

Abstract
Recognition of molecular diversity of cell surface proteomes in disease is essential for the development of targeted therapies. Progress in targeted therapeutics requires establishing effective approaches for high-throughput identification of agents specific for clinically relevant cell surface markers. Over the past decade, a number of platform strategies have been developed to screen polypeptide libraries for ligands targeting receptors selectively expressed in the context of various cell surface proteomes. Streamlined procedures for identification of ligand-receptor pairs that could serve as targets in disease diagnosis, profiling, imaging and therapy have relied on the display technologies, in which polypeptides with desired binding profiles can be serially selected, in a process called biopanning, based on their physical linkage with the encoding nucleic acid. These technologies include virus/phage display, cell display, ribosomal display, mRNA display and covalent DNA display (CDT), with phage display being by far the most utilized. The scope of this review is the recent advancements in the display technologies with a particular emphasis on molecular mapping of cell surface proteomes with peptide phage display. Prospective applications of targeted compounds derived from display libraries in the discovery of targeted drugs and gene therapy vectors are discussed.
Keywords: Combinatorial peptide libraries, Vascular cell surface proteomics, Targetomics, Targeted therapies, Display scaffold, Phage display, Viral display, Bacterial display, Yeast display, Cell display, Ribosome display, mRNA display, Covalent DNA display
1. Introduction
Strategies for identification of druggable disease markers and development of approaches for their ligand-directed targeting are required for drug design. In the first part of this review, we will discuss the development of the display technologies for high-throughput identification of agents targeting cell surface receptors. The second part includes a brief overview of libraries and display scaffolds used in ligand selection for clinically useful ligands, with a particular emphasis on phage peptide display. Finally, we will address current applications of individual display technologies to the emerging field, which we define as “targetomics”.
2. Therapeutic needs and “targetomics”
Human disease is often caused and/or associated with alterations in protein expression [1], [2], [3], [4]. The evidence for subject-specific heterogeneity in protein expression profile abnormalities illustrates the need for methodology to profile individual patients for disease markers toward personalized treatment [5], [6], [7]. Identification of reliable disease markers may enable the design of targeted therapies, as well as approaches to predict clinical behavior of affected tissues in a pathologically indistinguishable (but perhaps biologically diverse and heterogeneous) patient population. These issues are particularly important in considering malignant diseases. Until recently, approaches to cancer biomedicine have relied on empirical tissue culture and animal models. Both, the lack of validated molecular profiles indicative of an aggressive cancer phenotype and the absence of validated intermediate endpoints have made tumors difficult to diagnose and manage despite the occasional availability of serum markers along with pathological grade and clinical staging. Without reliable ways of forecasting which tumors will progress, in many cases cancers have been treated aggressively on the chance of cure, but often at the price of potentially severe treatment-associated toxicity. There is a clear need for identification of improved biomarkers of both tumor progression and metastasis potential and mechanisms in cancer. The characterization of molecular diversity in cancer is essential for the development of targeted therapies [3], [8], [9]. Ultimately, it may be possible to personalize the guiding of imaging or therapeutic compounds to tumor or to vascular targets in cancer patients [6]. Early identification of targets, optimized regimens tailored to molecular profile of individual cancer patients, and identification of new cancer-related addresses may also result in revisiting or salvaging of drug candidates that are ineffective or too toxic.
2.1. Cancer and other diseases with altered cell surface proteins
Studies in cancer have recently yielded a broad range of molecular targets upregulated on the surface of tumor and tumor-associated cells [10], [11], [12]. Numerous markers have been identified and characterized for tumor cells, the surrounding stroma, and tumor endothelium [2], [10], [13]. Analysis of the NCI-60 cell line panel, which includes carcinomas of several origins (kidney, breast, colon, lung, prostate, and ovarian), tumors of the central nervous system, malignant melanomas, leukemias and lymphomas, has served to illustrate such marker heterogeneity in tumor cells [14], [15]. In parallel with the NCI-60, gene and protein expression analysis, pharmacological sensitivity of the cells to over 105 different chemical compounds has been registered [15], [16]. Indeed, for some genes, correlation of expression data to drug sensitivity profiles has uncovered mechanistic basis for the activity of certain drugs [17], [18], [19], [20], [21], [22]. Thus, conventional genomic and proteomic approaches have already identified several potential tumor markers and drug targets. However, despite such advances, correlation between drug activity and gene expression profiles has not yet been established for most of the compounds tested [21], [23], [24]. This may suggest the existence of unknown factors and the need to develop alternative methodologies to discover “druggable” molecular targets.
2.2. Personalized diagnostics, imaging and therapy targeting
Over the past decade, drug development has largely focused on approaches to selectively deliver reagents to the cells primarily responsible for or affected by disease. Characterization of molecular diversity at the disease cell surface level—represented by membrane-associated proteins that are often modified by lipids and carbohydrates—is the basis for the development of targeted therapies [2], [3], [8]. We have proposed that selectively expressed disease-specific receptors and corresponding ligands can be applied toward the development of systems for target-directed delivery of imaging agents, gene therapy vectors and cytotoxic drugs [8]. Natural or artificial ligands binding to surface receptors preferentially expressed on accessible cells may be used to direct therapeutics to sites of disease with potential for increased therapeutic windows [8], [25]. In general, tumor markers, exposed to the circulation, are likely to be found on endothelial and other blood vessel-forming cells [10], [26]. Coupling of tissue-homing compounds to a drug yields a targeted therapeutic that is likely to be more effective and less toxic than the parental drug [3], [8]. So far, molecules selected based on their homing to tumors have been used as carriers to guide the delivery of cytotoxic drugs [25], pro-apoptotic and cytotoxic peptides [27], [28], metalloprotease inhibitors [29], cytokines [30], imaging agents [31], [32], and genes [32], [33] in transgenic and xenograft mouse models of human disease. If proven feasible, target-directed therapeutic approaches will improve the treatment efficacy of various diseases by limiting the systemic exposure of other tissues to untoward effects.
2.3. The promises and challenges of the “omics” approach
The Genome Project [34], [35] has led to the identification of a large catalog of human genes. Technological advances have identified genes expressed in cancer and in other diseases by using nucleic acid arrays [36], [37] or serial analysis of mRNA expression (SAGE) in disease tissue samples [12]. Gene expression determined by high-throughput microarray analysis has been used to survey the variation in abundance of many thousands of distinct transcripts from the NCI-60 tumor cell panel [14], [15]. Such data provided functional insights about the corresponding gene products in malignant tumor cell transformation [15], [17], [38], [39]. Information-intensive genomic approaches have yielded candidate tumor markers to be validated at the protein level in prospective studies [40]. From the standpoint of systemic drug targeting through differentially expressed vascular receptors, another initiative has been the identification of genes that are differentially expressed in neoplastic—but not in normal—endothelium [12].
Proteomics, aimed at the systematic analysis of protein contents of individual cells, tissues or biological samples, has been used to profile the spectrum of proteomes in disease [1], [41]. Proteomic approaches based on two-dimensional polyacrylamide gel electrophoresis have served in the identification and characterization of tissue-specific and disease-specific proteins and in the elucidation of their functional roles [42]. Protein microarrays have also evolved to allow fast high-throughput profiling of proteins that constitute biological samples, although this technology is still challenged by technical constraints [43], [44]. Ultimately, the ability to perform high-throughput profiling of protein expression may lead to the construction of the proteome for every tissue, in its normal state and in disease [45].
Large initiatives to systematically register protein complexes that can form inside the cell have been recently streamlined to establish thousands of candidate physiological protein–protein interactions. Various assays, such as the yeast two-hybrid system, have been major contributors to protein interaction mapping [46], [47], [48], [49]. Construction of such “interactomes” operating within the context of the cell surface will help to identify ligand/receptor systems for the purposes of therapy targeting [50]. As technologies related to proteomics advance, new approaches for systematic molecular analysis of endothelium at the protein level are also surfacing [51], [52]. However, conventional methods for systematic protein expression and interaction analysis may well overlook potential diagnostic markers or targets because mere establishment of all proteins and their interactions present in a tissue does not necessarily take anatomical context into account. Some disease markers may be exposed in restricted locations in a way that makes them good drug targets irrespective of their expression level. Therefore, to generate a map of druggable receptors for disease-directed targeting, information derived from conventional protein profiling and binding assessment should be enhanced by integration with data from functional screening for circulation-accessible molecules.
2.4. Vascular cell surface proteome mapping
Studies by our group and others have advanced the concept of vascular proteomics defined here as the molecular phenotyping of cells forming blood vessels at the protein–protein interaction level [13], [53], [54], [55], [56]. Tissue-specific selectivity of expression for endothelial surface molecules has been uncovered in disease and during normal development [10], [25], [57]. Systematic profiling of selectively expressed ‘vascular addresses’ has revealed molecular targets that may be used to direct therapies to specific tissues [8], [26]. In the long term, continued attempts to explore endothelial molecular diversity will lead to the construction of a functional ligand-receptor vascular map for every tissue [13]. Methods for time-efficient probing of human vascular beds for differentially expressed vascular cell surface markers will make it possible to personalize vascular-targeted therapies for patients [3]. The knowledge of relevant ligand-receptor interactions derived from display technology screens may become useful for the design of chemically synthesized peptidomimetic drug leads [58].
3. Polypeptide libraries as a source of targeting ligands
3.1. Combinatorial peptide libraries
Smith et al. [59] have first used libraries displaying short peptides on antibodies for the purpose of mapping antibody epitopes with phage display (see Section 4.1). Since then, random linear and cyclic peptide libraries [60], [61], antigen- or gene-fragment libraries [62], [63] or a combination of both [64], [65], [66] been used for the epitope mapping of monoclonal antibodies (mAbs). Combinatorial peptide libraries, which can be constructed to contain billions of unique peptide sequences, have been a useful source of molecules with different binding potential [67], [68]. Strategies for flexible and constrained random peptide library construction and screening approaches have been extensively discussed elsewhere [50], [69], [70].
Combinatorial peptide libraries comprised either by completely random peptides or by backbones randomized in defined positions have served to identify protein binding sites. Excellent reviews summarize the experience with using random and semi-random peptide libraries in the context of various display platforms [71], [72], [73], [74], [75]. Among the most commonly used libraries are those with the general structure CXNC, which express random 5 to 9 amino acid residue-long peptides flanked by cyclized cysteines on each side [76]. Such libraries expressing cyclic peptides on filamentous phage surface have been used by or group [77]. Other groups have also used similar libraries displayed on lytic phage surface [56].
As evidenced by numerous studies within the past decade, cell surface receptor proteomes can be mapped by screening peptide libraries [8], [57], [78], [79], which is discussed in detail in Section 4.1. Peptide libraries have also been selected for motifs mimicking epitopes of antibodies produced in disease. For example Mintz et al. panned a library displaying random peptides on autoantibodies of prostate cancer patients, which led to identification of Grp78 as an antigen overexpressed by human prostate cancer [80]. In another study, Vidal et al. found an HSP90-mimic peptide as a human ovarian cancer antigen by epitope mapping on the pool of antibodies from ascites purified from ovarian cancer patients [81]. Together, there seems to be a theme emerging, in which stress-response chaperons become immunogenic in cancer. As importantly, peptide ligands selected from unbiased screens without any predetermined notions about the nature of the cellular receptor repertoire have been used for the subsequent identification of the corresponding target cell surface receptors [77], [78], [82], [83], [84], [85]. As discussed above, peptide targeting specific receptors on the surface of cells implicated in disease can also be used for directing imaging agents, cytotoxics, or gene delivery vectors [3], [8]. Artificial inhibitory peptides produced from transgenes have also been used to disrupt signaling intracellular pathways of interest in the Drosophila model [86]. In the future, such artificial gene products could be developed into vectors for diagnostic or therapeutic intent. Peptides may have other desirable benefits over currently used antibodies, because of their increased tissue penetration and diffusion, rapid blood clearance, or simplicity of synthesis in vitro and quality control.
It has been serendipitously observed that cell surface-binding peptides selected from display libraries are often similar to domains found in naturally expressed proteins [87], [88]. For example, a peptide may mimic a ligand of a vascular receptor via a motif sufficient for receptor recognition. Initially, Koivunen et al. isolated peptides targeting alpha 5 beta 1 integrin via mimicking fibronectin, peptides targeting alpha v beta 3 and alpha v beta 5 integrins by mimicking vitronectin, and peptides targeting alpha IIb beta 3 integrin by mimicking fibrinogen [76]. The majority of integrin-binding peptides contained the three amino acid long (tripeptide) motif Arginine–Glycine–Aspartic Acid (RGD). Based on these observations, it has been proposed and proven that in many cases binding peptide motifs mimic interactions of cell surface receptor with their natural ligands. Examples of biochemical unit recognition and binding of ligand motifs other than those based on the RGD/integrins, include Asparagine–Glycine–Aspartic Acid (NGR) binding to aminopeptidase N/CD13 [25], [82], Glycine–Phenylalanine–Glutamic Acid (GFE) to membrane dipeptidase [25], [82], [89], [90], and Aspartic Acid–Proline–Leucine (RPL) to VEGF-1 [78]. Thus, a stretch of three residues appears to provide the minimal framework for structure formation and protein–protein interaction. As discussed in Section 4.1, our group has developed a bioinformatics platform based on the analysis of frequencies of tripeptide motifs within peptides selected in the screen for identification of the prototype receptor ligands mimicked by short peptides [85], [91], [92]. A number of comprehensive reviews summarize peptide motifs and their corresponding ligand/receptor systems identified by using phage display [67], [93], [94]. To interrogate early atherosclerotic lesions and explore plaque-associated endothelial cell proteome, Kelly et al. recently screened a phage-displayed random peptide library in ApoE-/-mice [95] to identify plaque-targeting peptides, similar to several proteins involved in atherosclerosis. Such findings may be translated into agents useful in early disease detection. The prospects for application of specific small peptides for specific needs of experimental targeted therapy have been critically evaluated over the past few years [3], [6], [96], [97]. Libraries of unnatural amide-linked oligomers encoded by RNA have also been designed to express and screen peptides consisting of unnatural amino acids [98], [99]. Techniques for peptide library construction constantly evolve [100]. Careful side-by-side comparison will be necessary to determine the best designs, which may actually be specific for individual display platforms. Recent approval of a number of peptides for clinical purposes reflects their recognition as promising therapeutic agents [101].
3.2. Immunological protein libraries
Molecules naturally involved in immune recognition have been heavily exploited in display technologies. The two basic types of immunological interaction include (i) binding of an antibody to an antigen and (ii) binding of a T cell receptor (TCR) to a major histocompatibility complex (MHC)-presented antigen-derived peptide [102], [103], [104]. The structural diversity of antibodies and TCRs has turned them into suitable platforms for construction of display libraries and as sources of specific and functional antigen-targeting molecules. In parallel, construction of MHC platform-displayed libraries has been initiated to facilitate detection of immune recognition mediated by TCRs. Such technologies may have future application in targeted medicine.
3.2.1. Antibody libraries
Presence of autoantibodies against various antigens has been revealed not only in infection, but also in autoimmune or allergic diseases [105], [106], [107] and in cancer [80], [108], [109], [110]. Factors that induce autoantibodies to self-antigens usually include protein mutations [111], [112], overexpression [113] or aberrant modifications [114] and cell localizations [114]. Serological identification of antigens by recombinant expression cloning (termed SEREX), which involves bacterial expression of cDNA libraries derived from tumor tissues and screening the recombinant proteins with autologous serum, has pioneered the combinatorial search for tumor antigens [115]. The list of disease antigens for which humoral immune response has been detected continues to grow, and is reviewed elsewhere [116], [117]. Several antibodies have been approved by the US Food and Drug Administration (FDA) and have been used as therapeutics in the clinic for several years [101], [118]. An emerging target for anti-cancer therapies is angiogenesis, the process of vasculature sprouting from existing blood vessels, which is regulated by vascular endothelial growth factor (VEGF) and its receptor [119]. A recent example of an antibody with clinical anti-cancer activity is against the VEGFR pathway [120]. FDA approval of Avastin (bevacizumab), an anti-VEGF humanized monoclonal antibody is a breakthrough for treatment of various cancers by targeting tumor-related angiogenesis [121]. Other drug-conjugated antibodies directed at cancer antigens include Rituxan and Zevalin (anti-CD20) for Non-Hodgkin Lymphoma, Herceptin (anti-HER2/neu) for metastatic breast cancer, Mylotarg (anti-CD33) for acute myeloid leukemia, Campath and Alemtuzumab (anti-CD52) for B-cell chronic lymphocytic leukemia (CLL), and Erbitux (cetuximab) (anti-EGFR) for advanced colorectal cancer, among many others. Despite the recent developments of approaches to streamlined generation of antibodies [122], [123] and their humanization, traditional techniques for raising antibodies with desired activities and specificities remain to pose limitations.
Screening of antibody libraries aimed at the isolation of prototype drugs targeting cancer and other diseases is one of the main approaches to target discovery and validation [124]. Display technologies have been instrumental in exploring the humoral immune response [125], [126] and antibody and immunogeneicity-based signatures in cancer have been systemically profiled [127], [128]. Interestingly, screening combinatorial antibody libraries from cancer patients for immunoglobulins that can identify metastatic tumor cells indicated that tumors induce antibodies against potential therapy targets, such as integrins [129]. Our group has identified stress-response chaperons as cancer antigens that induce humoral immune response [80], [81].
Various display platforms and vectors have been utilized for the display of antibodies in several different formats [130], [131], [132]. Originally, libraries of immunoglobulins fragments (Fab) and single chain antibodies (scFv) displayed on filamentous phage have been designed [133], [134]. Recombinant antibody engineering remains a dynamic field and new approaches to construct libraries of antibody-like molecules displaying antigen recognition motifs are in development. For example, a library displaying variable domains of camelid heavy chain antibody domains was selected for antagonistic anti-EGFR nanobodies, which efficiently inhibit EGF binding to the EGFR, EGF-mediated signaling, and delay the growth of tumor xenografts [135]. Affibodies, the backbone of which is based one of the IgG-binding domains of staphylococcal protein-A have been randomized to create libraries selected on such cancer epitopes as HER2 and TNF-alpha to create agents potentially useful for imaging and therapy targeting applications [136], [137].
In addition to dissection of the immune response against tumors, antibody display libraries have been recently used in a wide variety of application ranging from proteasome characterization in aging [138] to mapping immune response in infections [139], [140]. Recently, an scFv phage display library was used to screen vasculature as a proof-of-concept [55]. Selection was performed on the luminal endothelial cell plasma membranes isolated from tissues and enriched in proteins exposed to the bloodstream in vivo. Homing of the selected antibodies was confirmed by whole-body imaging.
Display technologies have accelerated the translation of antibodies into targeted therapeutics [118], [121], [124]. One of the FDA-approved antibodies, generated by using phage display, is Adalimumab (HUMIRA): a human anti-TNF IgG1 used against rheumatoid arthritis [141]. Many more antibodies selected by phage display are in clinical trials. Some examples include CAT-3888 (anti-CD22) against several leukaemias, CAT-354 (anti-IL-13) against asthma, CG-1008 (anti-TGF β) against idiopathic pulmonary fibrosis, MYO-029 (anti-GDF-8) against muscular dystrophy, ABT-874 (anti-IL12) against T cell-derived autoimmune diseases, as well as HGS-ETR1 (anti-Tumor Necrosis Factor Related Apoptosis-Inducing Ligand Receptor, TRAIL-R1) and HGS-ETR2 (anti-TRAIL-R2) against various cancers. Recently, antibodies blocking the immunosuppressive molecule CD200 were identified by panning a phage-displayed antibody library derived from rabbits immunized with primary chronic lymphocytic leukemia (CLL) cells [142]. Undoubtedly, phage and other display technologies have great potential for proteome-wide exploration of humoral immune response and for production of therapeutic antibodies [68], [143], [144].
3.2.2. TCR libraries
Progress in understanding of process of antigen presentation and recognition by TCRs expressed by antigen-specific T cells has led to the identification of several disease-specific self antigens that can be presented by MHC class I (MHC-I) on dendritic cell (DC) [103], [145] and elicit antigen-specific cytotoxic lymphocytes (CTL) to eliminate virus-infected or mutated cells [146], [147], [148]. Among such antigens are myeloid leukemia-associated PR1 derived from serine proteases proteinase 3 and neutrophil elastase [146], Wilm's tumor antigen (WT1) [149], overexpressed in several tumors, including myeloid and lymphoid leukemia. Disease-specific CTL can be stimulated in vitro by priming of the antigen-presenting cells (APC) pulsed with peptide antigen and in vivo by vaccination with peptide [150], [151], [152]. However, limited number of MHC-I presented disease-specific antigen peptides have been identified so far. In contrast to traditional hypothesis-driven investigations, the display strategies have a potential of high throughput discovery of disease-specific MHC-peptides and their counterparts: TCRs.
The first approach is the TCR display libraries, created by using single-chain variable domains of human TCR [153], [154], [155], [156]. In vivo, cytotoxic T lymphocytes use T cell receptors to recognize cell with abnormal antigen presentation, and activate antigen-specific cell killing. The specificity of TCR recognition toward MHC-presented antigens of altered cells makes them candidates for cell-specific drug delivery. Alternatively, TCR can be introduced in patients' T cells, which can be used for so called passive immunization. TCRs consist of alpha and beta chains, with each chain containing a variable (V) domain and a constant (C) domain. Similar to immunoglobulins' Fvs, V-domains of TCR contain hypervariable regions, which determine the specificity of T cell. The structure of the antigen-recognizing site of TCR is very similar to that of antibodies and, similar to antibodies, recombinant TCR can be expressed in bacteria as a single-chain TCR (scTCR), without loss of specificity for antigen [157]. Several groups have achieved scTCR expression in a display format [158] and made first steps toward selection of peptide-specific TCR from TCR libraries [159], [160]. However, due to low affinity of binding between TCR and MHC-I/peptide, further modifications of scTCR, like multimerization and mutations have been necessary for sensitive detection of target MHC-I/peptide. Yeast surface display of TCRs has also been shown possible by expression of scTCRs with permutated variable region residues proximal to the CD3 domain on yeast surface [155].
Another, more feasible strategy is the selection of antibodies against MHC/peptide complexes (or TCR-like antibodies) from naïve [161] or immunized [162] antibody libraries. Using this approach, a number of MHC/peptide antibodies with therapeutic or diagnostic potential have been obtained. For example, Krogsgaard et al. used an antibody isolated from immunized phage display library for visualization of myelin basic protein (MBP) T cell epitopes in multiple sclerosis lesions [162]. Others used a phage display-selected Fab fragment with MHC class I-restricted specificity to MAGE-A1 fused to the FcεRI signaling molecule for retargeting of primary human T lymphocytes [163]. Usage of TCR-like antibody molecules for studying MHC-I antigen presentation in health and disease, as well as for therapeutic purposes in cancer, infectious diseases, and autoimmune disorders, have been recently reviewed elsewhere [164].
3.2.3. MHC-peptide libraries
TCRs normally recognize antigens processed by the proteasome and presented in the grove of MHC-I or MHC-II molecules as linear peptides. Display of random peptides library in the context of MHC for the purpose of patient T cell repertoire surveillance would enable high throughput identification of MHC-restricted peptides as targets for cell-specific therapy. Because in vivo interaction of MHC-peptides with TCRs takes place in the context of APCs and T cells, and is orchestrated by a group of supporting cell surface molecule interactions (CD4/MHC-II, CD8/MHC-I, CD2/LFA-3, LFA-1/ICAM-1, among others) [103], [145], [165] it is challenging to mimic this supramolecular structure in the context of display systems. In addition, MHC-peptide library construction and screening has been hampered because MHC presentation must be accompanied by non-covalently bound beta-2 macroglobulin (β2M) [103]; therefore expression and folding of functionally active complex is Escherichia coli periplasm is technically difficult. Finally, the naturally low affinity of TCR binding to the MHC-peptide-β2M complex (Kd = 0.1–3 μM), in particular in the absence of stimulating molecules mediating efficient immunological synapse [166], makes selection of display libraries complex.
Nevertheless the possibility of expression of the MHC-peptide-β2M complexes as displayed on filamentous phage has been demonstrated [167]. In this study, folding of single chain MHC-peptide-β2M fusion was achieved, as evidenced by specific recognition with relevant conformation-specific monoclonal antibodies and, more importantly, with a unique “TCR-like” (i.e., peptide-specific, MHC-I-restricted) antibody. Unfortunately, the authors could not demonstrate interaction of displayed and folded single chain MHC/peptide complex with T cells. Consistent with low affinity of TCR binding to MHC/peptide increased avidity (achieved by tetramerization of MHC/peptide complex) allowed to visualize the interaction. In an attempt to design multivalent display of MHC-presented antigens, Le Doussal et al. has demonstrated bivalent fusion peptide –MHC-I (H-2Kd)-β2m presentation by a small fraction of phage particles [159]. Even with such a low ratio of bivalent phage, binding of Kd-displaying phage to a H2-Kd-restricted TCR immobilized on solid phase (as well as to Kd-restricted T cell hybridomas) has been shown. Still, no significant binding to relevant T cells was detected in the study. Therefore, further optimization of peptide-MHC display is still awaited for the TCR-mediated immune recognition to become a subject to productive screens for CTL antigens and their corresponding TCRs by using display technologies.
3.3. cDNA libraries
Display methods in general are limited in terms of the length of polypeptide translated from the library clones. Shorter fragments have a natural selective advantage because library units encoding them need less time for production and because they are less likely to pose constraints on the display unit function. These issues are relevant for virus/phage display methods (see Section 4.1), in which longer polypeptides have a higher chance of disrupting virion functions, such as assembly and infectivity. In addition, in the majority of phage display systems, proteins are generally expressed as C-terminal fusions with the N-terminus of the minor coat protein pIII for display on phage particles. Because full-length cDNAs generally contain several stop codons near their 3′ end, this approach cannot be used for their expression on the phage surface. These problems have impeded the progress in construction and use of full-length cDNA libraries in display applications. Instead, initial breakthroughs were achieved with libraries displaying protein fragments [168]. Such protein domain libraries have been developed for antibody epitope mapping to directly and unequivocally identify the antigen. Given these advantages over other techniques, this approach has been widely used and has served to screen the whole yeast proteome for protein domains binding the second src-homology (SH3) domain of Bem1p protein [169].
In certain cases, however, direct display of whole proteins has been proven feasible. Plasminogen-activator inhibitor 1 (PAI-1) was among the first to be displayed on the pIII protein of filamentous phage [170]. Phage-displayed PAI-1 retained its capacity to form equimolar complexes with its target serine protease tissue-type plasminogen activator (t-PA), as well as its ability to inhibit t-PA activity. Procedures for display cloning of cDNAs have evolved significantly since then, and monovalent phage display relying on helper phage (see Section 4.1) has made whole protein presentation generally possible [171], [172], [173], [174], [175], [176]. Considerable technical advances in systematic expression of cDNA libraries on the surface of bacteriophage has been achieved with the development of the Jun/Fos fusion approach [177], [178]. This system has been adopted for in vitro selection of catalytically active polypeptides [179]. An interesting attempt was undertaken to screen for extracellular proteins by using a cDNA library in which only proteins carrying the cell membrane localization domain can be displayed on filamentous phage [180]. Another perspective approach that may evolve as a tool for isolation of semi-natural molecules potentially superior to short peptides and antibodies is based on display of avimers: multidomain polypeptides shuffled from extracellular domains of receptors; in this study [181], avimers were multivalently displayed on phage, and selection of the library resulted in isolation of avimers with high avidity and the resulting sub-nanomolar affinity.
3.4. Scaffolds for polypeptide display
In addition to the widespread usage of immunological molecules, such as immunoglobulins and TCRs, naturally designed to display permutated polypeptide sequences, there are many other novel library frameworks that may be adopted for the presentation of polypeptides in display libraries [67]. Application of cell surface proteins as polypeptide presentation scaffolds is discussed in the corresponding sections for bacterial, yeast and mammalian cell surface display (see Section 4.2). Non-conventional scaffolds for protein display are discussed in an excellent recent review [74]. The key for selection of a library platform is the availability of exposed loops that link stable structural elements such as alpha-helices and beta-sheets. These loops are accessible for ligand binding and can accommodate sequence variation without changing the overall structure and stability of the underlying scaffold. Tertiary structures and rigidity of scaffolds can be modulated by the presence of stabilizing disulfide bonds linking spatially separated strands of the protein. Based on the architecture of their backbone, scaffold proteins can be classified into those consisting of: (i) alpha-helices, (ii) few secondary structures or irregular architecture of alpha-helices and beta-sheets, and (iii) predominantly beta-sheets. Examples of scaffolds with alpha-helical frameworks include Z-domain affibodies, immunity proteins of E. coli, cytochromes, and ankyrin repeat proteins and others. Examples of scaffolds with irregular structures include Kunitz domain proteins and PDZ domain proteins. Examples of scaffolds with beta-sheet frameworks include TCRs, CTLA proteins, knottins, and fibronectin type-III domains. Functional proteins have also evolved as attractive scaffolds for polypeptide display. For example, the beta-barrel green fluorescent protein (GFP), served as a combinatorial peptide library display scaffold, and peptides exhibiting anti-proliferative effects were selected [182]. Another useful function protein is executed by a series of small domains, termed protein transduction domains (PTDs), such as HIV-1 TAT protein, herpes simplex virus VP22 protein, or the third alpha-helix of Antennapedia homeodomain (penetratin), which promote the intracellular transport of peptides and proteins across cell membranes and into cells [183], [184]. Selection of cell membrane permeability libraries for PTD-displayed polypeptides could become an approach to identification of molecules with cell localization and/or function of interest, and labeling of these peptides for optical imaging, nuclear scintigraphy, or magnetic resonance imaging could have biological and medical applications [185].
4. Display technologies: applications in targetomics
The principle underlying display technologies is the ability to physically link phenotypes of polypeptides displayed on a certain platform to their corresponding genotype, which was first shown for phage display [59]. Typically, the phenotype registered in display assays is the physical binging, and it is serially selected [41], [186]. Although linking polypeptides to their encoding DNA extends to other functional selection methods, such as the yeast two-hybrid system [47], [48], [49], [187], assays for intracellular interaction are conventionally considered as a separate category. While display scaffolds presenting polypeptides in the library vary, the ability to link an individual polypeptide selected based on its particular properties to the coding nucleic acid (cDNA or mRNA) is the feature that makes it possible to identify, isolate and amplify the desired library members [188]. There are five major display technologies, which have evolved in parallel (Fig. 1 ). Here, we elaborate on individual display methods, with a special emphasis on filamentous phage display, since this is the major interest of our group. Within the first six months of year 2006 alone, close to 200 publications involving phage display have been published. Such popularity and versatility of phage display puts this technology into a separate category per se.
Fig. 1.
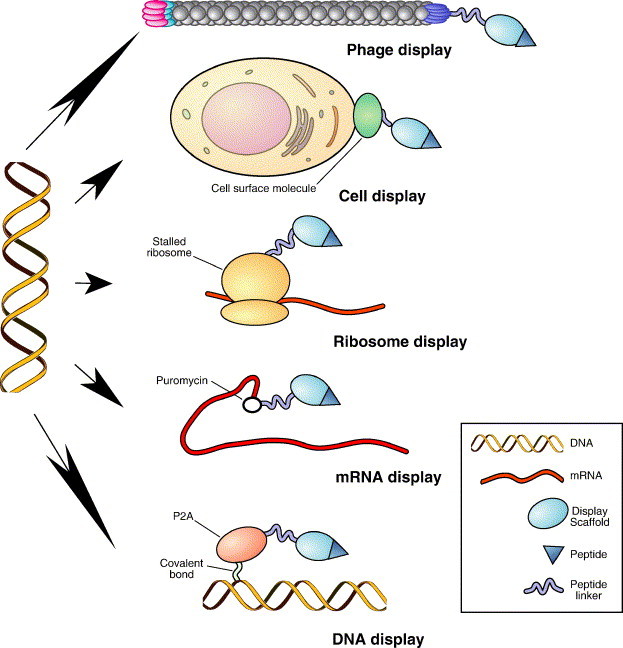
Schema of available display technologies. All display platforms are based on the ability to physically link the polypeptide produced by a library clone to its corresponding genotype. This allows one to recover the DNA encoding the clone selected based on the desired polypeptide phenotype, such as binding to the target. In phage/virus display, linkage between the gene and the encoded polypeptide is achieved by expression of the polypeptide as a fusion with a coat protein from DNA packaged in the same particle. In cell display, linkage between the gene and the encoded polypeptide is achieved by expression of the polypeptide as a fusion with a cell surface molecule from DNA, which the cell receives in order to be transformed. In ribosome display, linkage between the gene and the encoded polypeptide is achieved by stabilization of complexes between the ribosome, mRNA and the encoded polypeptide upon termination of elongation with a permissive marker, such as chloramphenicol or low temperature. In mRNA display, linkage between the gene and the encoded polypeptide is achieved by a puromycin molecule covalently bonding the mRNA 3′ and the translated polypeptide upon the ribosome stalling at the junction of mRNA and an engineered single-stranded DNA linker. In covalent DNA display, linkage between the gene and the encoded polypeptide is achieved by covalent bond that forms between the DNA-binding protein P2A (produced as a fusion with polypeptide) with the DNA encoding the fusion. Figure is not depicted in scale.
4.1. Virus/phage display
4.1.1. M13 pIII peptide phage display as a model technology
In 1985, Smith, Scott and colleagues established a method for displaying polypeptides on the surface of filamentous M13-derived bacteriophage (phage) [59]. This technique was originally developed to map epitope-binding sites of antibodies by panning random peptide-phage libraries on immobilized immunoglobulins. Since then, phage display has been used as a powerful method to register polypeptide binding with a diverse range of basic biological, as well as technical and medical applications [77], [186]. Biology of phage has been well reviewed [189], [190], [191], [192]. In contrast to the lytic bacteriophage species (e.g., T4), filamentous phages replicate and assemble without killing the E. coli host. The most abundant capsid component, is the major coat protein pVIII, which is represented by approximately 2700 molecules. There are 5 molecules of the minor pIII coat protein at one end of the phage virion, which, along with the 5 copies of pVI, another minor coat protein, is involved in bacterial cell binding and in the termination of phage particle assembly during morphogenesis. The other end of the phage is capped by five complexes of minor coat proteins pVII and pIX, which are required for initiation and maintenance of phage assembly in the host bacteria. In libraries used most commonly, polypeptides to be displayed are expressed as fusions with the phage coat protein pIII [59], [77], [189]. During the phage assembly process, the resulting fusion proteins are transported to the bacterial periplasm or inner cell membrane and are incorporated into the phage particle along with the single-stranded DNA (ssDNA) encoding the displayed fusion protein [189]. Thus, phage clones produced by host E. coli have peptide phenotypes coupled with their genotypes.
There are two basic formats of polypeptide display in phage libraries: polyvalent and monovalent [77], [189]. In polyvalent phage display, each copy of the capsid protein (such as pIII) displays the polypeptide. Libraries of that type are based on vectors derived directly from the phage genome, such as fd phage [193], and encode all the proteins needed for phage replication and assembly. Polyvalent phage display is commonly used for selection of libraries of short peptides, display of which does not interfere with the essential function of the pIII protein. Monovalent phage display is the solution for cDNA libraries, which encode proteins or protein domains too large for the pIII protein to retain its function. In monovalent display, particles mosaic for recombinant and wild-type capsid proteins are produced by a phagemid vector encoding the fusion protein and by a helper phage, respectively. The purpose of the helper phage is to provide all the proteins required for phagemid replication, ssDNA production and packaging, hence allowing “phage rescue”.
A big advantage of phage display, accounting for its widespread use, is the convenience with which libraries can be selected for target-specific binders. Rapid enrichment of library clones encoding binding polypeptides is achieved by phage library incubation with a target followed by removal of the non-reacting phage and amplification of binder clones in the host bacteria. Usually, three to five rounds of panning are sufficient to enrich for binding peptide sequences. The primary structure of the polypeptide corresponding to the clone is then deduced by sequencing the corresponding encoding DNA. In this manner, libraries with large diversity (approximately to 1010 unique individual sequences) can be created, amplified, stored, and screened against a target of interest.
Phage display normally enables isolation of target-specific polypeptides with affinity constants in the micromolar-to-nanomolar range. In general, multivalent phage display is more sensitive due to the comparatively higher avidity resulting from multiple copies of peptide displayed, and hence detects lower-affinity binding. When necessary, affinity maturation for individual clones can be performed through generation of secondary libraries of mutated peptides [68], [143], [194].
4.1.2. Phage display on coat proteins other than pIII
Although the pIII protein has been by far the most commonly used in phage display, alternative display systems utilizing the other four M13-based coat proteins: pVIII [190], [191], [192], pVI [158], [173], [175], [195], pVII [196], and pIX [197] have also been tested. Standing aside from other M13-based protein display systems, the pVIII-based platform, which features many more copies of this coat protein per particle, may offer specific advantages for imaging applications [198], [199]. Recently, a multivalent display system alternative to pIII on filamentous phage pVII minor coat protein was reported [196]. The authors show that pVII protein is apparently not structurally compromised by fusion with large polypeptides, such as scFv antibody fragments, thus demonstrating the feasibility of utilizing pVII as a fusion partner without the necessity of co-infecting helper phage required for pIII phagemid systems. There is slow but steady progress in the design of phagemid vectors that allow efficient display of correctly folded functional proteins on phage, and new systems are constantly added for applications involving specific types of displayed proteins [200].
4.1.3. Bacteriophage alternatives to M13
Although filamentous phage display has been used for display of various types of libraries [189], this system represents a limitation when one attempts to display proteins that do not fold properly in the periplasm. Because filamentous phage are assembled in the cytoplasmic membrane and are secreted from infected bacteria without cell lysis, correct transfer of the hybrid capsid proteins across the lipid bilayer of the inner membrane of E. coli is prevented [201]. To overcome that limitation, alternative bacteriophage display systems that assemble their capsid in the cytoplasm and are released via cell lysis have been developed by using lytic bacteriophage such as T4 [202], T7 [172], and P4 [201], [203]. Libraries of peptides displayed from lytic phage species have also been created and screened [204], [205], and T7 Select cDNA libraries (Novagen) are often used for isolation of interacting proteins [56], [206]. Recently, an ‘epitomics’ approach for global antibody/antigen interaction mapping has been developed based on T7 phage display of cDNA libraries [127], [128], [207].
Bacteriophage lambda is yet another alternative vector for the surface display of peptides and proteins [208]. There are features that make lambda phage potentially advantageous: ability to display multimeric proteins, lack of displayed fusion secretion requirement, and adjustable valency of the displayed fusions. Lambda display has been suggested as an alternative for poorly soluble proteins that tend to form inclusion bodies when expressed in bacteria [209]. Lambda-based systems are potentially attractive for protein display from cDNA libraries, and their input into functional proteomics progress is likely to increase [210]. There has been some follow up on the use of this system since its original inception, perhaps suggesting general application for lambda phage-displayed libraries [211], [212], [213].
4.1.4. Phage library selection on biological materials
Phage display has been routinely used to screen peptide or antibody libraries for ligands on purified and immobilized molecules in vitro [189]. Numerous research groups have employed phage display for selection of peptides binding to purified proteins of interest in projects aimed at identification of protein–protein interaction pairs and mapping of their interaction domains [6], [8], [88], [174], [214]. Recent examples from our group include the selections for random peptides binding to SPARC [215], [216], integrins [76], [184], caspases [217], metalloproteases [29], and their exploitation as agents directing drug delivery. Examples of peptides and antibodies identified in such screens on prospective drug targets have been reviewed elsewhere [6], [88], [96], [200], [218]. Several recent developments on phage display application to protein–protein interaction mapping are worth noting. The selectively infective phage technology (SIP) has been implemented for systematic registration of protein–protein interactions [219], [220], and attempts to map protein–ligand interactions by using whole genome phage libraries is underway [178]. Prediction and identification of protein interactions by using phage display in high-throughput is likely to complement and or synergize with other large-scale initiatives aiming at similar goals [87], [176]. Another example of interactome-type research is the screen for the membrane voltage-dependent anion-selective channel protein (VDAC) epitopes by using a phage library expressing liver cDNAs and VDAC reconstituted into liposomes and captured on a chip surface. Remarkably, at least 40 % of hits were already reported voltage-dependent anion channel (VDAC) ligands, and some of the 55 novel interactions functionally blocked the channel [221]. Successful identification of peptide epitopes sets the basis for subsequent utilization of phage display for mapping protein interaction domains (Kolonin et al., unpublished).
Over the past decade, approaches to efficiently select phage libraries on cultured cells have evolved [31], [222], [223]. One of the methods that enables quick and effective isolation of specific ligands from small numbers of cells is the biopanning and rapid analysis of selective interactive ligands (BRASIL) established by our group [78]. Recent examples of successful phage display selection on cells include the identification of peptides targeting vascular endothelial cells in gastric cancer [224] and of a VEGF-mimic [225]. It has been shown that phage particles displaying ligands binding to internalizing receptors, such as integrins, can undergo efficient uptake by targeted cells. This has been shown to be the case for both peptides [226] and antibodies [227] displayed on M13. Based on that notion, phage display technology has been successfully mobilized for targeted gene delivery into mammalian cells [32], [228], [229].
Encouraged by successful library selections on such complex biological systems as surfaces of cells, investigators have further pursued clinical utilization of phage display. Ex vivo protocols for selection and evaluation of libraries on biopsy specimens have been developed [230], [231]. In a recently reported procedure [232], tumor cell suspensions derived from biopsy specimens of seven human breast tumors following library subtraction on normal breast tissue were subjected to selection. As a result, peptides targeting phosphatidylserine, which binds to annexin V during apoptosis, were identified with the goal of developing new tools for apoptosis imaging.
In the past decade, we have pioneered a number of novel phage display applications [2], [8], [25], [26], [57], [77]. The subsequent research of our group has combined random peptide library selections with a platform strategy comprised by advanced molecular biology and bioinformatics, as well as utilization of clinically-annotated patient samples. This approach allows to quickly and efficiently identify functional cell surface receptors in conjunction with their ligands that can be directly used as prototypes for therapeutic targeting. For example, peptides targeting tumor receptors have been identified, functionally explored, and used in pre-clinical studies [6], [25], [32], [84], [96], [97], [199], [233], [234]. Below, we expand on several examples of recent developments that illustrate how this systematic strategy can be applied for clinical cell surface proteome mapping of human tumors and vascular beds.
One of the features of tumor vasculature is its fenestration that makes tumor cells directly accessible to circulation in many cancers [235], [236]. This presents the proteome of the tumor cells as potentially directly targetable with systemically administered therapeutics. To advance the construction of the cell surface ligand/receptor map of human cancers, the NCI-60 panel of human cancer cell lines from different histological origins and grades [14], [15] was recently systematically analyzed with phage display [91]. We used BRASIL [78] to select peptides binding to receptors expressed by the NCI-60 cells from a combinatorial library. The selected peptides were surveyed by using a comprehensive strategy (Fig. 2 ). By statistical analysis of peptide motif sequences, each NCI-60 cell line was assigned a unique set of peptide motifs that were isolated during the selection for cell surface binders. We showed that tumor cells can be grouped by profiles of their phage display-derived peptide ligands directed to differentially expressed cell surface receptors [91].
Fig. 2.
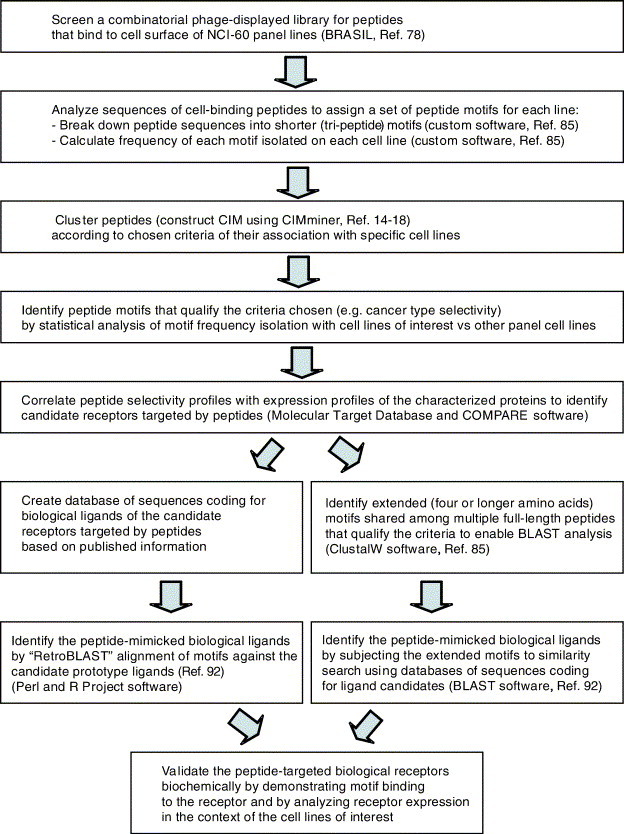
A comprehensive strategy for systematic characterization of ligand/receptor proteome functional on the surface of NCI-60 cancer cell lines by using ligand-mimicking peptides isolated from combinatorial phage display libraries. Identification of cell surface receptors through their soluble ligands enriches for receptors likely to be “targetable” (meaning, can be ligand-directed).
To further dissect cancer cell surface proteome, an approach for peptide-targeted receptor identification was designed. Analysis of selected peptide motif sequences grouped cells lines according to profiles of motif distribution within the NCI-60. Some of the peptide motifs were found within proteins known to bind the receptors that had NCI-60 expression profiles matching cell line recognition profiles of the peptides, and that are implicated in cancer. Candidate targeted cell surface molecules were identified, which included many tyrosine kinase receptors. As a proof of principle, epidermal growth factor receptor (EGFR), known to be upregulated in various cancers, was validated as a target of a pool of tripeptides, which were isolated as candidate mimics of biological EGFR ligands. Results of this feasibility study suggest that tumor cells can be grouped by profiles of their peptide ligands directed to differentially expressed cell surface receptors [91].
4.1.5. In vivo phage library selection
Our group has developed in vivo phage display: an approach to select for phage-displayed peptides that home to receptors differentially accessible in the vasculature of specific organs [2], [8], [25], [26], [27], [32], [57], [77], [82], [83], [85], [92], [237]. In vivo phage display selection is performed without preconceived notions about targeted receptor identity and identifies phage-displayed ligand peptides homing to an organ of interest in an unbiased and internally controlled process. The procedure involves intravenous administration of a phage-displayed random peptide library, which is let circulate for a certain period of time to allow distribution of the clones in the vascular system (Fig. 3 ). This is followed by perfusion (when possible and/or necessary) and washing of the tissue cells to remove nonspecifically bound phage clones, after which clones homing to selective vascular beds are recovered by host E. coli infection and amplified for subsequent rounds of selection. In vivo phage display has two main applications. First, library selection by using this approach identifies ligands targeting specific vascular beds. Second, the determination of molecular profiles of blood vessels in specific diseases enables identification of differentially expressed disease markers. Complementary approaches have been used to identify receptors for peptides homing to the vasculature of the lung [90], [238], breast [239], placenta [83], white adipose tissue [84], and other organs [79], [92]. Animal models have also been used to identify tumor-vascular molecular addresses with phage display [32], [82], [234], [238], [240], [241]. Systematic implementation of this strategy will eventually lead to the complete map of vascular ligand-receptor interactions.
Fig. 3.
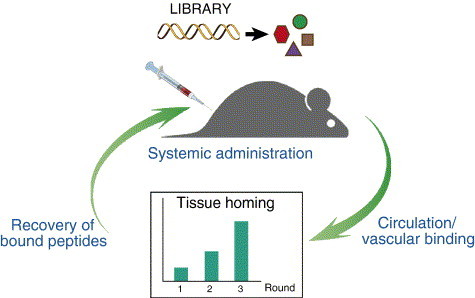
Schema of an in vivo display library selection. The library of displayed polypeptides is intravenously injected into an animal and let circulate to allow binding of polypeptides to differentially expressed receptors. After that, the blood vessels are perfused to remove unbound library and then the bound ligands are eluted from the extracted tissue. The library clones encoding the ligands are amplified and injected into an animal for a subsequent round of in vivo selection.
4.1.5.1. In vivo selections in animal models
Various peptide libraries have been screened for ligands selectively homing to tissues in mice, including brain and kidney [57], lung, skin, pancreas, intestine, uterus, adrenal gland and retina [89], lymph nodes [122], muscle [242], prostate [237], breast [239], placenta [83], and white adipose tissue [84]. This strategy also uncovered a vascular address system that allows angiogenesis-related targeting of tumor blood vessels [25], [27], [29], [82], [233], [234], [243], [244], [245], [246]. A mouse pancreatic cancer model was used to profile the progression of tumors from benign to metastatic state with phage-displayed peptides [247], [248]. In the past few years, animal models alternative to mice have been exploited, including rats [249], and insects [250]. An approach combining in vivo selection of a novel T7-displayed random peptide library with ex vivo bacterial two-hybrid hybridization has been used to identify candidate heart endothelial receptors [56].
Possible limitations in the evaluation of cross-species conservation of homing patterns of vascular-targeting peptides underline the need of designing approaches to directly profile vasculature on humans. In some cases, the data on homing ligands and their receptors generated in animal models have been useful for validation of corresponding drug targets in human disease. However, it has become clear that information derived from studies in mice cannot always be easily translated into real clinical applications because of the species-specific differences in expression of many vascular targets [237]. Inconsistent recapitulation of the results from the murine models to human biology may have correlated with frequent lack of immediate success in translational clinical trials. The uncovered cross-species variation in expression or presentation of tumor markers, with PSMA [251] and TEM7 [12], [252] serving as examples, could perhaps account for the lack of efficacy. A mere difference in tissue site, level, or time of expression of a vascular protein between mice and men may partially explain why pro-drugs identified by using mice undergo unexpected localization in humans and, therefore, are inappropriate for ligand-directed targeting. Thus, all in vivo phage display data derived from animal models must be carefully validated before being applied to clinical studies.
4.1.5.2. In vivo selections in humans
We have reasoned that in vivo selection of phage display random peptide libraries directly in patients would represent a major advance in the identification of clinically relevant vascular targeting probes and to facilitate development of targeted delivery of agents to the human vasculature [253], [254], [255]. As an initial step, we have selected a phage-displayed random peptide library by analyzing peptide motifs distribution to bone marrow, prostate, fat, muscle and skin in a cancer patient [85], [253], [254]. We performed a high throughput analysis of motifs recovered from five organs: bone marrow, white fat, skeletal muscle, prostate, and skin. For each organ, we identified frequently isolated short (tripeptide) motifs, which could direct binding in blood vessels upon phage library intravenous administration, contained within selected peptides. The large-scale survey of almost 50,000 tripeptides showed that the distribution of combinatorial peptides in human tissues was non-random [85]. Out of the 25 organ-homing tripeptides 11 were enriched in a single organ, whereas the others were enriched in multiple organs, consistent with the working hypothesis that some of them bind to tissue-specific endothelial markers and others bind to ubiquitous vascular cell surface molecules [85].
To identify candidate proteins mimicked by organ-homing motifs, we searched protein databases for sequences shared among multiple selected peptides. High-throughput analysis of the motif similarity revealed differentially expressed cell surface proteins to be validated. To demonstrate that this approach can identify functional ligand-receptor pairs, we showed that the motif GRRAGGS mimics interleukin (IL) 11 [85], and showed that this IL-11-mimic peptide binds to IL-11 receptor (IL-11Rα). A large follow-up study with well-annotated archival human tissues revealed that the expression of the IL-11Rα is increased in prostate cancer and in associated vasculature, in particular upon prostate cancer metastasis; IL-11Rα was also evaluated as a target in primary and metastatic prostate cancer by using morphological and functional analyses [226]. Importantly, although IL-11Rα, has been recognized as a target in human cancer, the specific requirements for ligand-receptor binding and molecular mechanisms that enable signal transduction were not as well understood. Based on our phage display selection data, we designed and validated an in-tandem strategy of binding assays and site-directed mutagenesis and NMR spectroscopy [225] and showed that the GRRAGGS sequence mimics a new functional protein-binding site within IL-11 (Cardó-Vila et al., submitted).
4.1.5.3. Synchronous serial in vivo selection
Because a single selection round of a large (∼ 109 unique peptide sequences) phage library usually does not sufficiently enrich for organ-homing peptides, until now, in vivo phage display screens have encompassed recovery of phage from the organ of interest in three-to-four rounds of library selection, utilizing one subject per each round [8], [57], [77]. Thus, systematic characterization of the vasculature by using this “conventional” in vivo setup has been rate-limited by the need to separately perform individual multi-round screens for motifs homing to each tissue studied. For instance, the reported patient screening [85] involved only a single round of selection and, therefore, required statistical analysis of very large numbers of phage clones (thousands, as opposed to hundreds in a conventional screen) to identify homing peptide motifs. For systematic vascular mapping, there are also practical limitations to the number of phage clones that can be routinely processed, thus requiring further methodology optimization. In order to meet such challenge, we have recently introduced a comprehensive strategy to use phage display for synchronous identification of organ-homing peptides for multiple tissues in a screen simultaneously [92]. This strategy allows one to survey an order of magnitude fewer peptide sequences than those analyzed in the original method [85] due to statistical power generated in subsequent selection rounds (Fig. 4 ). We evaluated our approach in mice by performing a screen to isolate peptides independently segregating to six different organs and developed a statistical analysis platform for identification of motifs selectively enriched in targeted tissues. Finally, to demonstrate the efficiency of this approach, we biochemically validated one of the identified pancreas-homing peptides as a mimic of peptide hormones that bind to prolactin receptor [92]. Improved statistical algorithms for analyzing peptide sequences isolated in serial synchronous selections by using Bayesian mixture models for complex high-dimensional count data are currently being developed (Ji et al., in press).
Fig. 4.
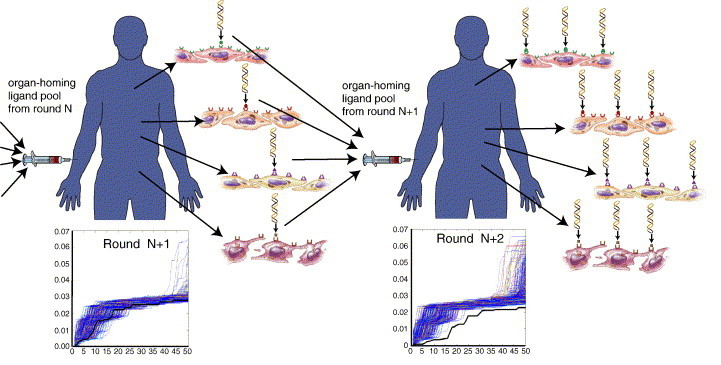
Scheme of a strategy for serial synchronous in vivo display library selection. In each round, the statistical significance (p-value) of recovery frequency is compared between tissue-homing peptides (black line) and nonspecific control peptides (background color lines). In each round, recovery frequencies are compared between tissue-homing peptides (black line) and nonspecific control peptides (background color lines). Progressively increased frequencies of peptides with every subsequent round of selection reflect the enrichment of peptides preferentially homing to the target organ.
The unprecedented initiative to screen combinatorial libraries in cancer patients represents a major step towards the ultimate goal of outlining a molecular map of the human vasculature. Ethics guidelines that provide a framework for patient-oriented research in end-of-life cancer patients have been established [253], [254], [256]. Screening libraries directly in patients will accelerate the development of drugs that target proteins differentially expressed in activated vasculature associated with human tumors and other diseases [257], [258]. The serial synchronous approach to in vivo library selection [92] outlines a practical direction for continuation of the human vascular mapping initiative and is currently being used to identify and validate tissue-specific molecular addresses in cancer patients (Kolonin et al., unpublished).
4.1.6. Phage display and developing biotechnology
Applications of phage display that have surfaced lately reflect the potential that phage display technology represents. An example of integration of phage display with advanced biotechniques is the use of real-time polymerase chain reaction (PCR) to facilitate identification of cell- and tissue-specific peptides. Quantitative PCR allows to quantify numbers of library clones bound to cells of interest, which was demonstrated for selections on live cells and on formalin-fixed, paraffin-embedded tissue sections [259], [260]. Real-time PCR was also adopted to monitor the changes in phage binding to cardiac vascular epitopes upon aging [261]. A method with potential for clinical diagnostics is phage display-mediated immuno-PCR (PD-IPCR), which is a modification of immuno-PCR (IPCR) performed with a recombinant phage particle instead of a monoclonal antibody [262]. In PD-IPCR, the phage-displayed scFv and the encoding phage DNA combine the detection and the template units. The sensitivity of this assay is a 1,000-fold over that achieved with conventional enzyme-linked immunosorbent assays (ELISAs).
Another recent illustration of the progress resulting from co-evolving technologies is the combination of phage display with laser capture microdissection (LCM) used to isolate cells of specific origin from sections of complex heterogeneous tissues such as biopsies or autopsies [260], [263], [264], [265]. Recovery of phage binding to cells of interest was successfully assisted by LCM in mice after in vivo library administration [260] , as well as after ex vivo application of phage to freshly collected oral squamous cell carcinomas [263]. Also, fluorochrome labeling of phage in display libraries has been introduced as a method for studying binding specificities, as well as phage display applications in flow cytometry and fluorescence microscopy [198], [266].
Combination of enzymatic assays with phage display technology has been useful in identification of peptides with a functional property of interest, such as enzyme substrates [71], [179], [267]. Similar approaches have been undertaken to the identification of peptides with inhibitory properties. For instance, random peptide libraries have been used as a source of Pseudomonas aeruginosa MurD amide ligase inhibitors, thus providing a prototype compound to target cell wall biosynthesis of this pathogen [268]. In another study, anti-protective antigen peptides were selected from a phage library by competitive panning with lethal factor precursor of the anthrax toxin [269]. The selected 12-mer peptides were synthesized in tetra-branched form and were modified for inhibitory efficacy, as prototypes of artificial anti-anthrax protective antigens. Similarly, enzymatically active kallikrein 2 (hK2), a serine protease that may mediate metastasis of prostate cancer, was subjected to combinatorial selection with phage libraries [270]. Several selected peptides were validated as inhibitors of hK2enzymatic activity and, therefore, as a prototype drug against prostate cancer. In another approach, phage display library based on connexin43 (Cx43)-binding RXP motif has been used to identify novel peptide sequences that bind Cx43 and modify Cx43 regulation [271]. Moreover, phage display has been used to functionally target specific cell compartments of non-protein nature. For example, a peptide library was selected for peptides that bind to liposomes in pH-dependent manner, some of which were able to cause leakage of liposome contents [272]. Lipopolysaccharides were also recently explored as targets of peptides displayed by phage libraries [273].
Phage display has been recently used for identifying adhesion molecules expressed by microbial pathogens that mediate infection [250]. Marchió et al. used a peptide library to identify an interaction between Tat and the HIV-1 gp120 envelope glycoprotein, which enhances virus attachment and entry into cells, as well as to map the interacting sites of both Tat and gp120 [274]. In another study, screening of phage-displayed libraries identified combinatorial peptides binding to the surface of Aspergillus fumigatus conidia and hyphae [275]. Various computational tools and algorithms have been generated to facilitate prediction of protein interaction sites based on phage display binding data [276]. Combination phage-displayed random peptide library selection with such computational predictions have, for instance, allowed mapping a neutralizing antibody epitope on the spike protein of severe acute respiratory syndrome (SARS) coronavirus [277], as well as to define cellular coronavirus receptor and thus approaches to development of SARS inhibitors [278].
Provided that phage has long been used as an anti-bacterial agent in the pre-antibiotic era [279], and recent FDA approval of bacteriophage preparation as a food additive on ready-to-eat meat and poultry products [280], vehicles based on phage may ultimately be proven particularly useful for treatment of infectious diseases. Display of a bacteria- or fungus-targeting moiety on phage surface can further improve the delivery of cytotoxic payload to pathogens [275], [281]. Application of phage-displayed random peptide libraries for identification of motifs facilitating transport across the gastrointestinal mucosal barrier [282], as well as transdermal transport through intact skin [283] may also accelerate translation of phage-based technologies in imaging and therapy.
Among the most exciting novel applications of phage display is based on its combination with the emerging field of nanotechnology [284], [285]. A possibly of using phage-displayed peptides and antibodies in high-throughput microarray formats has also been explored [127], [128]. Altogether, phage display technology has been and will likely remain instrumental in identification, validation, and prioritization of ligands to molecular targets expressed on the cell surface, as well as of peptide drug leads directed to these targets.
4.1.7. Eukaryotic virus display
Retroviruses are among the most efficient mammalian vectors in cell transduction for gene therapy purposes. Initially, to modulate retroviral host range, successful experiments to incorporate antibodies antibody-recognized epitopes into envelope proteins have been performed [286], [287]. Retroviral particles expressing the targeted envelope showed advanced cell infectivity. A small library containing laminin-binding human synthetic scFv displayed on retrovirus was also recently generated and selected, resulting in marked enrichment of antigen binders in a single selection cycle [288].
Baculovirus has also been developed as a display platform. Interestingly, both the virus itself and infected insect cells can present libraries of peptides. As discussed above (see Section 3.2), peptides presented by major histocompatibility complex MHC-I or MHC-II were displayed on baculovirus and were recognized by TCRs [289]. Presentation of pathogenic epitopes on capsid-like particles from hepatitis B virus, resulting in their enhanced immunogenicity, has also been reported [290]. Baculovirus envelope modification with display technology was also performed to direct viral tropism to tumor cells [291]; tumor-homing peptides originally identified by in vivo phage display were displayed on the baculoviral surface fused with the transmembrane anchor of vesicular stomatitis virus G protein. The targeted virions specifically bound to and internalized into human tumor cells. Display of tumor-targeting ligands appears to enhance baculovirus-mediated gene delivery [291].
Adeno-associated virus (AAV) has an advantage over other viruses because it has the ability to selectively integrate into the mammalian genome at a specific region, thus lacking pathogenicity related to genomic disruption and insertional mutagenesis [292]. Directing AAV to cells of interest with targeting adaptor peptides conjugated to an adenovirus-binding moiety [33], as well as incorporation of tumor-targeting peptides into recombinant AAV capsid for directing virus tropism [293] has been successful. As proof-of-concept, an AAV-based library displaying random peptides has been designed and screened on human coronary artery endothelial cells. Selected peptide motifs enhanced transduction in coronary endothelial cells but not in control cells, confirming AAV as a viable targeting vehicle [294]. Recently, AAV was also tested for in vivo display applications [249]. Peptides identified based on their homing to rat lungs and brain were incorporated into the VP3 region of the AAV-2 capsid. The resulting viruses displayed the peptides on the virion surface that retargeted the transgene-carrying viruses to the expected vascular beds in vivo [249]. Recently, for the first time, our group has also reported delivery of reporter and therapeutic genes in vivo by using a hybrid vector with genetic sic-elements merged from an M13 derived bacteriophage and adeno-associated virus [32]. We termed this new double-stranded AAV/single-stranded bacteriophage vector AAV/phage (AAVP). AAVP carrying a transgene cassette for its intracellular conversion from single stranded to double stranded DNA, was targeted to integrin-expressing cells and enabled tumor transduction and molecular genetic imaging in tumor models [32].
Adenovirus, one of the most generally accepted platforms for gene delivery, has been lagging behind other viral platforms in terms of providing a system for direct library display and selection. However, attempts to modify adenoviral capsid proteins to broaden adenovirus tropism by inserting targeting moieties are underway [295], [296] and may eventually identify viral surface protein sites useful for library peptide display. Combining these advances with optimization of assays assessing and quantifying gene transfer [231] will accelerate clinical implementation of virus-directed gene therapy.
4.2. Cell surface display
4.2.1. Bacterial display
Cell display systems feature transfection of cells with the library DNA and expression of library-encoded polypeptides as fusions with extracellular receptors (Fig. 1). Selection of a bacterially expressed library of scFv antibodies was introduced by using the periplasmic expression with cytometric screening (PECS) technology [266]. In this approach periplasmic localization of library proteins served as a dialysis bag to selectively retain scFv-bound, but not free, ligand. Fluorescent labeling of the ligand allows one to perform cytometric screening for cells containing antibody-antigen complexes. Since then, the ability to display polypeptides of interest as fusions to proteins of the bacterial envelope has also been tested as a potential alternative to viral/phage display [297]. Bacterial display can also serve to display functional enzymes, vaccine antigens and polypeptide libraries [298]. Outer membrane proteins of Gram-negative bacteria have been used as carrier proteins to present foreign peptide epitopes on bacterial cell surfaces [299]. Foreign gene products have been fused to surface-accessible regions of porins and other outer membrane proteins such as OmpA and the OmpC, PhoE, LamB, FhuA, and BtuB [299], [300]. Insertions, of more than 100 amino acid residues can be tolerated in certain cases. Another class of bacterial surface proteins for display is autotransporters, which contain the translocator domain mediating the outer membrane trafficking of the passenger [301]. Replacement of the natural passenger domain of autotransporters with heterologous proteins leads to their display at the bacterial surface by the translocator domain. Surface display of a human cathepsin G inhibitor indicated that bacterial peptide libraries can be subjected to functional screening based on target enzyme labeling [302]. Despite proof-of-principle experiments, side-by-side comparison of bacterial and phage display of the same peptide library in search of target-binding motifs indicated that the phage-based platform may be advantageous [303].
4.2.2. Yeast display
It has been shown that display on the cell wall of yeast Saccharomyces cerevisiae is possible through cell surface glycoproteins, ag-alpha-1 or aga2, which mediate interact to mating type-specific agglutination [304]. Yeast display provides an advantage over bacteria-based technologies in cases when cell-surface and secreted proteins require endoplasmic reticulum-specific post-translational processing for efficient folding and activity [305]. To date, yeast display has allowed presentation of numerous functional proteins on cell surface. For example, a hetero-oligomeric Fab fragment of a catalytic antibody was expressed with the tandem-linked C-terminal half of α-agglutinin in conjunction with expression of secreted Fd fragment of the heavy chain. The combined Fab fragment assembled on the yeast cell surface catalyzed hydrolysis of the corresponding substrate [306]. In addition to antibodies, yeast display has also been developed to express T cell receptors [155]. In a subsequent study, Holler et al. screened a yeast display library of V-alpha CDR3 mutants for higher affinity soluble monomeric TCR variants and selected TCR variants with affinities in low nanomolar range [153]. As an advantage over the wild-type TCR, high-affinity TCRs were capable of directly detecting peptide/MHC complexes on antigen-presenting cells. Recently, Jin et al. applied yeast display for molecular evolution of a human integrin protein and EGF; as a result, protein mutants with remarkable affinity for physiological interactors were identified [307].
4.2.3. Mammalian cell display
Although highly efficient, microbial display technologies are limited by protein folding, posttranslational modifications, and different codon usage from those in mammals. Establishment of approaches to display designed peptides of interest on the surface of mammalian cells will provide opportunities for high throughput analysis of interactions between peptides and proteins correctly folded and modified. An important breakthrough in mammalian display technology was an achievement of single-chain Fv expression on the cell surface display of 293T cells [194]. To prove the principle that affinity maturation is possible in mammalian systems, cells expressing an antibody with high affinity were efficiently enriched from a large excess of cells expressing a lower affinity antibody. Moreover, selection of a combinatory library randomized in an intrinsic antibody hotspot was also successful. Although this approach is challenged by the inherent limitations of library management in mammalian cells, development of targeting systems such as this will become relevant in biomedical applications.
4.3. In vitro display technologies
In vitro display systems may overcome some of the limitations of the more cumbersome cell-based display methods [308]. Their major potential advantage is the size of the libraries that can be displayed and therefore the diversity subject to selection, because in cell-free assays there is no transformation step and no limit for library diversity [309]. Over the decade since their inception, in vitro display libraries have been screened for specific high-affinity target-binding molecules including peptides, antibody fragments, and whole functional proteins presented by various scaffolds [308], [310].
4.3.1. Ribosome display
Ribosome (polysome) display was among the first cell-free in vitro display assays developed [311]. In ribosome display, selection is based on the ternary complexes formed as a result of in vitro translation of proteins and their attachment to the ribosome and to the encoding mRNA. Stabilization of mRNA-ribosome-polypeptide complexes in a rabbit reticulocyte translation system is achieved by modulation of magnesium ion concentration and by terminating polypeptide elongation either with chloramphenicol or with low temperature (Fig. 1). The immobilized mRNA is then amplified from the polysome complexes bound to the target for further rounds of selection. Ribosomal display has been proven a valuable method for selecting high affinity compounds from diverse libraries [308]. Applications of ribosomal display in biomarker identification, imaging and targeting are likely to further evolve [312]. For example, ribosome display was used for selection of DNA-binding proteins [313]: three different zinc finger DNA-binding protein libraries, containing randomized sequence in each finger, were selected on biotinylated target DNA fragments bound to streptavidin magnetic beads, leading to the isolation of semi-synthetic factors with potentially novel transcriptional activities. A ribosomally-displayed combinatorial library of ankyrin repeat proteins (DARPins) was selected for binders of MAP-kinases [314]. Remarkably, a single round of ribosome display selection lead to isolation of a specifically binding ligand with micromolar affinity. After subsequent selections, some of the enriched JNK1, JNK2 and p38-specific DARPins with nanomolar affinity were confirmed to be functional in the cellular cytoplasm. Moreover, it has been shown that affinity maturation of phage antibody populations by using ribosome display can generate high affinity antibodies with K(d)s in the picomolar range without the use of error prone mutagenesis, whereas the highest affinity antibody (scFv fragment) produced by phage display had K(d)s in the nanomolar range [143]. An improvement of cell-free translation system was achieved by combining recombinant E. coli protein factors with purified 70S ribosomes: higher selection-specific cDNA yields were observed [315]. Further attempts to prevent dissociation of the tripartite RNA-ribosome-polypeptide complexes may further improve the robustness of the system and may make ribosomal display a user-friendly method for combinatorial selection.
4.3.2. mRNA display
Covalent complexes between a polypeptide and its encoding mRNA can be generated by in vitro translation of synthetic mRNAs that carry puromycin, a peptidyl acceptor antibiotic, at their 3′ end [308]. During in vitro translation, ribosomes reach the junction between the mRNA and the engineered DNA linker and stall, which allows for puromycin entry of the ribosomal A-site and the formation of a stable amide bond with the encoded peptide (Fig. 1). As a proof-of-principle, fusions between an mRNA molecule and its encoded myc epitope peptide were enriched from a pool of random sequence mRNA-peptide fusions by immunoprecipitation [316]. In a continued effort of using mRNA display to isolate high-affinity ligands, various combinatorial or semi-combinatorial (randomized trypsin inhibitor EETI-II) peptide libraries have been selected and enriched for binding motifs [317]. A unidirectional nested deletion library displayed from mRNA was used to map epitopes of an anti-polyhistidine monoclonal antibody, leading to isolation of a high-affinity polyhistidine peptide, confirming the viability of this approach [75]. In addition, mRNA display library has been facilitated by the design of combinatorial libraries encoding peptides synthesized with unnatural amino acids [98], [99]. Such non-biological polymers may have potentially far-reaching advantages due to their extended spectra of binding targets and biological stability. However, the general applicability of this technology remains to be determined at this time.
4.3.3. Covalent DNA display
Covalent display technology (CDT) exploits an unusual property of a replication initiator protein from the E. coli bacteriophage P2, which replicates by attaching the P2A protein to its own DNA [308]. P2A is an endonuclease that initiates a rolling circle replication process by binding to the viral origin (ori) and introducing a single-strand discontinuity (nick) in the DNA, priming DNA synthesis by using the host replication machinery, and finally attaches to the molecule of DNA from which it has been expressed through a (still poorly understood) cis-activity. These features enable pools of polypeptides that are linked to P2A to be synthesized in vitro so that they also become covalently attached to their own coding DNA (Fig. 1). While potentially capable to avoid many of the problems limiting other display systems, CTD has as yet been underutilized. Recently, however, Reiersen et al. reported the suitability of CTD for library selection [318]. The covalent antibody display (CAD) was used for specific selection of anti-tetanus toxoid scFvs from a V-gene library with 50 million different members prepared from human lymphocytes. Although vulnerability of DNA and RNA complexes fundamental for in vitro display methods may pose a problem, in the future, in vitro display technologies could become instrumental not only for directed evolution of proteins, but also for in vivo selections and other applications currently limited to phage display.
5. Future directions
The future of display technologies will largely depend on improvement of screening approaches, vectors for library construction, and optimization of display scaffolds. Technical advances in methods to streamline display assays (see Section 4.1.6) may refine efficient strategies to select ligands of interest from display libraries.
The results from various display library screenings indicate that in many cases the selected peptide motifs target receptors via mimicking biological ligands of these receptors [78], [83], [85], [87], [88], [91], [92], [319]. Analysis of peptide motifs targeting an individual receptor provides a basis for rational drug design of targeted peptidomimetics, which is currently being undertaken based on the ligand-directed map of IL-11/IL-11Rα interaction mapped with in vivo phage display as a follow up of the human vascular mapping effort [85]. Investigational New Drug (IND) studies are planned for three peptidomimetic drugs from our group against cancer [80], [226], [233] and obesity [84]. Advancements in tools for peptide structure analysis open new possibilities for rapid and efficient identification and validation of receptors targeted by peptides in context-directed and functional screenings [41]. For example, Giordano et al. used NMR spectroscopy to understand the structural basis of the interaction between a VEGF-mimic mimic peptide and the VEGF receptors, which led to the identification of three key residues involved in ligand-receptor binding [225]. Rational anti-angiogenesis drug design based on this information is currently under development (Giordano et al., unpublished).
Integration of display technologies with nanotechnology is emerging as a field in which targeted nanoparticles will be pursued as multifunctional entities with diverse biomedical applications [284], [285]. Combination of display technologies with high-throughput screening approaches is likely to greatly advance the rate with which information is generated, similarly to the way it had been achieved for the genome project. Finally, development of in silico bioinformatics and systems biology tools for managing three-dimensional information-intensive data that come out of display selections is also underway [92], [320], [321], [322]. Combination of these efforts will become a major driving force for the continued application of display technologies for the needs of clinical proteomics and drug discovery.
Footnotes
This review is part of the Advanced Drug Delivery Reviews theme issue “2006 Supplementary Non-Thematic Collection", Vol. 58/15, 2006.
Contributor Information
Renata Pasqualini, Email: rpasqual@mdanderson.org.
Wadih Arap, Email: warap@mdanderson.org.
References
- 1.Engwegen J.Y., Gast M.C., Schellens J.H., Beijnen J.H. Clinical proteomics: searching for better tumour markers with SELDI-TOF mass spectrometry. Trends Pharmacol. Sci. 2006;27:251–259. doi: 10.1016/j.tips.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Pasqualini R., Arap W. Vascular targeting. In: Bertino J.R., editor. The Encyclopedia of Cancer. Academic Press; New Brunswick, NJ: 2002. pp. 525–530. [Google Scholar]
- 3.Ruoslahti E. Drug targeting to specific vascular sites. Drug Discov. Today. 2002;7:1138–1143. doi: 10.1016/s1359-6446(02)02501-1. [DOI] [PubMed] [Google Scholar]
- 4.Molldrem J.J., Komanduri K., Wieder E. Overexpressed differentiation antigens as targets of graft-versus-leukemia reactions. Curr. Opin. Hematol. 2002;9:503–508. doi: 10.1097/00062752-200211000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Arrell D.K., Neverova I., Van Eyk J.E. Cardiovascular proteomics: evolution and potential. Circ. Res. 2001;88:763–773. doi: 10.1161/hh0801.090193. [DOI] [PubMed] [Google Scholar]
- 6.Samoylova T.I., Morrison N.E., Globa L.P., Cox N.R. Peptide phage display: opportunities for development of personalized anti-cancer strategies. Anticancer Agents Med. Chem. 2006;6:9–17. doi: 10.2174/187152006774755492. [DOI] [PubMed] [Google Scholar]
- 7.Lu S., Wieder E., Komanduri K., Ma Q., Molldrem J.J. Vaccines in leukemia. Adv. Pharmacol. 2004;51:255–270. doi: 10.1016/S1054-3589(04)51011-6. [DOI] [PubMed] [Google Scholar]
- 8.Kolonin M.G., Pasqualini R., Arap W. Molecular addresses in blood vessels as targets for therapy. Curr. Opin. Chem. Biol. 2001;5:308–313. doi: 10.1016/s1367-5931(00)00207-6. [DOI] [PubMed] [Google Scholar]
- 9.Heslop H.E., Stevenson F.K., Molldrem J.J. Immunotherapy of hematologic malignancy. Hematology (Am. Soc. Hematol. Educ. Program) 2003:331–349. doi: 10.1182/asheducation-2003.1.331. [DOI] [PubMed] [Google Scholar]
- 10.Ruoslahti E. Specialization of tumour vasculature. Nat. Rev., Cancer. 2002;2:83–90. doi: 10.1038/nrc724. [DOI] [PubMed] [Google Scholar]
- 11.Vogelstein B., Kinzler K.W. Cancer genes and the pathways they control. Nat. Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 12.St Croix B., Rago C., Velculescu V., Traverso G., Romans K.E., Montgomery E., Lal A., Riggins G.J., Lengauer C., Vogelstein B., Kinzler K.W. Genes expressed in human tumor endothelium. Science. 2000;289:1197–1202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- 13.Kolonin M.G., Pasqualini R., Arap W. Mapping human vascular heterogeneity by in vivo phage display. In: Hoying J.B., editor. Genetics of Angiogenesis. BIOS Scientific Publishers Ltd; Oxford: 2003. [Google Scholar]
- 14.Monks A., Scudiero D., Skehan P., Shoemaker R., Paull K., Vistica D., Hose C., Langley J., Cronise P., Vaigro-Wolff A. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 15.Weinstein J.N., Myers T.G., O'Connor P.M., Friend S.H., Fornace A.J., Jr., Kohn K.W., Fojo T., Bates S.E., Rubinstein L.V., Anderson N.L., Buolamwini J.K., van Osdol W.W., Monks A.P., Scudiero D.A., Sausville E.A., Zaharevitz D.W., Bunow B., Viswanadhan V.N., Johnson G.S., Wittes R.E., Paull K.D. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275:343–349. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- 16.Monks A., Scudiero D.A., Johnson G.S., Paull K.D., Sausville E.A. The NCI anti-cancer drug screen: a smart screen to identify effectors of novel targets. Anticancer Drug Des. 1997;12:533–541. [PubMed] [Google Scholar]
- 17.Scherf U., Ross D.T., Waltham M., Smith L.H., Lee J.K., Tanabe L., Kohn K.W., Reinhold W.C., Myers T.G., Andrews D.T., Scudiero D.A., Eisen M.B., Sausville E.A., Pommier Y., Botstein D., Brown P.O., Weinstein J.N. A gene expression database for the molecular pharmacology of cancer. Nat. Genet. 2000;24:236–244. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- 18.Zaharevitz D.W., Holbeck S.L., Bowerman C., Svetlik P.A. COMPARE: a web accessible tool for investigating mechanisms of cell growth inhibition. J. Mol. Graph. Model. 2002;20:297–303. doi: 10.1016/s1093-3263(01)00126-7. [DOI] [PubMed] [Google Scholar]
- 19.Blower P.E., Yang C., Fligner M.A., Verducci J.S., Yu L., Richman S., Weinstein J.N. Pharmacogenomic analysis: correlating molecular substructure classes with microarray gene expression data. Pharmacogenomics J. 2002;2:259–271. doi: 10.1038/sj.tpj.6500116. [DOI] [PubMed] [Google Scholar]
- 20.Rabow A.A., Shoemaker R.H., Sausville E.A., Covell D.G. Mining the National Cancer Institute's tumor-screening database: identification of compounds with similar cellular activities. J. Med. Chem. 2002;45:818–840. doi: 10.1021/jm010385b. [DOI] [PubMed] [Google Scholar]
- 21.Wallqvist A., Rabow A.A., Shoemaker R.H., Sausville E.A., Covell D.G. Establishing connections between microarray expression data and chemotherapeutic cancer pharmacology. Mol. Cancer Ther. 2002;1:311–320. [PubMed] [Google Scholar]
- 22.Szakacs G., Annereau J.P., Lababidi S., Shankavaram U., Arciello A., Bussey K.J., Reinhold W., Guo Y., Kruh G.D., Reimers M., Weinstein J.N., Gottesman M.M. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cells. 2004;6:129–137. doi: 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 23.Brown J.M. NCI's anticancer drug screening program may not be selecting for clinically active compounds. Oncol. Res. 1997;9:213–215. [PubMed] [Google Scholar]
- 24.Wallqvist A., Rabow A.A., Shoemaker R.H., Sausville E.A., Covell D.G. Linking the growth inhibition response from the National Cancer Institute's anticancer screen to gene expression levels and other molecular target data. Bioinformatics. 2003;19:2212–2224. doi: 10.1093/bioinformatics/btg302. [DOI] [PubMed] [Google Scholar]
- 25.Arap W., Pasqualini R., Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 26.Hajitou A., Pasqualini R., Arap W. Vascular targeting: recent advances and therapeutic perspectives. Trends Cardiovasc. Med. 2006;16:80–88. doi: 10.1016/j.tcm.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellerby H.M., Arap W., Ellerby L.M., Kain R., Andrusiak R., Rio G.D., Krajewski S., Lombardo C.R., Rao R., Ruoslahti E., Bredesen D.E., Pasqualini R. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 1999;5:1032–1038. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- 28.Ellerby H.M., Lee S., Ellerby L.M., Chen S., Kiyota T., del Rio G., Sugihara G., Sun Y., Bredesen D.E., Arap W., Pasqualini R. An artificially designed pore-forming protein with anti-tumor effects. J. Biol. Chem. 2003;278:35311–35316. doi: 10.1074/jbc.M300474200. [DOI] [PubMed] [Google Scholar]
- 29.Koivunen E., Arap W., Valtanen H., Rainisalo A., Medina O.P., Heikkila P., Kantor C., Gahmberg C.G., Salo T., Konttinen Y.T., Sorsa T., Ruoslahti E., Pasqualini R. Tumor targeting with a selective gelatinase inhibitor. Nat. Biotechnol. 1999;17:768–774. doi: 10.1038/11703. [DOI] [PubMed] [Google Scholar]
- 30.Curnis F., Sacchi A., Borgna L., Magni F., Gasparri A., Corti A. Enhancement of tumor necrosis factor alpha antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13) Nat. Biotechnol. 2000;18:1185–1190. doi: 10.1038/81183. [DOI] [PubMed] [Google Scholar]
- 31.Hong F.D., Clayman G.L. Isolation of a peptide for targeted drug delivery into human head and neck solid tumors. Cancer Res. 2000;60:6551–6556. [PubMed] [Google Scholar]
- 32.Hajitou A., Trepel M., Lilley C.E., Soghomonyan S., Alauddin M.M., Marini F.C., III, Restel B.H., Ozawa M.G., Moya C.A., Rangel R., Sun Y., Zaoui K., Schmidt M., von Kalle C., Weitzman M.D., Gelovani J.G., Pasqualini R., Arap W. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell. 2006;125:385–398. doi: 10.1016/j.cell.2006.02.042. [DOI] [PubMed] [Google Scholar]
- 33.Trepel M., Grifman M., Weitzman M.D., Pasqualini R. Molecular adaptors for vascular-targeted adenoviral gene delivery. Hum. Gene Ther. 2000;11:1971–1981. doi: 10.1089/10430340050143408. [DOI] [PubMed] [Google Scholar]
- 34.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 35.Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., Smith H.O. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 36.Armstrong P.J., Johanning J.M., Calton W.C., Jr., Delatore J.R., Franklin D.P., Han D.C., Carey D.J., Elmore J.R. Differential gene expression in human abdominal aorta: aneurysmal versus occlusive disease. J. Vasc. Surg. 2002;35:346–355. doi: 10.1067/mva.2002.121071. [DOI] [PubMed] [Google Scholar]
- 37.Chen B.P., Li Y.S., Zhao Y., Chen K.D., Li S., Lao J., Yuan S., Shyy J.Y., Chien S. DNA microarray analysis of gene expression in endothelial cells in response to 24-h shear stress. Physiol. Genomics. 2001;7:55–63. doi: 10.1152/physiolgenomics.2001.7.1.55. [DOI] [PubMed] [Google Scholar]
- 38.Ross D.T., Scherf U., Eisen M.B., Perou C.M., Rees C., Spellman P., Iyer V., Jeffrey S.S., Van de Rijn M., Waltham M., Pergamenschikov A., Lee J.C., Lashkari D., Shalon D., Myers T.G., Weinstein J.N., Botstein D., Brown P.O. Systematic variation in gene expression patterns in human cancer cell lines. Nat. Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 39.Nishizuka S., Charboneau L., Young L., Major S., Reinhold W.C., Waltham M., Kouros-Mehr H., Bussey K.J., Lee J.K., Espina V., Munson P.J., Petricoin E., III, Liotta L.A., Weinstein J.N. Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc. Natl. Acad. Sci. U. S. A. 2003;100:14229–14234. doi: 10.1073/pnas.2331323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishizuka S., Chen S.T., Gwadry F.G., Alexander J., Major S.M., Scherf U., Reinhold W.C., Waltham M., Charboneau L., Young L., Bussey K.J., Kim S., Lababidi S., Lee J.K., Pittaluga S., Scudiero D.A., Sausville E.A., Munson P.J., Petricoin E.F., III, Liotta L.A., Hewitt S.M., Raffeld M., Weinstein J.N. Diagnostic markers that distinguish colon and ovarian adenocarcinomas: identification by genomic, proteomic, and tissue array profiling. Cancer Res. 2003;63:5243–5250. [PubMed] [Google Scholar]
- 41.Li M. Applications of display technology in protein analysis. Nat. Biotechnol. 2000;18:1251–1256. doi: 10.1038/82355. [DOI] [PubMed] [Google Scholar]
- 42.Myers T.G., Anderson N.L., Waltham M., Li G., Buolamwini J.K., Scudiero D.A., Paull K.D., Sausville E.A., Weinstein J.N. A protein expression database for the molecular pharmacology of cancer. Electrophoresis. 1997;18:647–653. doi: 10.1002/elps.1150180351. [DOI] [PubMed] [Google Scholar]
- 43.Sobek J., Bartscherer K., Jacob A., Hoheisel J.D., Angenendt P. Microarray technology as a universal tool for high-throughput analysis of biological systems. Comb. Chem. High Throughput Screen. 2006;9:365–380. doi: 10.2174/138620706777452429. [DOI] [PubMed] [Google Scholar]
- 44.MacBeath G. Protein microarrays and proteomics. Nat. Genet. 2002;32:526–532. doi: 10.1038/ng1037. (Suppl.) [DOI] [PubMed] [Google Scholar]
- 45.Cretich M., Damin F., Pirri G., Chiari M. Protein and peptide arrays: recent trends and new directions. Biomol. Eng. 2006;23:77–88. doi: 10.1016/j.bioeng.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Ramani A.K., Bunescu R.C., Mooney R.J., Marcotte E.M. Consolidating the set of known human protein–protein interactions in preparation for large-scale mapping of the human interactome. Genome Biol. 2005;6:40–45. doi: 10.1186/gb-2005-6-5-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giot L., Bader J.S., Brouwer C., Chaudhuri A., Kuang B., Li Y., Hao Y.L., Ooi C.E., B. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 48.Stanyon C.A., Liu G., Mangiola B.A., Patel N., Giot L., Kuang B., Zhang H., Zhong J., Finley R.L., Jr. A Drosophila protein-interaction map centered on cell-cycle regulators. Genome Biol. 2004;5:R96. doi: 10.1186/gb-2004-5-12-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uetz P., Finley R.L., Jr. From protein networks to biological systems. FEBS Lett. 2005;579:1821–1827. doi: 10.1016/j.febslet.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Pelletier J., Sidhu S. Mapping protein–protein interactions with combinatorial biology methods. Curr. Opin. Biotechnol. 2001;12:340–347. doi: 10.1016/s0958-1669(00)00225-1. [DOI] [PubMed] [Google Scholar]
- 51.Peale F.V., Jr., Gerritsen M.E. Gene profiling techniques and their application in angiogenesis and vascular development. J. Pathol. 2001;195:7–19. doi: 10.1002/path.888. [DOI] [PubMed] [Google Scholar]
- 52.Angenendt P., Glokler J., Murphy D., Lehrach H., Cahill D.J. Toward optimized antibody microarrays: a comparison of current microarray support materials. Anal. Biochem. 2002;309:253–260. doi: 10.1016/s0003-2697(02)00257-9. [DOI] [PubMed] [Google Scholar]
- 53.McIntosh D.P., Tan X.Y., Oh P., Schnitzer J.E. Targeting endothelium and its dynamic caveolae for tissue-specific transcytosis in vivo: a pathway to overcome cell barriers to drug and gene delivery. Proc. Natl. Acad. Sci. U. S. A. 2002;99:1996–2001. doi: 10.1073/pnas.251662398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Durr E., Yu J., Krasinska K.M., Carver L.A., Yates J.R., Testa J.E., Oh P., Schnitzer J.E. Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat. Biotechnol. 2004;22:892–985. doi: 10.1038/nbt993. [DOI] [PubMed] [Google Scholar]
- 55.Valadon P., Garnett J.D., Testa J.E., Bauerle M., Oh P., Schnitzer J.E. Screening phage display libraries for organ-specific vascular immunotargeting in vivo. Proc. Natl. Acad. Sci. U. S. A. 2006;103:407–412. doi: 10.1073/pnas.0506938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang L., Hoffman J.A., Ruoslahti E. Molecular profiling of heart endothelial cells. Circulation. 2005;112:1601–1611. doi: 10.1161/CIRCULATIONAHA.104.529537. [DOI] [PubMed] [Google Scholar]
- 57.Pasqualini R., Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380:364–366. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- 58.Dias R.L., Fasan R., Moehle K., Renard A., Obrecht D., Robinson J.A. Protein ligand design: from phage display to synthetic protein epitope mimetics in human antibody Fc-binding peptidomimetics. J. Am. Chem. Soc. 2006;128:2726–2732. doi: 10.1021/ja057513w. [DOI] [PubMed] [Google Scholar]
- 59.Smith G.P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 60.Miceli R.M., DeGraaf M.E., Fischer H.D. Two-stage selection of sequences from a random phage display library delineates both core residues and permitted structural range within an epitope. J. Immunol. Methods. 1994;167:279–287. doi: 10.1016/0022-1759(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 61.Parhami-Seren B., Keel T., Reed G.L. Sequences of antigenic epitopes of streptokinase identified via random peptide libraries displayed on phage. J. Mol. Biol. 1997;271:333–341. doi: 10.1006/jmbi.1997.1174. [DOI] [PubMed] [Google Scholar]
- 62.Christmann A., Wentzel A., Meyer C., Meyers G., Kolmar H. Epitope mapping and affinity purification of monospecific antibodies by Escherichia coli cell surface display of gene-derived random peptide libraries. J. Immunol. Methods. 2001;257:163–173. doi: 10.1016/s0022-1759(01)00461-6. [DOI] [PubMed] [Google Scholar]
- 63.Mullaney B.P., Pallavicini M.G. Protein–protein interactions in hematology and phage display. Exp. Hematol. 2001;29:1136–1146. doi: 10.1016/s0301-472x(01)00693-2. [DOI] [PubMed] [Google Scholar]
- 64.Stephen C.W., Helminen P., Lane D.P. Characterisation of epitopes on human p53 using phage-displayed peptide libraries: insights into antibody-peptide interactions. J. Mol. Biol. 1995;248:58–78. doi: 10.1006/jmbi.1995.0202. [DOI] [PubMed] [Google Scholar]
- 65.Fack F., Hugle-Dorr B., Song D., Queitsch I., Petersen G., Bautz E.K. Epitope mapping by phage display: random versus gene-fragment libraries. J. Immunol. Methods. 1997;206:43–52. doi: 10.1016/s0022-1759(97)00083-5. [DOI] [PubMed] [Google Scholar]
- 66.Coley A.M., Campanale N.V., Casey J.L., Hodder A.N., Crewther P.E., Anders R.F., Tilley L.M., Foley M. Rapid and precise epitope mapping of monoclonal antibodies against Plasmodium falciparum AMA1 by combined phage display of fragments and random peptides. Protein Eng. 2001;14:691–698. doi: 10.1093/protein/14.9.691. [DOI] [PubMed] [Google Scholar]
- 67.Uchiyama F., Tanaka Y., Minari Y., Tokui N. Designing scaffolds of peptides for phage display libraries. J. Biosci. Bioeng. 2005;99:448–456. doi: 10.1263/jbb.99.448. [DOI] [PubMed] [Google Scholar]
- 68.Irving R.A., Coia G., Roberts A., Nuttall S.D., Hudson P.J. Ribosome display and affinity maturation: from antibodies to single V-domains and steps towards cancer therapeutics. J. Immunol. Methods. 2001;248:31–45. doi: 10.1016/s0022-1759(00)00341-0. [DOI] [PubMed] [Google Scholar]
- 69.Nestler H.P. Combinatorial chemistry and fragment screening-two unlike siblings? Curr. Drug Discov. Technol. 2005;2:1–12. doi: 10.2174/1570163053175484. [DOI] [PubMed] [Google Scholar]
- 70.Eichler J. Synthetic peptide arrays and peptide combinatorial libraries for the exploration of protein–ligand interactions and the design of protein inhibitors. Comb. Chem. High Throughput Screen. 2005;8:135–143. doi: 10.2174/1386207053258497. [DOI] [PubMed] [Google Scholar]
- 71.Cortese R., Monaci P., Luzzago A., Santini C., Bartoli F., Cortese I., Fortugno P., Galfre G., Nicosia A., Felici F. Selection of biologically active peptides by phage display of random peptide libraries. Curr. Opin. Biotechnol. 1996;7:616–621. doi: 10.1016/s0958-1669(96)80072-3. [DOI] [PubMed] [Google Scholar]
- 72.McLafferty M.A., Kent R.B., Ladner R.C., Markland W. M13 bacteriophage displaying disulfide-constrained microproteins. Gene. 1993;128:29–36. doi: 10.1016/0378-1119(93)90149-w. [DOI] [PubMed] [Google Scholar]
- 73.Ladner R.C. Constrained peptides as binding entities. Trends Biotechnol. 1995;13:426–430. doi: 10.1016/S0167-7799(00)88997-0. [DOI] [PubMed] [Google Scholar]
- 74.Hosse R.J., Rothe A., Power B.E. A new generation of protein display scaffolds for molecular recognition. Protein Sci. 2006;15:14–27. doi: 10.1110/ps.051817606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ja W.W., Olsen B.N., Roberts R.W. Epitope mapping using mRNA display and a unidirectional nested deletion library. Protein Eng. Des. Sel. 2005;18:309–319. doi: 10.1093/protein/gzi038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koivunen E., Wang B., Ruoslahti E. Phage libraries displaying cyclic peptides with different ring sizes: ligand specificities of the RGD-directed integrins. Biotechnology (N. Y.) 1995;13:265–270. doi: 10.1038/nbt0395-265. [DOI] [PubMed] [Google Scholar]
- 77.Barbas C., Burton D., Silverman G., Scott J. Cold Spring Harbor Laboratory Press; New York, NY: 2001. Phage Display: A Laboratory Manual. [Google Scholar]
- 78.Giordano R.J., Cardó-Vila M., Lahdenranta J., Pasqualini R., Arap W. Biopanning and rapid analysis of selective interactive ligands. Nat. Med. 2001;7:1249–1253. doi: 10.1038/nm1101-1249. [DOI] [PubMed] [Google Scholar]
- 79.Koivunen E., Arap W., Rajotte D., Lahdenranta J., Pasqualini R. Identification of receptor ligands with phage display peptide libraries. J. Nucl. Med. 1999;40:883–888. [PubMed] [Google Scholar]
- 80.Mintz P.J., Kim J., Do K.A., Wang X., Zinner R.G., Cristofanilli M., Arap M.A., Hong W.K., Troncoso P., Logothetis C.J., Pasqualini R., Arap W. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat. Biotechnol. 2003;21:57–63. doi: 10.1038/nbt774. [DOI] [PubMed] [Google Scholar]
- 81.Vidal C.I., Mintz P.J., Lu K., Ellis L.M., Manenti L., Giavazzi R., Gershenson D.M., Broaddus R., Liu J., Arap W., Pasqualini R. An HSP90-mimic peptide revealed by fingerprinting the pool of antibodies from ovarian cancer patients. Oncogene. 2004;23:8859–8867. doi: 10.1038/sj.onc.1208082. [DOI] [PubMed] [Google Scholar]
- 82.Pasqualini R., Koivunen E., Kain R., Lahdenranta J., Sakamoto M., Stryhn A., Ashmun R.A., Shapiro L.H., Arap W., Ruoslahti E. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60:722–727. [PMC free article] [PubMed] [Google Scholar]
- 83.Kolonin M.G., Pasqualini R., Arap W. Teratogenicity induced by targeting a placental immunoglobulin transporter. Proc. Natl. Acad. Sci. U. S. A. 2002;99:13055–13060. doi: 10.1073/pnas.162468499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kolonin M.G., Saha P.K., Chan L., Pasqualini R., Arap W. Reversal of obesity by targeted ablation of adipose tissue. Nat. Med. 2004;10:625–632. doi: 10.1038/nm1048. [DOI] [PubMed] [Google Scholar]
- 85.Arap W., Kolonin M.G., Trepel M., Lahdenranta J., Cardó-Vila M., Giordano R.J., Mintz P.J., Ardelt P.U., Yao V.J., Vidal C.I., Chen L., Flamm A., Valtanen H., Weavind L.M., Hicks M.E., Pollock R.E., Botz G.H., Bucana C.D., Koivunen E., Cahill D., Troncoso P., Baggerly K.A., Pentz R.D., Do K.A., Logothetis C.J., Pasqualini R. Steps toward mapping the human vasculature by phage display. Nat. Med. 2002;8:121–127. doi: 10.1038/nm0202-121. [DOI] [PubMed] [Google Scholar]
- 86.Kolonin M.G., Finley R.L., Jr. Targeting cyclin-dependent kinases in Drosophila with peptide aptamers. Proc. Natl. Acad. Sci. U. S. A. 1998;95:14266–14271. doi: 10.1073/pnas.95.24.14266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smothers J.F., Henikoff S., Carter P. Tech.Sight. Phage display. Affinity selection from biological libraries. Science. 2002;298:621–622. doi: 10.1126/science.298.5593.621. [DOI] [PubMed] [Google Scholar]
- 88.Hartley O. The use of phage display in the study of receptors and their ligands. J. Recept. Signal Transduct. Res. 2002;22:373–392. doi: 10.1081/rrs-120014608. [DOI] [PubMed] [Google Scholar]
- 89.Rajotte D., Arap W., Hagedorn M., Koivunen E., Pasqualini R., Ruoslahti E. Molecular heterogeneity of the vascular endothelium revealed by in vivo phage display. J. Clin. Invest. 1998;102:430–437. doi: 10.1172/JCI3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rajotte D., Ruoslahti E. Membrane dipeptidase is the receptor for a lung-targeting peptide identified by in vivo phage display. J. Biol. Chem. 1999;274:11593–11598. doi: 10.1074/jbc.274.17.11593. [DOI] [PubMed] [Google Scholar]
- 91.Kolonin M.G., Bover L., Sun J., Zurita A.J., Do K.A., Lahdenranta J., Cardó-Vila M., Giordano R.J., Jaalouk D.E., Ozawa M.G., Moya C.A., Souza G.R., Staquicini F.I., Kunyiasu A., Scudiero D.A., Holbeck S.L., Sausville E.A., Arap W., Pasqualini R. Ligand-directed surface profiling of human cancer cells with combinatorial peptide libraries. Cancer Res. 2006;66:34–40. doi: 10.1158/0008-5472.CAN-05-2748. [DOI] [PubMed] [Google Scholar]
- 92.Kolonin M.G., Sun J., Do K.A., Vidal C.I., Ji Y., Baggerly K.A., Pasqualini R., Arap W. Synchronous selection of homing peptides for multiple tissues by in vivo phage display. FASEB J. 2006;20:979–981. doi: 10.1096/fj.05-5186fje. [DOI] [PubMed] [Google Scholar]
- 93.Landon L.A., Deutscher S.L. Combinatorial discovery of tumor targeting peptides using phage display. J. Cell. Biochem. 2003;90:509–517. doi: 10.1002/jcb.10634. [DOI] [PubMed] [Google Scholar]
- 94.Watt P.M. Screening for peptide drugs from the natural repertoire of biodiverse protein folds. Nat. Biotechnol. 2006;24:177–183. doi: 10.1038/nbt1190. [DOI] [PubMed] [Google Scholar]
- 95.Kelly K.A., Nahrendorf M., Yu A.M., Reynolds F., Weissleder R. In vivo phage display selection yields atherosclerotic plaque targeted peptides for imaging. Mol. Imaging Biol. 2006;8:201–207. doi: 10.1007/s11307-006-0043-6. [DOI] [PubMed] [Google Scholar]
- 96.Brissette R., Prendergast J.K., Goldstein N.I. Identification of cancer targets and therapeutics using phage display. Curr. Opin. Drug Discov. Dev. 2006;9:363–369. [PubMed] [Google Scholar]
- 97.Craig R., Li S. Function and molecular mechanism of tumor-targeted peptides for delivering therapeutic genes and chemical drugs. Mini Rev. Med. Chem. 2006;6:757–764. doi: 10.2174/138955706777698615. [DOI] [PubMed] [Google Scholar]
- 98.Josephson K., Hartman M.C., Szostak J.W. Ribosomal synthesis of unnatural peptides. J. Am. Chem. Soc. 2005;127:11727–11735. doi: 10.1021/ja0515809. [DOI] [PubMed] [Google Scholar]
- 99.Frankel A., Millward S.W., Roberts R.W. Encodamers: unnatural peptide oligomers encoded in RNA. Chem. Biol. 2003;10:1043–1050. doi: 10.1016/j.chembiol.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 100.Scholle M.D., Kehoe J.W., Kay B.K. Efficient construction of a large collection of phage-displayed combinatorial peptide libraries. Comb. Chem. High Throughput Screen. 2005;8:545–551. doi: 10.2174/1386207054867337. [DOI] [PubMed] [Google Scholar]
- 101.Rothe A., Hosse R.J., Power B.E. In vitro display technologies reveal novel biopharmaceutics. FASEB J. 2006;20:1599–1610. doi: 10.1096/fj.05-5650rev. [DOI] [PubMed] [Google Scholar]
- 102.Zinkernagel R.M., Bachmann M.F., Kundig T.M., Oehen S., Pirchet H., Hengartner H. On immunological memory. Annu. Rev. Immunol. 1996;14:333–367. doi: 10.1146/annurev.immunol.14.1.333. [DOI] [PubMed] [Google Scholar]
- 103.van der Merwe P.A., Davis S.J. Molecular interactions mediating T cell antigen recognition. Annu. Rev. Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 104.Krogsgaard M., Li Q.J., Sumen C., Huppa J.B., Huse M., Davis M.M. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 2005;434:238–243. doi: 10.1038/nature03391. [DOI] [PubMed] [Google Scholar]
- 105.Kitamura T., Tanaka N., Watanabe J., Uchida, Kanegasaki S., Yamada Y., Nakata K. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1999;190:875–880. doi: 10.1084/jem.190.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vynios D.H., Tsagaraki I., Grigoreas G.H., Samiotaki M., Panayotou G., Kyriakopoulou D., Georgiou P., Korbakis D., Panayotou A., Nanouri K., Assouti M., Andonopoulos A.P. Autoantibodies against aggrecan in systemic rheumatic diseases. Biochimie. 2006;88:767–773. doi: 10.1016/j.biochi.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 107.Stadler B.M., Miescher S., Horn M., Pachlopnik J., Stadler M., Kricek F., Vogel M. Allergic manifestations as the results of a conditional autoimmune response. Int. Arch. Allergy Immunol. 2001;124:411–413. doi: 10.1159/000053773. [DOI] [PubMed] [Google Scholar]
- 108.Bairey O., Blickstein D., Monselise Y., Lahav J., Stark P., Prokocimer M., Nativ H.M., Kirgner I., Pazgal I., Shaklai M. Antiphospholipid antibodies may be a new prognostic parameter in aggressive non-Hodgkin's lymphoma. Eur. J. Haematol. 2006;76:384–391. doi: 10.1111/j.1600-0609.2005.00620.x. [DOI] [PubMed] [Google Scholar]
- 109.Bachelot T., Ratel D., Menetrier-Caux C., Wion D., Blay J.Y., Berger F. Autoantibodies to endostatin in patients with breast cancer: correlation to endostatin levels and clinical outcome. Br. J. Cancer. 2006;94:1066–1070. doi: 10.1038/sj.bjc.6603037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Molldrem J.J., Lee P.P., Kant S., Wieder E., Jiang W., Lu S., Wang C., Davis M.M. Chronic myelogenous leukemia shapes host immunity by selective deletion of high-avidity leukemia-specific T cells. J. Clin. Invest. 2003;111:639–647. doi: 10.1172/JCI16398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Clave E., Molldrem J., Hensel N., Raptis A., Barrett A.J. Donor-recipient polymorphism of the proteinase 3 gene: a potential target for T-cell alloresponses to myeloid leukemia. J. Immunother. 1999;22:1–6. doi: 10.1097/00002371-199901000-00001. [DOI] [PubMed] [Google Scholar]
- 112.Engelhorn M.E., Guevara-Patino J.A., Noffz G., Hooper A.T., Lou O., Gold J.S., Kappel B.J., Houghton A.N. Autoimmunity and tumor immunity induced by immune responses to mutations in self. Nat. Med. 2006;12:198–206. doi: 10.1038/nm1363. [DOI] [PubMed] [Google Scholar]
- 113.Bowcock A.M. The genetics of psoriasis and autoimmunity. Annu. Rev. Genomics Hum. Genet. 2005;6:93–122. doi: 10.1146/annurev.genom.6.080604.162324. [DOI] [PubMed] [Google Scholar]
- 114.Cravens P.D., Lipsky P.E. Dendritic cells, chemokine receptors and autoimmune inflammatory diseases. Immunol. Cell Biol. 2002;80:497–505. doi: 10.1046/j.1440-1711.2002.01118.x. [DOI] [PubMed] [Google Scholar]
- 115.Tureci O., Usener D., Schneider S., Sahin U. Identification of tumor-associated autoantigens with SEREX. Methods Mol. Med. 2005;109:137–154. doi: 10.1385/1-59259-862-5:137. [DOI] [PubMed] [Google Scholar]
- 116.Imafuku Y., Omenn G.S., Hanash S. Proteomics approaches to identify tumor antigen directed autoantibodies as cancer biomarkers. Dis. Markers. 2004;20:149–153. doi: 10.1155/2004/829450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ditzel H.J. Human antibodies in cancer and autoimmune disease. Immunol. Res. 2000;21:185–193. doi: 10.1385/IR:21:2-3:185. [DOI] [PubMed] [Google Scholar]
- 118.Kim S.J., Park Y., Hong H.J. Antibody engineering for the development of therapeutic antibodies. Mol. Cells. 2005;20:17–29. [PubMed] [Google Scholar]
- 119.Lahdenranta J., Pasqualini R., Schlingemann R.O., Hagedorn M., Stallcup W.B., Bucana C.D., Sidman R.L., Arap W. An anti-angiogenic state in mice and humans with retinal photoreceptor cell degeneration. Proc. Natl. Acad. Sci. U. S. A. 2001;98:10368–10373. doi: 10.1073/pnas.181329198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yazici S., Kim S.J., Busby J.E., He J., Thaker P., Yokoi K., Fan D., Fidler I.J. Dual inhibition of the epidermal growth factor and vascular endothelial growth factor phosphorylation for antivascular therapy of human prostate cancer in the prostate of nude mice. Prostate. 2005;65:203–215. doi: 10.1002/pros.20283. [DOI] [PubMed] [Google Scholar]
- 121.Gatto B., Cavalli M. From proteins to nucleic acid-based drugs: the role of biotech in anti-VEGF therapy. Anticancer Agents Med. Chem. 2006;6:287–301. doi: 10.2174/187152006777698178. [DOI] [PubMed] [Google Scholar]
- 122.Trepel M., Arap W., Pasqualini R. Modulation of the immune response by systemic targeting of antigens to lymph nodes. Cancer Res. 2001;61:8110–8112. [PubMed] [Google Scholar]
- 123.Pasqualini R., Arap W. Hybridoma-free generation of monoclonal antibodies. Proc. Natl. Acad. Sci. U. S. A. 2004;101:257–259. doi: 10.1073/pnas.0305834101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Laffly E., Sodoyer R. Monoclonal and recombinant antibodies, 30 years after. Hum. Antibodies. 2005;14:33–55. [PubMed] [Google Scholar]
- 125.Wolf P., Gierschner D., Buhler P., Wetterauer U., Elsasser-Beile U. A recombinant PSMA-specific single-chain immunotoxin has potent and selective toxicity against prostate cancer cells. Cancer Immunol. Immunother. 2006;11:1367–1373. doi: 10.1007/s00262-006-0131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sharon J., Liebman M.A., Williams B.R. Recombinant polyclonal antibodies for cancer therapy. J. Cell. Biochem. 2005;96:305–313. doi: 10.1002/jcb.20536. [DOI] [PubMed] [Google Scholar]
- 127.Chatterjee M., Mohapatra S., Ionan A., Bawa G., Ali-Fehmi R., Wang X., Nowak J., Ye B., Nahhas F.A., Lu K., Witkin S.S., Fishman D., Munkarah A., Morris R., Levin N.K., Shirley N.N., Tromp G., Abrams J., Draghici S., Tainsky M.A. Diagnostic markers of ovarian cancer by high-throughput antigen cloning and detection on arrays. Cancer Res. 2006;66:1181–1190. doi: 10.1158/0008-5472.CAN-04-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang X., Yu J., Sreekumar A., Varambally S., Shen R., Giacherio D., Mehra R., Montie J.E., Pienta K.J., Sanda M.G., Kantoff P.W., Rubin M.A., Wei J.T., Ghosh D., Chinnaiyan A.M. Autoantibody signatures in prostate cancer. N. Engl. J. Med. 2005;353:1224–1235. doi: 10.1056/NEJMoa051931. [DOI] [PubMed] [Google Scholar]
- 129.Felding-Habermann B., Lerner R.A., Lillo A., Zhuang S., Weber M.R., Arrues S., Gao C., Mao S., Saven A., Janda K.D. Combinatorial antibody libraries from cancer patients yield ligand-mimetic Arg-Gly-Asp-containing immunoglobulins that inhibit breast cancer metastasis. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17210–17215. doi: 10.1073/pnas.0407869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Feldhaus M.J., Siegel R.W. Yeast display of antibody fragments: a discovery and characterization platform. J. Immunol. Methods. 2004;290:69–80. doi: 10.1016/j.jim.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 131.Pini A., Bracci L. Phage display of antibody fragments. Curr. Protein Pept. Sci. 2000;1:155–169. doi: 10.2174/1389203003381397. [DOI] [PubMed] [Google Scholar]
- 132.Clackson T., Hoogenboom H.R., Griffiths A.D., Winter G. Making antibody fragments using phage display libraries. Nature. 1991;352:624–628. doi: 10.1038/352624a0. [DOI] [PubMed] [Google Scholar]
- 133.Barbas C.F., III, Kang A.S., Lerner R.A., Benkovic S.J. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc. Natl. Acad. Sci. U. S. A. 1991;88:7978–7982. doi: 10.1073/pnas.88.18.7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McCafferty J., Griffiths A.D., Winter G., Chiswell D.J. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348:552–554. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 135.Roovers R.C., Laeremans T., Huang L., De Taeye S., Verkleij A.J., Revets H., de Haard H.J., van Bergen En Henegouwen P.M. Efficient inhibition of EGFR signalling and of tumour growth by antagonistic anti-EGFR Nanobodies. Cancer Immunol. Immunother. 2006:35–41. doi: 10.1007/s00262-006-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Orlova A., Magnusson M., Eriksson T.L., Nilsson M., Larsson B., Hoiden-Guthenberg I., Widstrom C., Carlsson J., Tolmachev V., Stahl S., Nilsson F.Y. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006;66:4339–4348. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 137.Hogbom M., Eklund M., Nygren P.A., Nordlund P. Structural basis for recognition by an in vitro evolved affibody. Proc. Natl. Acad. Sci. U. S. A. 2003;100:3191–3196. doi: 10.1073/pnas.0436100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gonzalez-Dosal R., Sorensen M.D., Clark B.F., Rattan S.I., Kristensen P. Phage-displayed antibodies for the detection of glycated proteasome in aging cells. Ann. N.Y. Acad. Sci. 2006;1067:474–478. doi: 10.1196/annals.1354.068. [DOI] [PubMed] [Google Scholar]
- 139.Sorensen M.D., Sorensen B., Gonzalez-Dosal R., Melchjorsen C.J., Weibel J., Wang J., Jun C.W., Huanming Y., Kristensen P. Severe acute respiratory syndrome (SARS): development of diagnostics and antivirals. Ann. N.Y. Acad. Sci. 2006;1067:500–505. doi: 10.1196/annals.1354.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Duan J., Ji X., Feng J., Han W., Zhang P., Cao W., Guo X., Qi C., Yang D., Jin G., Gao G., Yan X. A human neutralizing antibody against a conformational epitope shared by oligomeric SARS S1 protein. Antivir. Ther. 2006;11:117–123. [PubMed] [Google Scholar]
- 141.Bongartz T., Sutton A.J., Sweeting M.J., Buchan I., Matteson E.L., Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295:2275–2285. doi: 10.1001/jama.295.19.2275. [DOI] [PubMed] [Google Scholar]
- 142.McWhirter J.R., Kretz-Rommel A., Saven A., Maruyama T., Potter K.N., Mockridge C.I., Ravey E.P., Qin F., Bowdish K.S. Antibodies selected from combinatorial libraries block a tumor antigen that plays a key role in immunomodulation. Proc. Natl. Acad. Sci. U. S. A. 2006;103:1041–1046. doi: 10.1073/pnas.0510081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Groves M., Lane S., Douthwaite J., Lowne D., Gareth Rees D., Edwards B., Jackson R.H. Affinity maturation of phage display antibody populations using ribosome display. J. Immunol. Methods. 2006;313:129–139. doi: 10.1016/j.jim.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 144.Liu B., Huang L., Sihlbom C., Burlingame A., Marks J.D. Towards proteome-wide production of monoclonal antibody by phage display. J. Mol. Biol. 2002;315:1063–1073. doi: 10.1006/jmbi.2001.5276. [DOI] [PubMed] [Google Scholar]
- 145.Krogsgaard M., Davis M.M. How T cells ‘see’ antigen. Nat. Immunol. 2005;6:239–245. doi: 10.1038/ni1173. [DOI] [PubMed] [Google Scholar]
- 146.Molldrem J.J., Lee P.P., Wang C., Felio K., Kantarjian H.M., Champlin R.E., Davis M.M. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat. Med. 2000;6:1018–1023. doi: 10.1038/79526. [DOI] [PubMed] [Google Scholar]
- 147.Kurbegov D., Molldrem J.J. Immunity to chronic myelogenous leukemia. Hematol. Oncol. Clin. North. Am. 2004;18:733–752. doi: 10.1016/j.hoc.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 148.Kondo Y., Molldrem J.J. Immune-induced cytopenia: bone marrow failure syndrome. Curr. Hematol. Rep. 2004;3:178–183. [PubMed] [Google Scholar]
- 149.Oka Y., Udaka K., Tsuboi A., Elisseeva O.A., Ogawa H., Aozasa K., Kishimoto T., Sugiyama H. Cancer immunotherapy targeting Wilms' tumor gene WT1 product. J. Immunol. 2000;164:1873–1880. doi: 10.4049/jimmunol.164.4.1873. [DOI] [PubMed] [Google Scholar]
- 150.Molldrem J.J., Clave E., Jiang Y.Z., Mavroudis D., Raptis A., Hensel N., Agarwala V., Barrett A.J. Cytotoxic T lymphocytes specific for a nonpolymorphic proteinase 3 peptide preferentially inhibit chronic myeloid leukemia colony-forming units. Blood. 1997;90:2529–2534. [PubMed] [Google Scholar]
- 151.Molldrem J.J. Vaccination for leukemia. Biol. Blood Marrow Transplant. 2006;12:13–18. doi: 10.1016/j.bbmt.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 152.S. Rusakiewicz, J.J. Molldrem, Immunotherapeutic peptide vaccination with leukemia-associated antigens. Curr. Opin. Immunol. Electronic publication ahead of print. [DOI] [PubMed]
- 153.Holler P.D., Holman P.O., Shusta E.V., O'Herrin S., Wittrup K.D., Kranz D.M. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5387–5392. doi: 10.1073/pnas.080078297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Li Y., Moysey R., Molloy P.E., Vuidepot A.L., Mahon T., Baston E., Dunn S., Liddy N., Jacob J., Jakobsen B.K., Boulter J.M. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 155.Kieke M.C., Shusta E.V., Boder E.T., Teyton L., Wittrup K.D., Kranz D.M. Selection of functional T cell receptor mutants from a yeast surface-display library. Proc. Natl. Acad. Sci. U. S. A. 1999;96:5651–5656. doi: 10.1073/pnas.96.10.5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Onda T., LaFace D., Baier G., Brunner T., Honma N., Mikayama T., Altman A., Green D.R. A phage display system for detection of T cell receptor–antigen interactions. Mol. Immunol. 1995;32:1387–1397. doi: 10.1016/0161-5890(95)00098-4. [DOI] [PubMed] [Google Scholar]
- 157.Schlueter C.J., Schodin B.A., Tetin S.Y., Kranz D.M. Specificity and binding properties of a single-chain T cell receptor. J. Mol. Biol. 1996;256:859–869. doi: 10.1006/jmbi.1996.0132. [DOI] [PubMed] [Google Scholar]
- 158.Weidanz J.A., Card K.F., Edwards A., Perlstein E., Wong H.C. Display of functional alphabeta single-chain T-cell receptor molecules on the surface of bacteriophage. J. Immunol. Methods. 1998;221:59–76. doi: 10.1016/s0022-1759(98)00153-7. [DOI] [PubMed] [Google Scholar]
- 159.Le Doussal J., Piqueras B., Dogan I., Debre P., Gorochov G. Phage display of peptide/major histocompatibility complex. J. Immunol. Methods. 2000;241:147–158. doi: 10.1016/s0022-1759(00)00211-8. [DOI] [PubMed] [Google Scholar]
- 160.Boulter J.M., Jakobsen B.K. Stable, soluble, high-affinity, engineered T cell receptors: novel antibody-like proteins for specific targeting of peptide antigens. Clin. Exp. Immunol. 2005;142:454–460. doi: 10.1111/j.1365-2249.2005.02929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Andersen P.S., Stryhn A., Hansen B.E., Fugger L., Engberg J., Buus S. A recombinant antibody with the antigen-specific, major histocompatibility complex-restricted specificity of T cells. Proc. Natl. Acad. Sci. U. S. A. 1996;93:1820–1824. doi: 10.1073/pnas.93.5.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Krogsgaard M., Wucherpfennig K.W., Cannella B., Hansen B.E., Svejgaard A., Pyrdol J., Ditzel H., Raine C., Engberg J., Fugger L. Visualization of myelin basic protein (MBP) T cell epitopes in multiple sclerosis lesions using a monoclonal antibody specific for the human histocompatibility leukocyte antigen (HLA)-DR2-MBP 85–99 complex. J. Exp. Med. 2000;191:1395–1412. doi: 10.1084/jem.191.8.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Willemsen R.A., Ronteltap C., Chames P., Debets R., Bolhuis R.L. T cell retargeting with MHC class I-restricted antibodies: the CD28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J. Immunol. Methods. 2005;174:7853–7858. doi: 10.4049/jimmunol.174.12.7853. [DOI] [PubMed] [Google Scholar]
- 164.Denkberg G., Reiter Y. Recombinant antibodies with T-cell receptor-like specificity: novel tools to study MHC class I presentation. Autoimmun. Rev. 2006;5:252–257. doi: 10.1016/j.autrev.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 165.Ma Q., Shimaoka M., Lu C., Jing H., Carman C.V., Springer T.A. Activation-induced conformational changes in the I domain region of lymphocyte function-associated antigen 1. J. Biol. Chem. 2002;277:10638–10641. doi: 10.1074/jbc.M112417200. [DOI] [PubMed] [Google Scholar]
- 166.Wild M.K., Cambiaggi A., Brown M.H., Davies E.A., Ohno H., Saito T., van der Merwe P.A. Dependence of T cell antigen recognition on the dimensions of an accessory receptor-ligand complex. J. Exp. Med. 1999;190:31–41. doi: 10.1084/jem.190.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Vest Hansen N., Ostergaard Pedersen L., Stryhn A., Buus S. Phage display of peptide/major histocompatibility class I complexes. Eur. J. Immunol. 2001;31:32–38. doi: 10.1002/1521-4141(200101)31:1<32::aid-immu32>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 168.Irving M.B., Pan O., Scott J.K. Random-peptide libraries and antigen-fragment libraries for epitope mapping and the development of vaccines and diagnostics. Curr. Opin. Chem. Biol. 2001;5:314–324. doi: 10.1016/S1367-5931(00)00208-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hertveldt K., Robben J., Volckaert G. Whole genome phage display selects for proline-rich Boi polypeptides against Bem1p. Biotechnol. Lett. 2006;28:1233–1239. doi: 10.1007/s10529-006-9082-y. [DOI] [PubMed] [Google Scholar]
- 170.Pannekoek H., van Meijer M., Schleef R.R., Loskutoff D.J., Barba C.F., III Functional display of human plasminogen-activator inhibitor 1 (PAI-1) on phages: novel perspectives for structure-function analysis by error-prone DNA synthesis. Gene. 1993;128:135–140. doi: 10.1016/0378-1119(93)90164-x. [DOI] [PubMed] [Google Scholar]
- 171.Sche P.P., McKenzie K.M., White J.D., Austin D.J. Display cloning: functional identification of natural product receptors using cDNA-phage display. Chem. Biol. 1999;6:707–716. doi: 10.1016/s1074-5521(00)80018-6. [DOI] [PubMed] [Google Scholar]
- 172.Danner S., Belasco J.G. T7 phage display: a novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc. Natl. Acad. Sci. U. S. A. 2001;98:12954–12959. doi: 10.1073/pnas.211439598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sioud M., Hansen M.H. Profiling the immune response in patients with breast cancer by phage-displayed cDNA libraries. Eur. J. Immunol. 2001;31:716–725. doi: 10.1002/1521-4141(200103)31:3<716::aid-immu716>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 174.Walter G., Konthur Z., Lehrach H. High-throughput screening of surface displayed gene products. Comb. Chem. High Throughput Screen. 2001;4:193–205. doi: 10.2174/1386207013331228. [DOI] [PubMed] [Google Scholar]
- 175.Jespers L.S., Messens J.H., De Keyser A., Eeckhout D., Van den Brande I., Gansemans Y.G., Lauwereys M.J., Vlasuk G.P., Stanssens P.E. Surface expression and ligand-based selection of cDNAs fused to filamentous phage gene VI. Biotechnology (N. Y.) 1995;13:378–382. doi: 10.1038/nbt0495-378. [DOI] [PubMed] [Google Scholar]
- 176.Hertveldt K., Dechassa M.L., Robben J., Volckaert G. Identification of Gal80p-interacting proteins by Saccharomyces cerevisiae whole genome phage display. Gene. 2003;307:141–149. doi: 10.1016/s0378-1119(03)00454-2. [DOI] [PubMed] [Google Scholar]
- 177.Crameri R., Suter M. Display of biologically active proteins on the surface of filamentous phages: a cDNA cloning system for selection of functional gene products linked to the genetic information responsible for their production. Gene. 1993;137:69–75. doi: 10.1016/0378-1119(93)90253-y. [DOI] [PubMed] [Google Scholar]
- 178.Palzkill T., Huang W., Weinstock G.M. Mapping protein–ligand interactions using whole genome phage display libraries. Gene. 1998;221:79–83. doi: 10.1016/s0378-1119(98)00425-9. [DOI] [PubMed] [Google Scholar]
- 179.Brunet E., Chauvin C., Choumet V., Jestin J.L. A novel strategy for the functional cloning of enzymes using filamentous phage display: the case of nucleotidyl transferases. Nucleic Acids Res. 2002;30:e40. doi: 10.1093/nar/30.9.e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rosander A., Bjerketorp J., Frykberg L., Jacobsson K. Phage display as a novel screening method to identify extracellular proteins. J. Microbiol. Methods. 2002;51:43–55. doi: 10.1016/s0167-7012(02)00052-0. [DOI] [PubMed] [Google Scholar]
- 181.Silverman J., Liu Q., Bakker A., To W., Duguay A., Alba B.M., Smith R., Rivas A., Li P., Le H., Whitehorn E., Moore K.W., Swimmer C., Perlroth V., Vogt M., Kolkman J., Stemmer W.P. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat. Biotechnol. 2005;23:1556–1561. doi: 10.1038/nbt1166. [DOI] [PubMed] [Google Scholar]
- 182.Hitoshi Y., Gururaja T., Pearsall D.M., Lang W., Sharma P., Huang B., Catalano S.M., McLaughlin J., Pali E., Peelle B., Vialard J., Janicot M., Wouters W., Luyten W., Bennett M.K., Anderson D.C., Payan D.G., Lorens J.B., Bogenberger J., Demo S. Cellular localization and antiproliferative effect of peptides discovered from a functional screen of a retrovirally delivered random peptide library. Chem. Biol. 2003;10:975–987. doi: 10.1016/j.chembiol.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 183.Deshayes S., Morris M.C., Divita G., Heitz F. Interactions of primary amphipathic cell penetrating peptides with model membranes: consequences on the mechanisms of intracellular delivery of therapeutics. Curr. Pharm. Des. 2005;11:3629–3638. doi: 10.2174/138161205774580741. [DOI] [PubMed] [Google Scholar]
- 184.Cardó-Vila M., Arap W., Pasqualini R. Alpha v beta 5 integrin-dependent programmed cell death triggered by a peptide mimic of annexin V. Mol. Cell. 2003;11:1151–1162. doi: 10.1016/s1097-2765(03)00138-2. [DOI] [PubMed] [Google Scholar]
- 185.Bullok K.E., Gammon S.T., Violini S., Prantner A.M., Villalobos V.M., Sharma V., Piwnic-Worms D. Permeation peptide conjugates for in vivo molecular imaging applications. Mol. Imaging. 2006;5:1–15. [PubMed] [Google Scholar]
- 186.Scott J.K., Smith G.P. Searching for peptide ligands with an epitope library. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 187.Zhong J., Zhang H., Stanyon C.A., Tromp G., Finley R.L., Jr. A strategy for constructing large protein interaction maps using the yeast two-hybrid system: regulated expression arrays and two-phase mating. Genome Res. 2003;13:2691–2699. doi: 10.1101/gr.1134603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ma D., Li M. Applications of display technologies to proteomic analyses. J. Cell. Biochem., Suppl. 2001;37:34–41. doi: 10.1002/jcb.10076. [DOI] [PubMed] [Google Scholar]
- 189.Kehoe J.W., Kay B.K. Filamentous phage display in the new millennium. Chem. Rev. 2005;105:4056–4072. doi: 10.1021/cr000261r. [DOI] [PubMed] [Google Scholar]
- 190.Hill H.R., Stockley P.G. Phage presentation. Mol. Microbiol. 1996;20:685–892. doi: 10.1111/j.1365-2958.1996.tb02508.x. [DOI] [PubMed] [Google Scholar]
- 191.Iannolo G., Minenkova O., Petruzzelli R., Cesareni G. Modifying filamentous phage capsid: limits in the size of the major capsid protein. J. Mol. Biol. 1995;248:835–844. doi: 10.1006/jmbi.1995.0264. [DOI] [PubMed] [Google Scholar]
- 192.Cabilly S. The basic structure of filamentous phage and its use in the display of combinatorial peptide libraries. Mol. Biotechnol. 1999;12:143–148. doi: 10.1385/MB:12:2:143. [DOI] [PubMed] [Google Scholar]
- 193.Zacher A.N., III, Stock C.A., Golden J.W., II, Smith G.P. A new filamentous phage cloning vector: fd-tet. Gene. 1980;9:127–140. doi: 10.1016/0378-1119(80)90171-7. [DOI] [PubMed] [Google Scholar]
- 194.Ho M., Nagata S., Pastan I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc. Natl. Acad. Sci. U. S. A. 2006;103:9637–9642. doi: 10.1073/pnas.0603653103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hufton S.E., Moerkerk P.T., Meulemans E.V., de Bruine A., Arends J.W., Hoogenboom H.R. Phage display of cDNA repertoires: the pVI display system and its applications for the selection of immunogenic ligands. J. Immunol. Methods. 1999;231:39–51. doi: 10.1016/s0022-1759(99)00139-8. [DOI] [PubMed] [Google Scholar]
- 196.Kwasnikowski P., Kristensen P., Markiewicz W.T. Multivalent display system on filamentous bacteriophage pVII minor coat protein. J. Immunol. Methods. 2005;307:135–143. doi: 10.1016/j.jim.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 197.Gao C., Mao S., Kaufmann G., Wirsching P., Lerner R.A., Janda K.D. A method for the generation of combinatorial antibody libraries using pIX phage display. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12612–12616. doi: 10.1073/pnas.192467999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jaye D.L., Geigerman C.M., Fuller R.E., Akyildiz A., Parkos C.A. Direct fluorochrome labeling of phage display library clones for studying binding specificities: applications in flow cytometry and fluorescence microscopy. J. Immunol. Methods. 2004;295:119–127. doi: 10.1016/j.jim.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 199.Chen L., Zurita A.J., Ardelt P.U., Giordano R.J., Arap W., Pasqualini R. Design and validation of a bifunctional ligand display system for receptor targeting. Chem. Biol. 2004;11:1081–1091. doi: 10.1016/j.chembiol.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 200.Paschke M. Phage display systems and their applications. Appl. Microbiol. Biotechnol. 2006;70:2–11. doi: 10.1007/s00253-005-0270-9. [DOI] [PubMed] [Google Scholar]
- 201.Castagnoli L., Zucconi A., Quondam M., Rossi M., Vaccaro P., Panni S., Paoluzi S., Santonico E., Dente L., Cesareni G. Alternative bacteriophage display systems. Comb. Chem. High Throughput Screen. 2001;4:121–133. doi: 10.2174/1386207013331174. [DOI] [PubMed] [Google Scholar]
- 202.Jiang J., Abu-Shilbayeh L., Rao V.B. Display of a PorA peptide from Neisseria meningitidis on the bacteriophage T4 capsid surface. Infect. Immun. 1997;65:4770–4777. doi: 10.1128/iai.65.11.4770-4777.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lindqvist B.H., Naderi S. Peptide presentation by bacteriophage P4. FEMS Microbiol. Rev. 1995;17:33–39. doi: 10.1111/j.1574-6976.1995.tb00185.x. [DOI] [PubMed] [Google Scholar]
- 204.Malys N., Chang D.Y., Baumann R.G., Xie D., Black L.W. A bipartite bacteriophage T4 SOC and HOC randomized peptide display library: detection and analysis of phage T4 terminase (gp17) and late sigma factor (gp55) interaction. J. Mol. Biol. 2002;319:289–304. doi: 10.1016/S0022-2836(02)00298-X. [DOI] [PubMed] [Google Scholar]
- 205.Sokoloff A.V., Bock I., Zhang G., Sebestyen M.G., Wolff J.A. The interactions of peptides with the innate immune system studied with use of T7 phage peptide display. Mol. Ther. 2000;2:131–139. doi: 10.1006/mthe.2000.0110. [DOI] [PubMed] [Google Scholar]
- 206.Krumpe L.R., Atkinson A.J., Smythers G.W., Kandel A., Schumacher K.M., McMahon J.B., Makowski L., Mori T. T7 lytic phage-displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries. Proteomics. 2006;6:4210–4222. doi: 10.1002/pmic.200500606. [DOI] [PubMed] [Google Scholar]
- 207.Draghici S., Chatterjee M., Tainsky M.A. Epitomics: serum screening for the early detection of cancer on microarrays using complex panels of tumor antigens. Expert Rev. Mol. Diagn. 2005;5:735–743. doi: 10.1586/14737159.5.5.735. [DOI] [PubMed] [Google Scholar]
- 208.Sternberg N., Hoess R.H. Display of peptides and proteins on the surface of bacteriophage lambda. Proc. Natl. Acad. Sci. U. S. A. 1995;92:1609–1613. doi: 10.1073/pnas.92.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kong B., Ma W.J. Display of aggregation-prone ligand binding domain of human PPAR gamma on surface of bacteriophage lambda. Acta Pharmacol. Sin. 2006;27:91–99. doi: 10.1111/j.1745-7254.2006.00253.x. [DOI] [PubMed] [Google Scholar]
- 210.Santi E., Capone S., Mennuni C., Lahm A., Tramontano A., Luzzago A., Nicosia A. Bacteriophage lambda display of complex cDNA libraries: a new approach to functional genomics. J. Mol. Biol. 2000;296:497–508. doi: 10.1006/jmbi.1999.3471. [DOI] [PubMed] [Google Scholar]
- 211.Hoess R.H. Bacteriophage lambda as a vehicle for peptide and protein display. Curr. Pharm. Biotechnol. 2002;3:23–28. doi: 10.2174/1389201023378481. [DOI] [PubMed] [Google Scholar]
- 212.Zanghi C.N., Lankes H.A., Bradel-Tretheway B., Wegman J., Dewhurst S. A simple method for displaying recalcitrant proteins on the surface of bacteriophage lambda. Nucleic Acids Res. 2005;33:160–165. doi: 10.1093/nar/gni158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Niwa M., Fukuoka K., Fujimoto T., Maruyama I.N. Efficient isolation of cDNA clones encoding rheumatoid arthritis autoantigens by lambda phage surface display. J. Biotechnol. 2004;114:55–58. doi: 10.1016/j.jbiotec.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 214.Romanov V.I. Phage display selection and evaluation of cancer drug targets. Curr. Cancer Drug Targets. 2003;3:119–129. doi: 10.2174/1568009033482010. [DOI] [PubMed] [Google Scholar]
- 215.Kzhyshkowska J., Workman G., Cardó-Vila M., Arap W., Pasqualini R., Gratchev A., Krusell L., Goerdt S., Sage E.H. Novel function of alternatively activated macrophages: stabilin-1-mediated clearance of SPARC. J. Immunol. 2006;176:5825–5832. doi: 10.4049/jimmunol.176.10.5825. [DOI] [PubMed] [Google Scholar]
- 216.Barker T.H., Baneyx G., Cardó-Vila M., Workman G.A., Weaver M., Menon P.M., Dedhar S., Rempel S.A., Arap W., Pasqualini R., Vogel V., Sage E.H. SPARC regulates extracellular matrix organization through its modulation of integrin-linked kinase activity. J. Biol. Chem. 2005;280:36483–36493. doi: 10.1074/jbc.M504663200. [DOI] [PubMed] [Google Scholar]
- 217.Tamm I., Trepel M., Cardó-Vila M., Sun Y., Welsh K., Cabezas E., Swatterthwait A., Arap W., Reed J.C., Pasqualini R. Peptides targeting caspase inhibitors. J. Biol. Chem. 2003;278:14401–14405. doi: 10.1074/jbc.M210133200. [DOI] [PubMed] [Google Scholar]
- 218.Landon L.A., Zou J., Deutscher S.L. Is phage display technology on target for developing peptide-based cancer drugs? Curr. Drug Discov. Technol. 2004;1:113–132. doi: 10.2174/1570163043335108. [DOI] [PubMed] [Google Scholar]
- 219.Jung S., Arndt K.M., Muller K.M., Pluckthun A. Selectively infective phage (SIP) technology: scope and limitations. J. Immunol. Methods. 1999;231:93–104. doi: 10.1016/s0022-1759(99)00143-x. [DOI] [PubMed] [Google Scholar]
- 220.Rudert F., Woltering C., Frisch C., Rottenberger C., Ilag L.L. A phage-based system to select multiple protein–protein interactions simultaneously from combinatorial libraries. FEBS Lett. 1998;440:135–140. doi: 10.1016/s0014-5793(98)01413-6. [DOI] [PubMed] [Google Scholar]
- 221.Roman I., Figys J., Steurs G., Zizi M. Hunting interactomes of a membrane protein: obtaining the largest set of VDAC-interacting protein epitopes. Mol. Cell Proteomics. 2006:1535–9476. doi: 10.1074/mcp.T600009-MCP200. (Print) [DOI] [PubMed] [Google Scholar]
- 222.Barry M.A., Dower W.J., Johnston S.A. Toward cell-targeting gene therapy vectors: selection of cell-binding peptides from random peptide-presenting phage libraries. Nat. Med. 1996;2:299–305. doi: 10.1038/nm0396-299. [DOI] [PubMed] [Google Scholar]
- 223.Ivanenkov V.V., Felici F., Menon A.G. Targeted delivery of multivalent phage display vectors into mammalian cells. Biochim. Biophys. Acta. 1999;1448:463–472. doi: 10.1016/s0167-4889(98)00163-3. [DOI] [PubMed] [Google Scholar]
- 224.Liang S., Lin T., Ding J., Pan Y., Dang D., Guo C., Zhi M., Zhao P., Sun L., Hong L., Shi Y., Yao L., Liu J., Wu K., Fan D. Screening and identification of vascular-endothelial-cell-specific binding peptide in gastric cancer. J. Mol. Med. 2006;9:764–773. doi: 10.1007/s00109-006-0064-2. [DOI] [PubMed] [Google Scholar]
- 225.Giordano R.J., Anobom C.D., Cardó-Vila M., Kalil J., Valente A.P., Pasqualini R., Almeida F.C., Arap W. Structural basis for the interaction of a vascular endothelial growth factor mimic peptide motif and its corresponding receptors. Chem. Biol. 2005;12:1075–1083. doi: 10.1016/j.chembiol.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 226.Zurita A.J., Troncoso P., Cardó-Vila M., Logothetis C.J., Pasqualini R., Arap W. Combinatorial screenings in patients: the interleukin-11 receptor alpha as a candidate target in the progression of human prostate cancer. Cancer Res. 2004;64:435–439. doi: 10.1158/0008-5472.can-03-2675. [DOI] [PubMed] [Google Scholar]
- 227.Gao C., Mao S., Ronca F., Zhuang S., Quaranta V., Wirsching P., Janda K.D. De novo identification of tumor-specific internalizing human antibody-receptor pairs by phage-display methods. J. Immunol. Methods. 2003;274:185–197. doi: 10.1016/s0022-1759(02)00522-7. [DOI] [PubMed] [Google Scholar]
- 228.Poul M.A., Marks J.D. Targeted gene delivery to mammalian cells by filamentous bacteriophage. J. Mol. Biol. 1999;288:203–211. doi: 10.1006/jmbi.1999.2678. [DOI] [PubMed] [Google Scholar]
- 229.Larocca D., Kassner P.D., Witte A., Ladner R.C., Pierce G.F., Baird A. Gene transfer to mammalian cells using genetically targeted filamentous bacteriophage. FASEB J. 1999;13:727–734. doi: 10.1096/fasebj.13.6.727. [DOI] [PubMed] [Google Scholar]
- 230.Ardelt P.U., Wood C.G., Chen L., Mintz P.J., Moya C., Arap M.A., Wright K.C., Pasqualini R., Arap W. Targeting urothelium: ex vivo assay standardization and selection of internalizing ligands. J. Urol. 2003;169:1535–1540. doi: 10.1097/01.ju.0000055477.37115.66. [DOI] [PubMed] [Google Scholar]
- 231.Arap M.A., Lahdenranta J., Hajitou A., Marini F.C., III, Wood C.G., Wright K.C., Fueyo J., Arap W., Pasqualini R. Model of unidirectional transluminal gene transfer, Mol. Ther. 2004;9:305–310. doi: 10.1016/j.ymthe.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 232.Shukla G.S., Krag D.N. Selection of tumor-targeting agents on freshly excised human breast tumors using a phage display library. Oncol. Rep. 2005;13:757–764. [PubMed] [Google Scholar]
- 233.Arap M.A., Lahdenranta J., Mintz P.J., Hajitou A., Sarkis A.S., Arap W., Pasqualini R. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cells. 2004;6:275–284. doi: 10.1016/j.ccr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 234.Marchió S., Lahdenranta J., Schlingemann R.O., Valdembri D., Wesseling P., Arap M.A., Hajitou A., Ozawa M.G., Trepel M., Giordano R.J., Nanus D.M., Dijkman H.B., Oosterwijk E., Sidman R.L., Cooper M.D., Bussolino F., Pasqualini R., Arap W. Aminopeptidase A is a functional target in angiogenic blood vessels. Cancer Cells. 2004;5:151–162. doi: 10.1016/s1535-6108(04)00025-x. [DOI] [PubMed] [Google Scholar]
- 235.Ozawa M.G., Yao V.J., Chanthery Y.H., Troncoso P., Uemura A., Varner A.S., Kasman I.M., Pasqualini R., Arap W., McDonald D.M. Angiogenesis with pericyte abnormalities in a transgenic model of prostate carcinoma. Cancer. 2005;104:2104–2115. doi: 10.1002/cncr.21436. [DOI] [PubMed] [Google Scholar]
- 236.Hashizume H., Baluk P., Morikawa S., McLean J.W., Thurston G., Roberge S., Jain R.K., McDonald D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000;156:1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Arap W., Haedicke W., Bernasconi M., Kain R., Rajotte D., Krajewski S., Ellerby H.M., Bredesen D.E., Pasqualini R., Ruoslahti E. Targeting the prostate for destruction through a vascular address. Proc. Natl. Acad. Sci. U. S. A. 2002;99:1527–1531. doi: 10.1073/pnas.241655998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Oh Y., Mohiuddin I., Sun Y., Putnam J.B., Hong W.K., Arap W., Pasqualini R. Phenotypic diversity of the lung vasculature in experimental models of metastases. Chest. 2005;128:596S–600S. doi: 10.1378/chest.128.6_suppl.596S. [DOI] [PubMed] [Google Scholar]
- 239.Essler M., Ruoslahti E. Molecular specialization of breast vasculature: a breast-homing phage-displayed peptide binds to aminopeptidase P in breast vasculature. Proc. Natl. Acad. Sci. U. S. A. 2002;99:2252–2257. doi: 10.1073/pnas.251687998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Zhang L., Giraudo E., Hoffman J.A., Hanahan D., Ruoslahti E. Lymphatic zip codes in premalignant lesions and tumors. Cancer Res. 2006;66:5696–5706. doi: 10.1158/0008-5472.CAN-05-3876. [DOI] [PubMed] [Google Scholar]
- 241.Pilch J., Brown D.M., Komatsu M., Jarvinen T.A., Yang M., Peters D., Hoffman R.M., Ruoslahti E. Peptides selected for binding to clotted plasma accumulate in tumor stroma and wounds. Proc. Natl. Acad. Sci. U. S. A. 2006;103:2800–2804. doi: 10.1073/pnas.0511219103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Samoylova T.I., Smith B.F. Elucidation of muscle-binding peptides by phage display screening. Muscle Nerve. 1999;22:460–466. doi: 10.1002/(sici)1097-4598(199904)22:4<460::aid-mus6>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 243.Pasqualini R., Koivunen E., Ruoslahti E. Alpha v integrins as receptors for tumor targeting by circulating ligands. Nat. Biotechnol. 1997;15:542–546. doi: 10.1038/nbt0697-542. [DOI] [PubMed] [Google Scholar]
- 244.Yao V.J., Ozawa M.G., Varner A.S., Kasman I.M., Chanthery Y.H., Pasqualini R., Arap W., McDonald D.M. Antiangiogenic therapy decreases integrin expression in normalized tumor blood vessels. Cancer Res. 2006;66:2639–2649. doi: 10.1158/0008-5472.CAN-05-1824. [DOI] [PubMed] [Google Scholar]
- 245.Curnis F., Arrigoni G., Sacchi A., Fischetti L., Arap W., Pasqualini R., Corti A. Differential binding of drugs containing the NGR Motif to CD13 isoforms in yumor vessels, epithelia, and myeloid cells. Cancer Res. 2002;62:867–874. [PubMed] [Google Scholar]
- 246.Bhagwat S.V., Lahdenranta J., Giordano R., Arap W., Pasqualini R., Shapiro L.H. CD13/APN is activated by angiogenic signals and is essential for capillary tube formation. Blood. 2001;97:652–659. doi: 10.1182/blood.v97.3.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Joyce J.A., Laakkonen P., Bernasconi M., Bergers G., Ruoslahti E., Hanahan D. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cells. 2003;4:393–403. doi: 10.1016/s1535-6108(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 248.Hoffman J.A., Giraudo E., Singh M., Zhang L., Inoue M., Porkka K., Hanahan D., Ruoslahti E. Progressive vascular changes in a transgenic mouse model of squamous cell carcinoma. Cancer Cells. 2003;4:383–391. doi: 10.1016/s1535-6108(03)00273-3. [DOI] [PubMed] [Google Scholar]
- 249.Work L.M., Buning H., Hunt E., Nicklin S.A., Denby L., Britton N., Leike K., Odenthal M., Drebber U., Hallek M., Baker A.H. Vascular bed-targeted in vivo gene delivery using tropism-modified adeno-associated viruses. Mol. Ther. 2006;13:683–693. doi: 10.1016/j.ymthe.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 250.Mullen L.M., Nair S.P., Ward J.M., Rycroft A.N., Henderson B. Phage display in the study of infectious diseases. Trends Microbiol. 2006;14:141–147. doi: 10.1016/j.tim.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Bacich D.J., Pinto J.T., Tong W.P., Heston W.D. Cloning, expression, genomic localization, and enzymatic activities of the mouse homolog of prostate-specific membrane antigen/NAALADase/folate hydrolase. Mamm. Genome. 2001;12:117–123. doi: 10.1007/s003350010240. [DOI] [PubMed] [Google Scholar]
- 252.Carson-Walter E.B., Watkins D.N., Nanda A., Vogelstein B., Kinzler K.W., St Croix B. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res. 2001;61:6649–6655. [PubMed] [Google Scholar]
- 253.Pentz R.D., Cohen C.B., Wicclair M., DeVita M.A., Flamm A.L., Youngner S.J., Hamric A.B., McCabe M.S., Glover J.J., Kittiko W.J., Kinlaw K., Keller J., Asch A., Kavanagh J.J., Arap W. Ethics guidelines for research with the recently dead. Nat. Med. 2005;11:1145–1149. doi: 10.1038/nm1105-1145. [DOI] [PubMed] [Google Scholar]
- 254.Pentz R.D., Flamm A.L., Pasqualini R., Logothetis C.J., Arap W. Revisiting ethical guidelines for research with terminal wean and brain-dead participants. Hastings Cent. Rep. 2003;33:20–26. [PubMed] [Google Scholar]
- 255.Pentz R.D., Flamm A.L. The newly and nearly dead. Hastings Cent. Rep. 2003;33:4. [PubMed] [Google Scholar]
- 256.Krag D.N., Shukla G.S., Shen G.P., Pero S., Ashikaga T., Fuller S., Weaver D.L., Burdette-Radoux S., Thomas C. Selection of tumor-binding ligands in cancer patients with phage display libraries. Cancer Res. 2006;66:7724–7733. doi: 10.1158/0008-5472.CAN-05-4441. [DOI] [PubMed] [Google Scholar]
- 257.Narasimhan K. Zip codes: deciphering vascular addresses. Nat. Med. 2002;8:116. doi: 10.1038/nm0202-116. [DOI] [PubMed] [Google Scholar]
- 258.Folkman J. Looking for a good endothelial address. Cancer Cells. 2002;1:113–115. doi: 10.1016/s1535-6108(02)00038-7. [DOI] [PubMed] [Google Scholar]
- 259.Jaye D.L., Nolte F.S., Mazzucchelli L., Geigerman C., Akyildiz A., Parkos C.A. Use of real-time polymerase chain reaction to identify cell- and tissue-type-selective peptides by phage display. Am. J. Pathol. 2003;162:1419–1429. doi: 10.1016/S0002-9440(10)64275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Yao V.J., Ozawa M.G., Trepel M., Arap W., McDonald D.M., Pasqualini R. Targeting pancreatic islets with phage display assisted by laser pressure catapult microdissection. Am. J. Pathol. 2005;166:625–636. doi: 10.1016/S0002-9440(10)62283-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ballard V.L., Holm J.M., Edelberg J.M. A quantitative PCR-based approach for rapid phage display analysis: a foundation for high throughput vascular proteomic profiling. Physiol. Genomics. 2006;26:202–208. doi: 10.1152/physiolgenomics.00025.2006. [DOI] [PubMed] [Google Scholar]
- 262.Guo Y.C., Zhou Y.F., Zhang X.E., Zhang Z.P., Qiao Y.M., Bi L.J., Wen J.K., Liang M.F., Zhang J.B. Phage display mediated immuno-PCR. Nucleic Acids Res. 2006;34:e62. doi: 10.1093/nar/gkl260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Lu H., Jin D., Kapila Y.L. Application of laser capture microdissection to phage display peptide library screening. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endo. 2004;98:692–697. doi: 10.1016/j.tripleo.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 264.Shukla G.S., Krag D.N. Phage display selection for cell-specific ligands: development of a screening procedure suitable for small tumor specimens. J. Drug Target. 2005;13:7–18. doi: 10.1080/10611860400020464. [DOI] [PubMed] [Google Scholar]
- 265.Eltoum I.A., Siegal G.P., Frost A.R. Microdissection of histologic sections: past, present, and future. Adv. Anat. Pathol. 2002;9:316–322. doi: 10.1097/00125480-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 266.Chen G., Hayhurst A., Thomas J.G., Harvey B.R., Iverson B.L., Georgiou G. Isolation of high-affinity ligand-binding proteins by periplasmic expression with cytometric screening (PECS) Nat. Biotechnol. 2001;19:537–542. doi: 10.1038/89281. [DOI] [PubMed] [Google Scholar]
- 267.Sugimura Y., Hosono M., Wada F., Yoshimura T., Maki M., Hitomi K. Screening for the preferred substrate sequence of transglutaminase using a phage-displayed peptide library: identification of peptide substrates for TGASE 2 and Factor XIIIA. J. Biol. Chem. 2006;281:17699–17706. doi: 10.1074/jbc.M513538200. [DOI] [PubMed] [Google Scholar]
- 268.Paradis-Bleau C., Beaumont M., Boudreault L., Lloyd A., Sanschagrin F., Bugg T.D., Levesque R.C. Selection of peptide inhibitors against the Pseudomonas aeruginosa MurD cell wall enzyme. Peptides. 2006;27:1693–1700. doi: 10.1016/j.peptides.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 269.Pini A., Runci Y., Falciani C., Lelli B., Brunetti J., Pileri S., Fabbrini M., Lozzi L., Ricci C., Bernini A., Tonello F., Dal Molin F., Neri P., Niccolai N., Bracci L. Stable peptide inhibitors prevent binding of lethal and oedema factors to protective antigen and neutralize anthrax toxin in vivo. Biochem. J. 2006;395:157–163. doi: 10.1042/BJ20051747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Hekim C., Leinonen J., Narvanen A., Koistinen H., Zhu L., Koivunen E., Vaisanen V., Stenman U.H. Novel peptide inhibitors of human kallikrein 2. J. Biol. Chem. 2006;281:12555–12560. doi: 10.1074/jbc.M600014200. [DOI] [PubMed] [Google Scholar]
- 271.Shibayama J., Lewandowski R., Kieken F., Coombs W., Shah S., Sorgen P.L., Taffet S.M., Delmar M. Identification of a novel peptide that interferes with the chemical regulation of connexin43. Circ. Res. 2006;98:1365–1372. doi: 10.1161/01.RES.0000225911.24228.9c. [DOI] [PubMed] [Google Scholar]
- 272.Hirosue S., Weber T. pH-Dependent lytic peptides discovered by phage display. Biochemistry. 2006;45:6476–6487. doi: 10.1021/bi052488s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Guo X., Chen R.R. An improved phage display procedure for identification of lipopolysaccharide-binding peptides. Biotechnol. Prog. 2006;22:601–604. doi: 10.1021/bp050315o. [DOI] [PubMed] [Google Scholar]
- 274.Marchió S., Alfano M., Primo L., Gramaglia D., Butini L., Gennero L., De Vivo E., Arap W., Giacca M., Pasqualini R., Bussolino F. Cell surface-associated Tat modulates HIV-1 infection and spreading through a specific interaction with gp120 viral envelope protein. Blood. 2005;105:2802–2811. doi: 10.1182/blood-2004-06-2212. [DOI] [PubMed] [Google Scholar]
- 275.Lionakis M.S., Lahdenranta J., Sun J., Liu W., Lewis R.E., Albert N.D., Pasqualini R., Arap W., Kontoyiannis D.P. Development of a ligand-directed approach to study the pathogenesis of invasive aspergillosis. Infect. Immun. 2005;73:7747–7758. doi: 10.1128/IAI.73.11.7747-7758.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Halperin I., Wolfson H., Nussinov R. SiteLight: binding-site prediction using phage display libraries. Protein Sci. 2003;12:1344–1359. doi: 10.1110/ps.0237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Tarnovitski N., Matthews L.J., Sui J., Gershoni J.M., Marasco W.A. Mapping a neutralizing epitope on the SARS coronavirus spike protein: computational prediction based on affinity-selected peptides. J. Mol. Biol. 2006;359:190–201. doi: 10.1016/j.jmb.2006.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Kontoyiannis D.P., Pasqualini R., Arap W. Aminopeptidase N inhibitors and SARS. Lancet. 2003;361:1558. doi: 10.1016/S0140-6736(03)13186-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Barrow P.A., Soothill J.S. Bacteriophage therapy and prophylaxis: rediscovery and renewed assessment of potential. Trends Microbiol. 1997;5:268–271. doi: 10.1016/S0966-842X(97)01054-8. [DOI] [PubMed] [Google Scholar]
- 280.Hudson J.A., Billington C., Carey-Smith G., Greening G. Bacteriophages as biocontrol agents in food. J. Food Prot. 2005;68:426–437. doi: 10.4315/0362-028x-68.2.426. [DOI] [PubMed] [Google Scholar]
- 281.Yacoby I., Shamis M., Bar H., Shabat D., Benhar I. Targeting antibacterial agents by using drug-carrying filamentous bacteriophages. Antimicrob. Agents Chemother. 2006;50:2087–2097. doi: 10.1128/AAC.00169-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Duerr D.M., White S.J., Schluesener H.J. Identification of peptide sequences that induce the transport of phage across the gastrointestinal mucosal barrier. J. Virol. Methods. 2004;116:177–180. doi: 10.1016/j.jviromet.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 283.Chen Y., Shen Y., Guo X., Zhang C., Yang W., Ma M., Liu S., Zhang M., Wen L.P. Transdermal protein delivery by a coadministered peptide identified via phage display. Nat. Biotechnol. 2006;24:455–460. doi: 10.1038/nbt1193. [DOI] [PubMed] [Google Scholar]
- 284.Lee S.W., Mao C., Flynn C.E., Belcher A.M. Ordering of quantum dots using genetically engineered viruses. Science. 2002;296:892–895. doi: 10.1126/science.1068054. [DOI] [PubMed] [Google Scholar]
- 285.Souza G.R., Christianson D.R., Staquicini F.I., Ozawa M.G., Snyder E.Y., Sidman R.L., Miller J.H., Arap W., Pasqualini R. Networks of gold nanoparticles and bacteriophage as biological sensors and cell-targeting agents. Proc. Natl. Acad. Sci. U. S. A. 2006;103:1215–1220. doi: 10.1073/pnas.0509739103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Chu T.H., Martinez I., Sheay W.C., Dornburg R. Cell targeting with retroviral vector particles containing antibody-envelope fusion proteins. Gene Ther. 1994;1:292–299. [PubMed] [Google Scholar]
- 287.Russell S.J., Hawkins R.E., Winter G. Retroviral vectors displaying functional antibody fragments. Nucleic Acids Res. 1993;21:1081–1085. doi: 10.1093/nar/21.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Urban J.H., Schneider R.M., Compte M., Finger C., Cichutek K., Alvarez-Vallina L., Buchholz C.J. Selection of functional human antibodies from retroviral display libraries. Nucleic Acids Res. 2005;33:e35. doi: 10.1093/nar/gni033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Crawford F., Jordan K.R., Stadinski B., Wang Y., Huseby E., Marrack P., Slansky J.E., Kappler J.W. Use of baculovirus MHC/peptide display libraries to characterize T-cell receptor ligands. Immunol. Rev. 2006;210:156–170. doi: 10.1111/j.0105-2896.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 290.Nassal M., Skamel C., Kratz P.A., Wallich R., Stehle T., Simon M.M. A fusion product of the complete Borrelia burgdorferi outer surface protein A (OspA) and the hepatitis B virus capsid protein is highly immunogenic and induces protective immunity similar to that seen with an effective lipidated OspA vaccine formula. Eur. J. Immunol. 2005;35:655–665. doi: 10.1002/eji.200425449. [DOI] [PubMed] [Google Scholar]
- 291.Makela A.R., Matilainen H., White D.J., Ruoslahti E., Oker-Blom C. Enhanced baculovirus-mediated transduction of human cancer cells by tumor-homing peptides. J. Virol. 2006;80:6603–6611. doi: 10.1128/JVI.00528-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Lieber A. AAV display-homing in on the target. Nat. Biotechnol. 2003;21:1011–1013. doi: 10.1038/nbt0903-1011. [DOI] [PubMed] [Google Scholar]
- 293.Grifman M., Trepel M., Speece P., Gilbert L.B., Arap W., Pasqualini R., Weitzman M.D. Incorporation of tumor-targeting peptides into recombinant adeno-associated virus capsids. Mol. Ther. 2001;3:964–975. doi: 10.1006/mthe.2001.0345. [DOI] [PubMed] [Google Scholar]
- 294.Muller O.J., Kaul F., Weitzman M.D., Pasqualini R., Arap W., Kleinschmidt J.A., Trepel M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 2003;21:1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 295.Douglas J.T., Miller C.R., Kim M., Dmitriev I., Mikheeva G., Krasnykh V., Curiel D.T. A system for the propagation of adenoviral vectors with genetically modified receptor specificities. Nat. Biotechnol. 1999;17:470–475. doi: 10.1038/8647. [DOI] [PubMed] [Google Scholar]
- 296.Fontana L., Nuzzo M., Urbanelli L., Monaci P. General strategy for broadening adenovirus tropism. J. Virol. 2003;77:11094–11104. doi: 10.1128/JVI.77.20.11094-11104.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Georgiou G., Stathopoulos C., Daugherty P.S., Nayak A.R., Iverson B.L., Curtiss R., III Display of heterologous proteins on the surface of microorganisms: from the screening of combinatorial libraries to live recombinant vaccines. Nat. Biotechnol. 1997;15:29–34. doi: 10.1038/nbt0197-29. [DOI] [PubMed] [Google Scholar]
- 298.Samuelson P., Gunneriusson E., Nygren P.A., Stahl S. Display of proteins on bacteria. J. Biotechnol. 2002;96:129–154. doi: 10.1016/s0168-1656(02)00043-3. [DOI] [PubMed] [Google Scholar]
- 299.Lang H. Outer membrane proteins as surface display systems. Int. J. Med. Microbiol. 2000;290:579–585. doi: 10.1016/S1438-4221(00)80004-1. [DOI] [PubMed] [Google Scholar]
- 300.Etz H., Minh D.B., Schellack C., Nagy E., Meinke A. Bacterial phage receptors, versatile tools for display of polypeptides on the cell surface. J. Bacteriol. 2001;183:6924–6935. doi: 10.1128/JB.183.23.6924-6935.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Lee S.Y., Choi J.H., Xu Z. Microbial cell-surface display. Trends Biotechnol. 2003;21:45–52. doi: 10.1016/s0167-7799(02)00006-9. [DOI] [PubMed] [Google Scholar]
- 302.Jose J., Betscheider D., Zangen D. Bacterial surface display library screening by target enzyme labeling: identification of new human cathepsin G inhibitors. Anal. Biochem. 2005;346:258–267. doi: 10.1016/j.ab.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 303.Lunder M., Bratkovic T., Doljak B., Kreft S., Urleb U., Strukelj B., Plazar N. Comparison of bacterial and phage display peptide libraries in search of target-binding motif. Appl. Biochem. Biotechnol. 2005;127:125–131. doi: 10.1385/abab:127:2:125. [DOI] [PubMed] [Google Scholar]
- 304.Boder E.T., Wittrup K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 305.Kondo A., Ueda M. Yeast cell-surface display-applications of molecular display. Appl. Microbiol. Biotechnol. 2004;64:28–40. doi: 10.1007/s00253-003-1492-3. [DOI] [PubMed] [Google Scholar]
- 306.Lin Y., Tsumuraya T., Wakabayashi T., Shiraga S., Fujii I., Kondo A., Ueda M. Display of a functional hetero-oligomeric catalytic antibody on the yeast cell surface. Appl. Microbiol. Biotechnol. 2003;62:226–232. doi: 10.1007/s00253-003-1283-x. [DOI] [PubMed] [Google Scholar]
- 307.Jin M., Song G., Carman C.V., Kim Y.S., Astrof N.S., Shimaoka M., Wittrup D.K., Springer T.A. Directed evolution to probe protein allostery and integrin I domains of 200,000-fold higher affinity. Proc. Natl. Acad. Sci. U. S. A. 2006;103:5758–5763. doi: 10.1073/pnas.0601164103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.FitzGerald K. In vitro display technologies — new tools for drug discovery. Drug Discov. Today. 2000;5:253–258. doi: 10.1016/s1359-6446(00)01501-4. [DOI] [PubMed] [Google Scholar]
- 309.Lipovsek D., Pluckthun A. In-vitro protein evolution by ribosome display and mRNA display. J. Immunol. Methods. 2004;290:51–67. doi: 10.1016/j.jim.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 310.He M., Khan F. Ribosome display: next-generation display technologies for production of antibodies in vitro. Expert Rev. Proteomics. 2005;2:421–430. doi: 10.1586/14789450.2.3.421. [DOI] [PubMed] [Google Scholar]
- 311.Mattheakis L.C., Bhatt R.R., Dower W.J. An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc. Natl. Acad. Sci. U. S. A. 1994;91:9022–9026. doi: 10.1073/pnas.91.19.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Rothe A., Hosse R.J., Power B.E. Ribosome display for improved biotherapeutic molecules. Expert Opin. Biol. Ther. 2006;6:177–187. doi: 10.1517/14712598.6.2.177. [DOI] [PubMed] [Google Scholar]
- 313.Ihara H., Mie M., Funabashi H., Takahashi F., Sawasaki T., Endo Y., Kobatake E. In vitro selection of zinc finger DNA-binding proteins through ribosome display. Biochem. Biophys. Res. Commun. 2006;345:1149–1154. doi: 10.1016/j.bbrc.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 314.Amstutz P., Koch H., Binz H.K., Deuber S.A., Pluckthun A. Rapid selection of specific MAP kinase-binders from designed ankyrin repeat protein libraries. Protein Eng. Des. Sel. 2006;19:219–229. doi: 10.1093/protein/gzl004. [DOI] [PubMed] [Google Scholar]
- 315.Villemagne D., Jackson R., Douthwaite J.A. Highly efficient ribosome display selection by use of purified components for in vitro translation. J. Immunol. Methods. 2006;313:140–148. doi: 10.1016/j.jim.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 316.Roberts R.W., Szostak J.W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. U. S. A. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Baggio R., Burgstaller P., Hale S.P., Putney A.R., Lane M., Lipovsek D., Wright M.C., Roberts R.W., Liu R., Szostak J.W., Wagner R.W. Identification of epitope-like consensus motifs using mRNA display. J. Mol. Recognit. 2002;15:126–134. doi: 10.1002/jmr.567. [DOI] [PubMed] [Google Scholar]
- 318.Reiersen H., Lobersli I., Loset G.A., Hvattum E., Simonsen B., Stacy J.E., McGregor D., Fitzgerald K., Welschof M., Brekke O.H., Marvik O.J. Covalent antibody display-an in vitro antibody-DNA library selection system. Nucleic Acids Res. 2005;33:e10. doi: 10.1093/nar/gni010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Laumonier C., Segers J., Laurent S., Michel A., Coppee F., Belayew A., Elst L.V., Muller R.N. A new peptidic vector for molecular imaging of apoptosis, identified by phage display technology. J. Biomol. Screen. 2006;11:537–545. doi: 10.1177/1087057106288220. [DOI] [PubMed] [Google Scholar]
- 320.Ghosh S., Nie A., An J., Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr. Opin. Chem. Biol. 2006;10:194–202. doi: 10.1016/j.cbpa.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 321.Rognan D. Development and virtual screening of target libraries. J. Physiol. (Paris) 2006;99:232–244. doi: 10.1016/j.jphysparis.2005.12.084. [DOI] [PubMed] [Google Scholar]
- 322.Klebe G. Recent developments in structure-based drug design. J. Mol. Med. 2000;78:269–281. doi: 10.1007/s001090000084. [DOI] [PubMed] [Google Scholar]