet flanked by two α-helices (Figures 6A and B). Ninety-one percent of the modeled ACF residues fell into favored regions of the Ramachandran plot, with an additional 5.4% in allowed regions. Compared to the equivalent residues in HuD, S132 of ACF is conserved while S154, T176, and S188 are substituted with glutamine, serine and lysine, respectively. ACF residues T49/T50 and S368 fall outside the boundaries of the model, and therefore could not be evaluated in this context.
In HuD, single-stranded RNA contacts the β-sheets and loops of RRMs 1 and 2 (Figure 6B), whereas the flanking α-helices are more solvent exposed and therefore accessible to protein-protein interactions (Figure 6B and [35, 36]). It is important to note that while the position and orientation of each residue within the modeled ACF RRMs can be evaluated, the relative spatial orientations of each RRM cannot be deduced without empirical structural information for ACF. Hence, although multiple tandem RRM structures exist and the individual RRM fold is highly conserved, the conformation of the inter-RRM linker varies greatly among structures [37, 38]. Nonetheless, since comparative modeling of a single RRM does not rely upon knowledge of the inter-domain linker, there is confidence that S154 resides in a solvent exposed region of the second helix of RRM 2 (Figure 6B). As such, the hydroxyl side chain of this residue should be accessible to protein kinases and phosphatases. Likewise, T176 and S171 are located within the loop joining β-strands 2 and 3 of RRM 1 (Figure 6B). In contrast, S132 is located in the linker between RRMs 1 and 2, which suggests solvent accessibility, but precludes rigorous modeling (Figure 6B).
Discussion
ApoB mRNA editing requires the coordinated assembly of a multi-protein editosome [14] that governs substrate and site specificity [39], the proportion of substrate edited [10] and possibly export of the edited product from the nucleus. Thus, regulation of editosome assembly and trafficking of editing complexes is necessary for efficient editing. ACF is an obligate component of the editing machinery and is the site-specific RNA-binding protein [39, 40] that docks APOBEC-1 to cytidine 6666. Recently, phosphorylation of ACF [26] was identified as a mechanism whereby apoB mRNA editing can be regulated. Hyper-phosphorylation of serine residues, in active nuclear 27S editosomes, was implicated in ethanol-stimulated editing [26].
To better understand the role of ACF phosphorylation in basal and metabolically stimulated editing, the sites of phosphorylation and the enzymes responsible were characterized. The expression of all mutants was validated to be at similar levels through the selection of McArdle cell lines stably expressing each variant. While it is likely that overexpressed wtACF or mutant ACF competed with endogenous ACF leading to the observed changes in editing activity, the overall abundance and subcellular distribution of endogenous ACF was unchanged in response to ectopic expression of phosphorylation site mutants. We have demonstrated in this report that mutation of S154 and S368 to alanine inhibited the ability of McArdle cells to regulate (increase) editing activity in response to ethanol. Mutation of these residues to aspartic acid fully stimulated editing activity even in the absence of ethanol treatment.
S154 and S368 were predicted as phosphorylation sites, potentially targeted by either PKA or PKC. Protein kinase activator studies demonstrated that only PKC activators stimulated editing activity and enhanced ACF phosphorylation in hepatocytes. The role of PKC in ACF phosphorylation was further supported by in vitro protein kinase assays in which liver expressed PKC isoforms phosphorylated recombinant ACF, whereas PKA had to be used at 10-fold higher concentrations to affect ACF phosphorylation. Thus our data support the role of PKC phosphorylation of ACF64, particularly PKC isoforms α β II and ζ, in ethanol regulated hepatic editing activity.
Immunological data indicated that rat liver ACF possesses both serine and threonine phosphorylation sites although only serine phosphorylation has been observed by radiolabeling during metabolic stimulation [26]. We believe this discrepancy to be due to inherent differences in the experimental systems used. Detection of phosphorylated residues by antibodies does not require phosphate turnover (unlike metabolic labeling with 32P). These data suggest that threonine phosphorylation is subject to slower turnover than serine phosphorylation in rat primary hepatocytes and/or that phosphothreonine is significantly less abundant. Our mutagenesis studies were designed to identify sites of phosphorylation that affect ACF64 function during metabolically stimulated editing. Given that threonine residues were not identified in this mutagenic screen, our collective data only support the role of regulated ACF64 serine phosphorylation in the metabolically modulation of apoB mRNA editing. Phosphorylated threonines are likely constitutive or subject to slow phosphate turnover. ACF threonine phosphorylation however could play an inhibitory role, by maintaining editing at low levels under basal metabolic conditions. Consistent with this possibility are data showing that inhibition of protein synthesis stimulated editing in primary hepatocytes [25]. This could be due to reduced protein synthesis of a negative regulator of editing, such as a protein kinase active on ACF threonines. If constitutively expressed, a protein kinase with activity on ACF T49/T50 and T176 for example could inhibit editing activity. The phenotype of S145D and S368D suggested that ethanol stimulated phosphorylation of S154 and/or S368 will override the inhibitory effects of threonine phosphorylation and activate editing activity without necessarily requiring dephosphorylation of phosphothreonine sites. We therefore predict that at steady state hepatic ACF64 will be phosphorylated at different sites and to varying degrees depending on the metabolic demand. ACF containing multiple phosphorylations was predicted by two-dimensional gel analysis [26].
Hepatic ethanol-stimulated editing [41, 42] coincides with hyper-phosphorylation of ACF [26]. Our results demonstrate that liver expressed PKC isozymes α β II and ζ have the greatest ability to phosphorylate recombinant ACF. PKC θ over expression has been reported to modulate editing in rat hepatoma cells, whereas PKC isozymes α β II, ε and ζ were unable to modulate editing [43]. However, the authors did not provide direct evidence that individual isoforms phosphorylated proteins involved in editing. Moreover, PKC θ is not a physiologically relevant isozyme since it is not expressed in liver [44]. Further, the relative expression level of each PKC isoform was not indicated and therefore, non-selective phosphorylation of proteins may have been induced experimentally by overexpression of protein kinase. In this regard, although PKA is predicted to phosphorylate ACF and is regulated by ethanol [45], PKA had very low activity in vitro on recombinant ACF64 and PKA-specific activators did not modulate editing in primary hepatocytes (Table 2). Neither direct activation of PKA with cAMP analogs nor indirect activation via adenylate cyclase stimulated editing. Conversely, activation of PKC with indolactam V significantly enhanced apoB editing (Table 2) and PKC isozymes phosphorylated purified ACF in vitro. Our data is in agreement with results previously published demonstrating that activation of PKC induces editing in intestinally derived Caco-2 cells [43].
Although phosphorylated ACF has only been recovered in the nucleus of hepatocytes, the cellular domain in which ACF is phosphorylated remains to be determined. In vivo, PKC substrate specificity is believed to be imparted by the subcellular distribution of the enzyme and substrate [46]. Therefore, PKC interaction with and phosphorylation of ACF in the perinuclear environment, at some point during nuclear import or at some point in time subsequent to nuclear import remain formal possibilities.
ACF is a single polypeptide that comprises three tandem RRMs [6]. The protein itself has been implicated in apoB mRNA-binding, APOBEC-1 binding and protection of edited apoB mRNA from NMD [6, 17]. In general, each RRM is composed of four antiparallel β-sheets flanked by two α-helices (reviewed in [47]). RNA recognition occurs through the β– strands and their connecting loops. Other complex embellishments to the RRM fold have been documented that allow protein-protein interactions at the α-helices [36, 48, 49], or β-strands [50, 51]-, although the nature of the ACF-APOBEC-1 interaction is unknown at present. In HuD, ssRNA makes contact primarily to the β-sheets of RRMs 1 and 2 (Figure 6A, red residues), whereas the outward facing αhelices are exposed to solvent (Figure 6B) [35]. Based on ACF sequence homology to HuD and the conserved structure of the RRM fold, S154 of ACF is predicted to localize within the second helix of RRM 2, where it would be accessible to protein kinases, but would not be expected to contact RNA or affect RNA-binding when phosphorylated. As such, it is plausible that this residue could be involved in protein-protein interactions (Figures 6A and B). The three-dimensional model for the position and orientation of S154 within RRM 2 is consistent with previous studies demonstrating that treatment of extracts with calf-intestinal alkaline phosphatase reduced co-immunopurification of APOBEC-1 with ACF, but did not affect ACF’s ability to bind to apoB RNA [26]. Regulation of protein-protein interactions by changes in the phosphorylation state of one or more proteins has been well documented [52, 53]. Additionally, phosphorylation may alter tertiary fold promoting protein-protein interactions as is the case with CREB and CREB binding protein [54].
Another residue of interest in ACF is S171, which is a glutamine in HuD. This residue is located in the loop betweenβ–strands 2 and 3 of RRM 2. Loop 3 is the most genetically divergent region among RNP proteins and is important for substrate specificity [47, 55]. Q171 of HuD is 4 Å from the non-bridging phosphate oxygen of U5, suggesting that the introduction of negative charge in this area could affect substrate specificity irrespective of the phosphorylation state. Editing activities for the S171D mutant were 15% and 28% greater than untreated McArdle cells in the absence and presence of ethanol, respectively. The introduction of negative charge by aspartic acid mutation in S171D may have contributed to the slight elevation in editing activity, the data suggest that it is unlikely that S171 is an ethanol regulated phosphorylation site.
Our mutagenesis analyses predicted that some serine/threonine residues in and of themselves are important for ACF structure and also editing. The introduction or ablation of negative charge at these residues could affect protein folding, protein-protein interactions, protein-RNA interaction and thus function independent of protein phosphorylation. In this case, mutagenesis of these sites to alanine and aspartic acid would disrupt editing. Case in point, S132 is conserved in rat and human ACF as well as in HuD and is located in between RRMs 1 and 2. The data suggested that mutation to alanine at this site was tolerated since editing was comparable to wild type ACF64. However, mutation to aspartic acid was detrimental. Although serine residues are polar, the R group is uncharged at pH 7.0. Mutation to alanine, a nonpolar amino acid, with only a methyl moiety as the R group would be less likely to interfere with protein folding. To the contrary, substitution with negatively charged aspartic acid could alter protein folding. Therefore, S132 is unlikely to be a phosphorylation site.
Limited structural information precludes modeling outside of ACF RRMs 1 and 2. Consequently, mutations amino terminal to amino acid 55 (T49/T50) and those carboxy terminal to amino acid 223 (i.e. S368) cannot be evaluated in the context of this model. However, S368 is located within ACF’s nuclear localization sequence (amino acids 360–401) [33]. ACF is a nucleocytoplasmic shuttling protein related to hnRNP proteins [33] and its subcellular distribution is sensitive to metabolic perturbations [33]. Significantly, phospho-ACF accumulates in the nucleus of hepatocytes treated with ethanol or insulin [14]. The S368 to alanine mutation was refractory to ethanol, whereas the aspartic acid mutation stimulated editing in untreated McArdle cells to levels comparable to cells treated with ethanol. These data suggest that phosphorylation of S368 may modulate ACF nucleocytoplasmic shuttling. In fact, several examples exist in the literature where phosphorylation mediated nuclear localization ([53, 56, 57] reviewed in [58]). The coordinated action of PP1 [26] and PKC could regulate that subcellular distribution of ACF, and therefore the proportion of apoB mRNA that is edited.
In conclusion, the data suggest a model of editing regulation in McArdle cells whereby ACF is phosphorylated minimally on serine and threonine residues in basal McArdle cells. Metabolic stimulation of editing by ethanol may be accompanied by phosphorylation of S154 and S368 by PKC. Protein phosphatase 1 activity has been implicated in removing phosphate from metabolically regulated sites of ACF phosphorylation, enhancing export of ACF from the nucleus [26] and reducing editing activity. Our findings suggest that phosphorylation of ACF by PKC in response to ethanol is part of the mechanism for nuclear import of ACF and activation of editing activity. In this regard, PKC and PP1 activities on ACF are predicted to act in concert to modulate the overall phosphorylation status of ACF, regulating its protein interactions and subcellular distribution.
Acknowledgments
We thank Dr. N. Ballatori for assistance in the preparation of rat primary hepatocytes and Jenny M.L. Smith for the preparation of Tables and Figures. This work was supported in part by a Public Health Services Grant DK43739 awarded to HCS and RR15934 and GM63162 awarded to JEW. DML was supported through a Toxicology Training Grant 5T32 ES07026, a Grant in Aid from the American Liver Foundation and Sigma Xi. CM was supported by a Training in Cellular, Biochemical and Molecular Sciences Training Grant 5T32 GM068411.
Abbreviations
- APOBEC-1
apolipoprotein B editing catalytic subunit 1
- ACF
APOBEC-1 complementation factor
- apoB
apolipoprotein B
- RRM
RNA recognition motif
- PP1
protein phosphatase 1
- PKC
protein kinase C
- PKA
protein kinase A
- NT
N-terminal
- CT
C-terminal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- 1.Johnson DF, Poksay KS, Innerarity TL. The mechanism for apo-B mRNA editing is deamination. Biochem Biophys Res Commun. 1993;195:1204–10. doi: 10.1006/bbrc.1993.2172. [DOI] [PubMed] [Google Scholar]
- 2.Powell LM, Wallis SC, Pease RJ, Edwards YH, Knott TJ, Scott J. A novel form of tissue-specific RNA processing produces apolipoprotein- B48 in intestine. Cell. 1987;50:831–40. doi: 10.1016/0092-8674(87)90510-1. [DOI] [PubMed] [Google Scholar]
- 3.Chen SH, Habib G, Yang CY, Gu ZW, Lee BR, Weng SA, Silberman SR, Cai SJ, Deslypere JP, Rosseneu M, et al. Apolipoprotein B-48 is the product of a messenger RNA with an organ- specific in-frame stop codon. Science. 1987;238:363–6. doi: 10.1126/science.3659919. [DOI] [PubMed] [Google Scholar]
- 4.Lau PP, Zhu HJ, Baldini HA, Charnsangavej C, Chan L. Dimeric structure of a human apo B mRNA editing protein and cloning and chromosomal localization of its gene. Proc Natl Acad Sci USA. 1994;91:8522–8526. doi: 10.1073/pnas.91.18.8522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260:1816–1819. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- 6.Mehta A, Kinter MT, Sherman NE, Driscoll DM. Molecular cloning of apobec-1 complementation factor, a novel RNA- binding protein involved in the editing of apolipoprotein B mRNA. Mol Cell Biol. 2000;20:1846–54. doi: 10.1128/mcb.20.5.1846-1854.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greeve J, Altkemper I, Dieterich JH, Greten H, Windler E. Apolipoprotein B mRNA editing in 12 different mammalian species: hepatic expression is reflected in low concentrations of apoB- containing plasma lipoproteins. J Lipid Res. 1993;34:1367–83. [PubMed] [Google Scholar]
- 8.Backus JW, Eagleton MJ, Harris SG, Sparks CE, Sparks JD, Smith HC. Quantitation of endogenous liver apolipoprotein B mRNA editing. Biochem Biophys Res Commun. 1990;170:513–8. doi: 10.1016/0006-291x(90)92121-f. [DOI] [PubMed] [Google Scholar]
- 9.Lau PP, Xiong W, Zhu HJ, Chen SH, Chan L. Apo B mRNA editing is an intranuclear event that occurs posttranscriptionally coincident with splicing and polyadenylation. J Biol Chem. 1991;266:20550–20554. [PubMed] [Google Scholar]
- 10.Sowden M, Hamm JK, Spinelli S, Smith HC. Determinants involved in regulating the proportion of edited apolipoprotein B RNAs. Rna. 1996;2:274–88. [PMC free article] [PubMed] [Google Scholar]
- 11.Smith HC, Kuo SR, Backus JW, Harris SG, Sparks CE, Sparks JD. In vitro apolipoprotein B mRNA editing: identification of a 27S editing complex. Proc Natl Acad Sci U S A. 1991;88:1489–93. doi: 10.1073/pnas.88.4.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith HC. Analysis of protein complexes assembled on apolipoprotein B mRNA for mooring sequence-dependent RNA editing. Methods. 1998;15:27–39. doi: 10.1006/meth.1998.0603. [DOI] [PubMed] [Google Scholar]
- 13.Harris SG, Sabio I, Mayer E, Steinberg MF, Backus JW, Sparks JD, Sparks CE, Smith HC. Extract-specific heterogeneity in high-order complexes containing apolipoprotein B mRNA editing activity and RNA-binding proteins. J Biol Chem. 1993;268:7382–92. [PubMed] [Google Scholar]
- 14.Sowden MP, Ballatori N, De Mesy Jensen KL, Hamilton Reed L, Smith HC. The editosome for cytidine to uridine mRNA editing has a native complexity of 27S: identification of intracellular domains containing active and inactive editing factors. J Cell Science. 2002;115:1027–1039. doi: 10.1242/jcs.115.5.1027. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Smith HC. Multiple protein domains determine the cell type-specific nuclear distribution of the catalytic subunit required for apolipoprotein B mRNA editing. Proc Natl Acad Sci U S A. 1997;94:13075–80. doi: 10.1073/pnas.94.24.13075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blanc V, Henderson JO, Kennedy S, Davidson NO. Mutagenesis of apobec-1 complementation factor reveals distinct domains that modulate RNA binding, protein-protein interaction with apobec-1, and complementation of C to U RNA-editing activity. J Biol Chem. 2001;276:46386–93. doi: 10.1074/jbc.M107654200. [DOI] [PubMed] [Google Scholar]
- 17.Chester A, Somasekaram A, Tzimina M, Jarmuz A, Gisbourne J, O’keefe R, Scott J, Navaratnam N. The apolipoprotein B mRNA editing complex performs a multifunctional cycle and suppresses nonsense-mediated decay. Embo J. 2003;22:3971–82. doi: 10.1093/emboj/cdg369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Funahashi TF, Giannoni AM, Depaoli SF, Skarosi, Davidson NO. Tissue-specific, developmental and nutritional regulation of the gene encoding the catalytic subunit of the rat apoB mRNA editing enzyme: functional role in the modulation of apoB mRNA editing. J Lipid Res. 1995;36:414–428. [PubMed] [Google Scholar]
- 19.Demmer LA, Levin MS, Elovson J, Reuben MA, Lusis AJ, Gordon JI. Tissue-specific expression and developmental regulation of the rat apolipoprotein B gene. Proc Natl Acad Sci U S A. 1986;83:8102–6. doi: 10.1073/pnas.83.21.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson AP, Tennyson GE, Hoeg JM, Sviridov DD, Brewer HB., Jr Ontogenetic regulation of apolipoprotein B mRNA editing during human and rat development in vivo. Arterioscler Thromb. 1992;12:468–73. doi: 10.1161/01.atv.12.4.468. [DOI] [PubMed] [Google Scholar]
- 21.Wu JH, Semenkovish CF, Chen SH, Li WH, Chan L. Apolipoprotein B mRNA editing: validation of a sensitive assay and developmental biology of RNA editing in the rat. J Biol Chem. 1990;265:12312–12316. [PubMed] [Google Scholar]
- 22.Huguchi K, Kitagawa K, Kogishi K, Takeda T. Developmental and age-related changes in apoB mRNA editing in mice. J Lipid Res. 1992;33:1753–1764. [PubMed] [Google Scholar]
- 23.Jiao S, Moberly JB, Schonfeld G. Editing of apolipoprotein B messenger RNA in differentiated Caco-2 cells. J Lipid Res. 1990;31:695–700. [PubMed] [Google Scholar]
- 24.Von Wronski MA, Hirano KI, Cagen LM, Wilcox HG, Raghow R, Thorngate FE, Heimberg M, Davidson NO, Elam MB. Insulin increases expression of apobec-1, the catalytic subunit of the apolipoprotein B mRNA editing complex in rat hepatocytes. Metabolism. 1998;47:869–73. doi: 10.1016/s0026-0495(98)90128-7. [DOI] [PubMed] [Google Scholar]
- 25.Giangreco A, Sowden MP, Mikityansky I, Smith HC. Ethanol stimulates apolipoprotein B mRNA editing in the absence of de novo RNA or protein synthesis. Biochem Biophys Res Commun. 2001;289:1162–7. doi: 10.1006/bbrc.2001.6082. [DOI] [PubMed] [Google Scholar]
- 26.Lehmann DM, Galloway CA, Sowden MP, Smith HC. Metabolic regulation of apoB mRNA editing is associated with phosphorylation of APOBEC-1 complementation factor. Nucleic Acids Res. 2006;34:3299–308. doi: 10.1093/nar/gkl417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galloway CA, Sowden MP, Smith HC. Increasing the yield of soluble recombinant protein expressed in E. coli by induction during late log phase. Biotechniques. 2003;34:524–6. 528–530. doi: 10.2144/03343st04. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa Y, Irie K, Ohigashi H, Hayashi H, Wender PA. Synthesis and PKC isozyme surrogate binding of indothiolactam-V, a new thioamide analogue of tumor promoting indolactam-V. Bioorg Med Chem Lett. 2000;10:2087–90. doi: 10.1016/s0960-894x(00)00411-x. [DOI] [PubMed] [Google Scholar]
- 29.Higashi K, Hoshino M, Nomura T, Saso K, Ito M, Hoek JB. Interaction of protein phosphatases and ethanol on phospholipase C-mediated intracellular signal transduction processes in rat hepatocytes: role of protein kinase A. Alcohol Clin Exp Res. 1996;20:320A–324A. [PubMed] [Google Scholar]
- 30.Bouscarel B, Matsuzaki Y, Le M, Gettys TW, Fromm H. Changes in G protein expression account for impaired modulation of hepatic cAMP formation after BDL. Am J Physiol. 1998;274:G1151–9. doi: 10.1152/ajpgi.1998.274.6.G1151. [DOI] [PubMed] [Google Scholar]
- 31.Liu HX, Zhang M, Krainer AR. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998;12:1998–2012. doi: 10.1101/gad.12.13.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta A, Driscoll DM. Identification of Domains in APOBEC-1 Complementation Factor Required for RNA Binding and Apolipoprotein B mRNA editing. RNA. 2002;8:69–82. doi: 10.1017/s1355838202015649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanc V, Kennedy S, Davidson NO. A novel nuclear localization signal in the auxiliary domain of apobec-1 complementation factor regulates nucleocytoplasmic import and shuttling. J Biol Chem. 2003;278:41198–204. doi: 10.1074/jbc.M302951200. [DOI] [PubMed] [Google Scholar]
- 34.Yang X, Gabuzda D. Mitogen-activated protein kinase phosphorylates and regulates the HIV-1 Vif protein. J Biol Chem. 1998;273:29879–87. doi: 10.1074/jbc.273.45.29879. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Tanaka Hall TM. Structural basis for recognition of AU-rich element RNA by the HuD protein. Nat Struct Biol. 2001;8:141–5. doi: 10.1038/84131. [DOI] [PubMed] [Google Scholar]
- 36.Selenko P, Gregorovic G, Sprangers R, Stier G, Rhani Z, Kramer A, Sattler M. Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/mBBP. Mol Cell. 2003;11:965–76. doi: 10.1016/s1097-2765(03)00115-1. [DOI] [PubMed] [Google Scholar]
- 37.Deo RC, Bonanno JB, Sonenberg N, Burley SK. Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell. 1999;98:835–45. doi: 10.1016/s0092-8674(00)81517-2. [DOI] [PubMed] [Google Scholar]
- 38.Handa N, Nureki O, Kurimoto K, Kim I, Sakamoto H, Shimura Y, Muto Y, Yokoyama S. Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature. 1999;398:579–85. doi: 10.1038/19242. [DOI] [PubMed] [Google Scholar]
- 39.Backus JW, Smith HC. Three distinct RNA sequence elements are required for efficient apolipoprotein B (apoB) RNA editing in vitro. Nucleic Acids Res. 1992;20:6007–14. doi: 10.1093/nar/20.22.6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mehta A, Driscoll DM. A sequence-specific RNA-binding protein complements apobec-1 To edit apolipoprotein B mRNA. Mol Cell Biol. 1998;18:4426–32. doi: 10.1128/mcb.18.8.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Mater D, Sowden MP, Cianci J, Sparks JD, Sparks CE, Ballatori N, Smith HC. Ethanol increases apolipoprotein B mRNA editing in rat primary hepatocytes and McArdle cells. Biochem Biophys Res Commun. 1998;252:334–9. doi: 10.1006/bbrc.1998.9647. [DOI] [PubMed] [Google Scholar]
- 42.Lau PP, Cahill DJ, Zhu HJ, Chan L. Ethanol modulates apolipoprotein B mRNA editing in the rat. J Lipid Res. 1995;36:2069–78. [PubMed] [Google Scholar]
- 43.Chen Z, Eggerman TL, Patterson AP. Phosphorylation is a regulatory mechanism in apolipoprotein B mRNA editing. Biochem J. 2001;357:661–672. doi: 10.1042/0264-6021:3570661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osada S, Mizuno K, Saido TC, Suzuki K, Kuroki T, Ohno S. A new member of the protein kinase C family, nPKC theta, predominantly expressed in skeletal muscle. Mol Cell Biol. 1992;12:3930–8. doi: 10.1128/mcb.12.9.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoek JB, Kholodenko BN. The intracellular signaling network as a target for ethanol. Alcohol Clin Exp Res. 1998;22:224S–230S. doi: 10.1111/j.1530-0277.1998.tb04007.x. [DOI] [PubMed] [Google Scholar]
- 46.Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–8. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 47.Varani G, Nagai K. RNA recognition by RNP proteins during RNA processing. Annu Rev Biophys Biomol Struct. 1998;27:407–45. doi: 10.1146/annurev.biophys.27.1.407. [DOI] [PubMed] [Google Scholar]
- 48.Kielkopf CL, Rodionova NA, Green MR, Burley SK. A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell. 2001;106:595–605. doi: 10.1016/s0092-8674(01)00480-9. [DOI] [PubMed] [Google Scholar]
- 49.Price SR, Evans PR, Nagai K. Crystal structure of the spliceosomal U2B″-U2A′ protein complex bound to a fragment of U2 small nuclear RNA. Nature. 1998;394:645–50. doi: 10.1038/29234. [DOI] [PubMed] [Google Scholar]
- 50.Fribourg S, Gatfield D, Izaurralde E, Conti E. A novel mode of RBD-protein recognition in the Y14-Mago complex. Nat Struct Biol. 2003;10:433–9. doi: 10.1038/nsb926. [DOI] [PubMed] [Google Scholar]
- 51.Kadlec J, Izaurralde E, Cusack S. The structural basis for the interaction between nonsense-mediated mRNA decay factors UPF2 and UPF3. Nat Struct Mol Biol. 2004;11:330–7. doi: 10.1038/nsmb741. [DOI] [PubMed] [Google Scholar]
- 52.Tacke R, Chen Y, Manley JL. Sequence-specific RNA binding by an SR protein requires RS domain phosphorylation: creation of an SRp40-specific splicing enhancer. Proc Natl Acad Sci U S A. 1997;94:1148–53. doi: 10.1073/pnas.94.4.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu Y. Regulation of p53 responses by post-translational modifications. Cell Death Differ. 2003;10:400–3. doi: 10.1038/sj.cdd.4401182. [DOI] [PubMed] [Google Scholar]
- 54.Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell. 1997;91:741–52. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 55.Keene JD, Tenenbaum SA. Eukaryotic mRNPs May Represent Posttranscriptional Operons. Mol Cell. 2002;9:1161–7. doi: 10.1016/s1097-2765(02)00559-2. [DOI] [PubMed] [Google Scholar]
- 56.Basu U, Si K, Deng H, Maitra U. Phosphorylation of mammalian eukaryotic translation initiation factor 6 and its Saccharomyces cerevisiae homologue Tif6p: evidence that phosphorylation of Tif6p regulates its nucleocytoplasmic distribution and is required for yeast cell growth. Mol Cell Biol. 2003;23:6187–99. doi: 10.1128/MCB.23.17.6187-6199.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yun CY, Velazquez-Dones AL, Lyman SK, Fu XD. Phosphorylation-dependent and -independent nuclear import of RS domain-containing splicing factors and regulators. J Biol Chem. 2003;278:18050–5. doi: 10.1074/jbc.M211714200. [DOI] [PubMed] [Google Scholar]
- 58.Jans DA. The regulation of protein transport to the nucleus by phosphorylation. Biochem J. 1995;311:705–16. doi: 10.1042/bj3110705. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delano WL. DeLano Scientific LLC; San Carlos, CA, USA.: [Google Scholar]