

Abstract
Cranberry (Vaccinium macrocarpon Ait., Ericaceae) juice has been used for urinary tract infections for approximately 50 years. Recent research suggests that this botanical blocks adherence of pathogenic E. coli to urinary tract cells, thus preventing infection. While current evidence indicates that proanthocyanidins are responsible for this activity, these compounds may not reach the urinary tract, thus further investigation is warranted. Fractionation of cranberry juice concentrate was guided by a recently published antiadherence assay, and the resulting fractions were phytochemically characterized. Two new coumaroyl iridoid glycosides, 10-p-trans- (1) and 10-p-cis-coumaroyl-1S-dihydromonotropein (2), and a depside, 2-O-(3,4-dihydroxybenzoyl)-2,4,6-trihydroxyphenylmethylacetate (3) were isolated, and although these compounds did not have antiadherent activity in isolation, they might constitute a new group of marker compounds for this active fraction of cranberry.
Cranberry (Vaccinium macrocarpon Ait., Ericaceae) juice, generally in the form of cranberry juice cocktail consisting of a mixture of cranberry juice, water and sugar, is a popular botanical dietary supplement1, 2 primarily used for the treatment and prevention of urinary tract infections, with documented human use for at least 47 years.3 This use is supported by several clinical trials,4–11 although some trials, notably for children with neuropathic bladder and catheterization, did not find cranberry to be effective.12, 13 Cranberry juice was originally believed to be active due to its acidifying effect on urine, and/or the increased excretion of the cranberry urinary metabolite hippuric acid.14, 15 However, subsequent research suggested that the bacteriostatic impact from acidification alone could not account for its demonstrated effects.16, 17
Inhibition of adherence of Escherichia coli to uroepithelial cells,18–20 rather than direct bacteriostatic or bactericidal activity, has been proposed as the mechanism of action. Specifically, there is support for inhibition of the papG fimbrial attachment of uropathogenic strains of E. coli to human cells21, 22 by cranberry’s A-type proanthocyanidin compounds, but not by a B-type dimer or the (–)-epicatechin monomer.18, 23 Proanthocyanidins, however, may not be assimilated in the gut, nor reach the urinary tract intact.24 The putative active cranberry A-type proanthocyanidin oligomers, containing a second link (carbon-oxygen) between at least two of their epicatechin monomer units, are chemically similar to B-type proanthocyanidins, which contain only single, carbon-carbon links between units. Knowledge about the metabolic route in humans of A-type proanthocyanidins is extremely sparse. With very limited recent exceptions for small amounts of dimers and possibly a trimer,25 evidence currently indicates that B-type proanthocyanidins, especially trimers and larger, are degraded in the gut, and/or not assimilated in any quantity, and do not reach the urinary tract intact.26–37 Whether this information can be applied to A-type proanthocyanidins is unknown, and should be verified before abandoning the search for other active cranberry constituents. Additionally, while urine after cranberry ingestion has been shown to be antiadherent,16, 17, 19 and while proanthocyanidin metabolites38 are possibly the active constituents, to date no researchers have elucidated any specific antiadherent cranberry compounds or metabolites thereof, proanthocyanidin or otherwise, found in urine after cranberry ingestion. It is also important to consider possible synergism between compounds, as seen for the antimutagenic activity of cranberry.39 Relying on one class of isolated active compounds and ignoring interactions from the remaining constituents, while tempting for reasons of feasibility, can generally be misleading in terms of explaining efficacy of herbals. Therefore, one aim of the present study was to remove proanthocyanidins and bactericidal benzoic acid and then further characterize the in vitro antiadherent fraction of cranberry juice. In this small but important step in the complete understanding of the use of this botanical for urinary tract infection, we present data on two new compounds that, while not active in isolation, are potential new (phytochemical) marker compounds for the bioactive cranberry fraction.
An assay using biologically relevant cells, and of sufficiently high throughput such that it can efficiently guide the fractionation of cranberry, was developed in our laboratory and recently published.40 We have used this assay in the bioactivity-guided fractionation of cranberry juice, and report here two new and one known compound (1–3) found in, and assisting in the identification of, the active fraction. Compounds 1 and 2 represent an isomeric pair of acylated dihydromonotropein iridoids and, together with previously reported congeners,41 underline the significance of this class of phytochemicals for the characterization of cranberry preparations. The third compound is the depside, 2-O-(3,4-dihydroxybenzoyl)-2,4,6-trihydroxyphenylmethylacetate (3), a methylated form42 of another depside (5) so far only reported in Papaver rhoeas.43
Cranberry juice, obtained as a frozen concentrate, was thawed, partially neutralized with aqueous ammonia (28% NH4OH), and fractionated over HP-20 resin. Two more levels of fractionation, guided by positive activity in the antiadhesion assay, resulted in an active fraction from which two new coumaroyl-iridoid glucosides (1, 2) and a depside (3) were isolated using reverse-phase HPLC. These compounds were identified by spectroscopic analysis, and by comparison of their data with literature values of the similar compounds dihydromonotropein (4) and a Papaver depside (5).41, 43
The deprotonated molecular ion of 1 at m/z 537.1629 (1) [M-H]− (high-resolution negative ion electrospray) corresponded to the molecular formula C25H30O13 (calc. 537.1608). The IR spectra showed the characteristic absorption bands of hydroxy groups, free acid or ester groups, and alkenes at 3308, 1684, and 1138 cm−1, respectively. UV absorptions at 215, 230 and 315 suggested that 1 had two independent conjugated systems, one of which was aromatic. In the 1H NMR spectrum of 1 (Table 1), AM spin system resonances at δH 7.668 and 6.385 (each 1H, d, J = 15.9 Hz; H-3″, H-2″ resp.) revealed the presence of a trans-ethylene group conjugated with an aromatic ring, and a typical AA′XX′ spin system for 1,4-disubstituted aromatic ring protons at δH 7.487 and 6.804 (each 2H, brddd, J = 8.7, 2.9, 2.0 Hz; H-5″/H-9″, and H-6″/H-8″, resp.). The spectrum also revealed a six-carbon sugar moiety with all-axial ring protons at δH 4.725 (1H, d, J = 7.9 Hz; H-1′), 3.238 (1H, dd, J = 9.0, 7.9 Hz; H-2′), 3.329 (1H, overlapped with solvent signals; H-3′), 3.352 (1H, t, 9.0; H-4′), 3.336 (1H, overlapped with solvent signals; H-5′), 3.846 (1H, dd, J =11.8, 1.6 Hz; H-6a′), and 3.662 (1H, dd, J = 11.8, 5.2 Hz; H-6b′). In addition to the observed resonances of characteristic spin systems and a sugar moiety, an olefinic proton was observed at δH 7.492 (1H, d, J = 1.4 Hz; H-3), a dioxymethine proton at δH 5.514 (1H, d, J = 5.3 Hz; H-1), methylene resonances at δH 4.256 and 4.182 (each 1H, d, J = 11.3 Hz; H-10), three proton resonances at δH 2.933 (1H, ddddd, J = 15.2, 9.2, 1.7, 1.4, 0.5 Hz; H-5), 2.319 (1H, dd, J = 9.2, 5.3 Hz; H-9), and 2.174 (1H, m; H-6a), and three overlapped multiplets in the range δH 1.831~1.653 accounting for the C-7 methylene protons and one of the two C-6 protons. The shielded H-6b at δH 1.704 overlapped with H-7, as revealed in the HMQC spectrum. The 13C and DEPT NMR spectra of 1 (Table 2) included resonances for a trans-ethylene group at δC 114.89 (d, C-2″) and 147.03 (d, C-3″), as well as two resonances representing four carbons of a 1,4-disubstituted aromatic ring at δC 131.32 (d, C-5″ and 9″) and 116.85 (d, C-6″ and 8″), and six oxygenated carbon resonances belonging to the sugar substituent at δC 100.88 (d, C-1′), 74.62 (d, C-2′), 77.88 (d, C-3′), 71.22 (d, C-4′), 78.40 (d, C-5′), and 62.49 (t, C-6′). In addition, the spectra showed an acyl group and a carbonyl group at δC 169.23 (s, C-1″) and 170.74 (s, C-11), a pair of resonances for olefinic carbons at δC 153.28 (d, C-3) and 112.47 (s, C-4), three methine carbons at δC 96.14 (d, C-1), 35.21 (d, C-5), and 46.87 (d, C-9), three methylene carbons at δC 31.57 (t, C-6), 37.47 (t, C-7), and 70.88 (t, C-10), and an oxygenated quaternary carbon at δC 81.66 (s, C-8). These NMR data suggested that 1 was likely an iridoid glycoside, as they were similar to those of the known iridoid glucoside, 6,7-dihydromonotropein (4) recently isolated from cranberry by Jensen et al.41
Table 1.
| pos. | 1b | 1c | 2b | 2c | 4d |
|---|---|---|---|---|---|
| 1 | 5.514 (d, 5.3) | 5.543 (d, 3.9) | 5.466 (d, 5.4) | 5.496 (d, 4.0) | 5.54 (d, 3.6) |
| 3 | 7.492 (d, 1.4) | 7.526 (br s) | 7.463 (d, 1.4) | 7.527 (br s) | 7.55 (br s) |
| 5 | 2.933 (ddddd, 15.2, 9.2, 1.7, 1.4, 0.5) | 2.915 (m) | 2.840 (ddddd, 15.2, 9.4, 2.0, 1.4, 0.5) | 2.828 (m) | 2.95 (m) |
| 6 | Ha: 1.704 (m) | 1.672 (m) | Ha: 1.630 (m) | 1.674 (m) | 2.08 – 1.63 (m w/H7) |
| Hb: 2.174 (m) | 2.091 (m) | Hb: 2.113 (m) | 2.073 (m) | ||
| 7 | 1.840~1.716 (2H, m) | 1.831~1.710 (2H, m) | 1.749~1.645 (2H, m) | 1.794~1.670 (2H, m) | 2.08 – 1.63 (m w/H6) |
| 9 | 2.319 (dd, 9.2, 5.3) | 2.353 (dd 9.3, 4.0) | 2.224 (dd, 9.4, 5.4) | 2.270 (dd, 9.2, 4.2) | 2.34 (dd, 9.3, 3.6) |
| 10 | 4.182 (d, 11.3) | 4.159 (d, 11.1) | 4.114 (d, 11.2) | 4.194 (d, 11.2) | 3.56 (d, 17.5) |
| 4.256 (d, 11.3) | 4.199 (d, 11.1) | 4.176 (d, 11.2) | 4.230 (d, 11.2) | 3.62 (d, 17.5) | |
| 1′ | 4.725 (d, 7.9) | 4.797 (overlappede) | 4.711 (d, 7.9) | 4.799 (overlappede) | 4.82 (d, 8.4) |
| 2′ | 3.238(dd, 9.0, 7.9) | 3.285 (dd, 9.1, 8.6) | 3.243 (dd, 9.0, 7.9) | 3.291 (dd, 9.2, 8.1) | 3.29 (t, 9.1, 8.1) |
| 3′ | 3.329 (overlappede) | 3.487 (t, 9.1) | 3.327 (overlappede) | 3.509 (dd, 9.5, 9.2) | 3.51 (t, 9.1) |
| 4′ | 3.352 (t, 9.0) | 3.389 (t, 8.6) | 3.349 (t, 9.0) | 3.388 (t, 9.5) | 3.41 (t, 9.1, 9.1) |
| 5′ | 3.336 (overlappede) | 3.436~3.406 (m) | 3.330 (overlappede) | 3.500~3.465 (m) | 3.54–3.48 (m) |
| 6′ | 3.662 (dd, 11.8, 5.2) | 3.638 (dd, 12.4, 5.2) | 3.667 (dd, 11.8, 5.1) | 3.708 (dd, 12.4, 5.5) | 3.74 (dd, 12.3, 5.7) |
| 3.846 (dd, 11.8, 1.6) | 3.788 (dd, 12.4, 2.1) | 3.860 (dd, 11.8, 1.4) | 3.897 (dd, 12.4, 2.2) | 3.92 (br d, 12.3) | |
| 2″ | 6.385 (d, 15.9) | 6.276 (d, 16.0) | 5.858 (d, 12.6) | 5.992 (d, 12.4) | - |
| 3″ | 7.668 (d, 15.9) | 7.570 (d, 16.0) | 6.914 (d, 12.6) | 7.143 (d, 12.4) | - |
| 5″, 9″ | 7.487 (2H, br ddd, 8.7, 2.9, 2.0) | 7.445 (d, 8.6) | 7.616 (2H, br ddd, 8.7, 3.0, 2.1) | 7.471 (d, 8.9) | - |
| 6″, 8″ | 6.804 (2H, br ddd, 8.7, 2.9, 2.0) | 6.875 (d, 8.6) | 6.750 (2H, br ddd, 8.7, 3.0, 2.1) | 6.903 (d, 8.9) | - |
Some proton data were deduced from HMQC.
Data were recorded on 360 MHz in CD3OD. The signals of CD3OD at δ 3.305 ppm were used as reference.
Data were recorded on 500 MHz in D2O. The signal of D2O at δ 4.800 ppm was used as reference.
Data were recorded on 300 MHz in D2O; from Jensen, et al. 2002.41
Signals overlapped with solvent signals.
Table 2.
| pos. | 1a | 2b | 4c |
|---|---|---|---|
| 1 | 96.14 d | 95.05 d | 95.6 d |
| 3 | 153.28 d | 152.50 d | 153.4 d |
| 4 | 112.47 s | 111.57 s | 112.4 s |
| 5 | 35.21 d | 32.40 d | 32.5 d |
| 6 | 31.57 t | 29.67 t | 30.1 t |
| 7 | 37.47 t | 35.61 t | 35.7 t |
| 8 | 81.66 s | 80.37 s | 82.8 s |
| 9 | 46.87 d | 45.66 d | 45.7 d |
| 10 | 70.88 t | 69.55 t | 68.2 t |
| 11 | 170.74 s | 171.25 s | 172 s |
| 1′ | 100.88 d | 98.88 d | 99.4 d |
| 2′ | 74.62 d | 72.72 d | 73.3 d |
| 3′ | 77.88 d | 75.72 d | 76.3 d |
| 4′ | 71.22 d | 70.14 d | 70.1 d |
| 5′ | 78.40 d | 76.38 d | 77.0 d |
| 6′ | 62.49 t | 60.78 d | 61.3 d |
| 1″ | 169.23 s | 169.22 s | - |
| 2″ | 114.89 d | 116.89 d | - |
| 3″ | 147.03 d | 144.32 d | - |
| 4″ | 127.13 s | 127.31 s | - |
| 5″, 9″ | 131.32 d | 131.51 d | - |
| 6″, 8″ | 116.85 d | 115.34 d | - |
| 7″ | 161.39 s | 156.83 s | - |
The primary differences in the NMR data of 1 with 4, in addition to the trans-coumaroyl signals in 1, were that the protons at δH 3.56 (1H, d, J = 17.5 Hz; H-10b) and 3.62 (1H, d, J = 17.5 Hz; H-10a) in 4 were shifted toward lower field to δH 4.182 (1H, d, J = 11.3) and 4.256 (1H, d, J = 11.3 Hz) in 1, and that the corresponding carbon at δC 68.2 (t, C-10) in 4 was also deshielded to δC 70.88 (t) in 1. This evidence indicated that the coumaroyl group in compound 1 was connected to the C-10 hydroxyl function of the iridoid skeleton. Additionally, the shift in the geminal coupling constants for H-10a and H-10b from 17.5 Hz in 4 to 11.3 Hz in 1 suggested that the hydroxyl proton had been replaced by a bulky substituent.
In the HMBC spectrum, correlations of the C-10 methylene protons resonating at δH 4.182 and 4.256 with the quaternary carbon at δC 169.23 (C-1″) were observed. This quaternary carbon was also correlated with two other protons at δH 6.385 and 7.668 (H-2″ and H-3″, respectively). This evidence confirmed that the coumaroyl group must be attached at position 10. Two fragments of m/z 163.0344 and 373.1023 in the negative ion tandem mass spectrum supported this conclusion.
The coupling constants of J1, ′2′ (7.9 Hz), J2′,3′ (9.0 Hz), J3′,4’ and J4′,5′ (9.0 Hz in CD3OD and 8.6 Hz in D2O) indicated that all of these protons are pyranose ring protons in an axial position, confirming that the sugar substituent was glucose. In the ROESY spectrum of 1, the correlations between H-1 and H-1′ and one of two C-7 protons indicated that H-1 is in an α orientation. Both H-5 and H-9 correlated with H-6 (δH 2.174) and with one of the two C-7 protons (δH 1.840~1.726). Moreover, correlations of H-10 (δH 4.182 and 4.256) with H-9 (δH 2.319), H-5 (δH 2.933), and H-7 (δH 1.840~1.716, 2H) were observed. NOE experiments provided evidence that these protons were in β orientations, i.e., 1 had the same relative configuration as 4. Consequently, compound 1 was deduced to be 10-trans-coumaroyl-6,7-dihydromonotropein.
Compound 2 was very similar to compound 1. Comparing the 1H, 13C, and DEPT NMR spectra of 2 with those of 1, the only difference was for the proton and carbon resonances within the coumaroyl moiety. In the 1H NMR spectra, the H-2″ and H-3″ signals were shifted upfield from δH 6.385 and 7.668 in 1 to δH 5.858 and 6.914 in 2, respectively. The coupling constant between H-2″ and H-3″ differed markedly from 15.9 Hz in 1 to 12.6 Hz in 2. Comparing the 13C NMR spectra, the chemical shift of C-2″ was shifted downfield from δC 114.89 in 1 to 116.89 in 2, while the chemical shift of C-3″ moved to higher field from δC 147.03 in 1 to 144.32 in 2. This evidence indicated that compound 2 had a cis-coumaroyl substituent, as opposed to the trans-coumaroyl seen in 1. Therefore, compound 2 was determined to be 10-cis-coumaroyl-6,7-dihydromonotropein.
Additional iridoid compounds are likely to be present in cranberry. A 1HNMR analysis of the parent fraction, from which the two iridoids 1 and 2 were isolated, reveals a total of four pairs of doublets between δH 4.6 and 4.1 with the typical coupling of C-10 acylated geminal protons (J = 11.1 to 11.4 Hz). While 1 and 2 account for one pair of doublets each, two pairs remain, for a possibility of at least two more compounds belonging to the same series of iridoids. At least one compound is highly likely to be a 10-O-substituted iridoid glycoside: a doublet can be clearly seen downfield from the H-10 resonances of both 1 and 2, at δH 4.329 (J = 11.4 Hz); resonances for the second C-10 proton for this third iridoid are likely to be found at around 4.2, overlapping with the resonances of 1 and 2 (likely a J = 11.4 Hz doublet at 4.205). While the low available sample mass limited analysis in the present study, further analysis of this fraction with regard to the presence of additional iridoid glycosides is clearly warranted (Figure 1).
Figure 1.
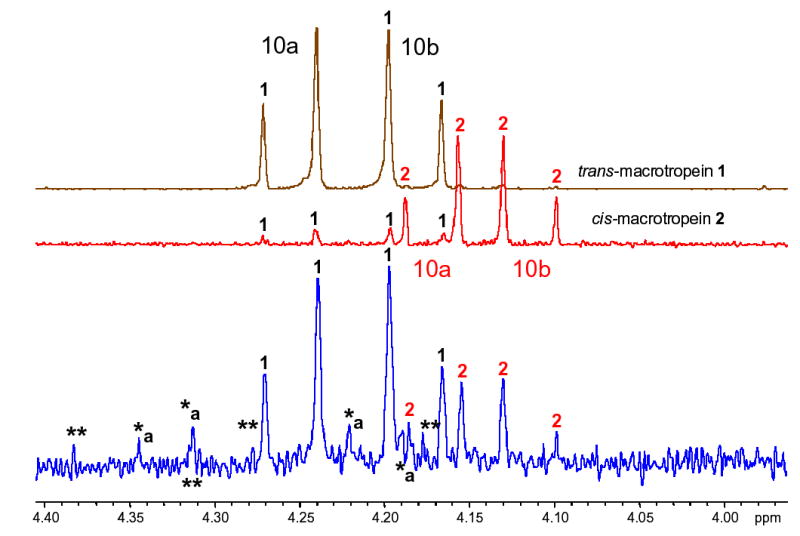
Additional resonances (*/**) in the 1H NMR spectrum of the parent fraction, from which 1 and 2 were isolated, revealed the presence of structurally related minor iridoids, indicated by the presence of pairs of doublets with the typical ~11 Hz geminal coupling of acylated C-10 oxymethylene resonances. E.g., the signals at δ 4.329 and 4.205 (*a) likely belong to a third major monotropein-type iridoid. This indicates that these iridoids are distinct phytochemical markers of the antiadherence active fraction of V. macrocarpon.
Compound 3 was isolated in a yield of 1.7 mg (13 ppm, dry weight) as an amorphous, colorless solid. The high-resolution negative ion electrospray mass spectrum of compound 3 revealed a deprotonated molecule at m/z: 333.0606 [M-H]−, corresponding to a molecular formula of C16H14O8 (calc. 333.0611). The negative ion tandem mass spectrum of compound 3 contained a fragment ion m/z 301, which corresponded to a loss of methanol and indicated the presence of a methoxy group in 3. The IR spectrum showed the characteristic absorption bands corresponding to a hydroxy group, acyl or carboxy groups, and alkenes at 3362, 1684, and 1138 cm−1, respectively. In the 1H NMR (Table 3) and 1H-1H COSY spectra of 3, only two aromatic spin systems were observed at lower field. One was a 1,2,4-trisubstituted ABX spin system with resonances at δH 7.529 (1H, dd, J = 8.0, 2.0 Hz; H-6′), 7.517 (1H, dd, J = 2.0, 0.4 Hz; H-2′), and 6.863 (1H, dd, J = 8.0, 0.4 Hz; H-5′), respectively. The other was a 1,2,3,5-tetrasubstituted AX spin system with resonances at δH 6.251 (1H, d, J = 2.3 Hz; H-3) and 6.135 (1H, d, J = 2.3 Hz; H-5). In addition to the above resonances, a methoxy and a similarly shifted methylene resonance were observed at δH 3.550 (3H, s; OMe) and 3.479 (2H, brs; H-7a, H-7b), respectively. The 13C and DEPT NMR resonances could be ascribed to a 1,2,3,5-tetrasubstituted aromatic ring containing resonances at δC 107.14 (s, C-1), 158.63 (s, C-2), 101.02 (d, C-3), 158.42 (s, C-4), 102.12 (d, C-5), and 152.40 (s, C-6), and to a 1,2,4-trisubstituted aromatic ring with carbons resonating at δC 121.65 (s, C-1′), 117.81 (d, C-2′), 146.45 (s, C-3′), 152.51 (s, C-4′), 116.09 (d, C-5′), and 124.37 (d, C-6′), respectively. In addition, there were two carbonyl resonances at δC 174.34 (s, C-8) and 166.29 (s, C-7′), a methylene at δC 29.89 (t, C-7), and one methoxy resonance at δC 52.33. The HMBC spectrum of 3 exhibited correlations between the respective protons and carbons, as illustrated in Figure 2. Compared to what is typical of a free carboxylic acid,43 C-7′ in 3 was shifted to a higher field, indicating that this is the site of esterification, which, by a process of elimination, must have occurred between the two rings of this depside. However, the C-7′ shift supplied only indirect evidence; additional support for this ester configuration was provided by MS data. The positive ion electrospray tandem mass spectrum contained the dominant fragment ion of m/z 137.0239 (C7H5O3) corresponding to the cleavage of the ester bond (see Figure 2). This ion further fragmented to lose CO, which further supported the assignment of the ester group. The NMR data were consistent with a similar compound recently discovered in Papaver rhoeas petals,43 with the exception of the 8.2 Hz coupling constant reported for H-2′. It is our understanding that this is a typographical error on the part of Hillenbrand et al.,43 because the presence of three ortho coupling constants is inconsistent with their tri- and para-substituted ring, and, additionally, a matching 2.2 Hz meta coupling constant (H-2′ with H-6′) is missing from their data. Compound 3 was thus deduced to be 2-O-(3,4-dihydroxybenzoyl)-2,4,6-trihydroxyphenylmethylacetate (3).42
Table 3.
NMR data of compounds 3 and 5.
| pos. | δH(3)a | δH(5)b | δC(3)a | δC(5)b |
|---|---|---|---|---|
| 1 | - | 107.14 s | 107.7 | |
| 2 | - | 158.63 s | 158.5 | |
| 3 | 6.251 (d, 2.3) | 6.18 (d 2.2) | 101.02 d | 101.2 |
| 4 | - | 158.42 s | 158.6 | |
| 5 | 6.135 (d, 2.3) | 6.27 (d 2.2) | 102.12 d | 102.1 |
| 6 | - | 152.40 s | 152.4 | |
| 7 | 3.479 (brs) | 3.47 s (2H) | 29.89 t | 30.3 |
| 8 | - | 174.34 s | 176.1 | |
| 1′ | - | 121.65 s | 121.8 | |
| 2′ | 7.517 (dd 2.0, 0.4) | 7.55 (d 8.2) | 117.81 d | 117.9 |
| 3′ | - | 146.45 s | 146.4 | |
| 4′ | - | 152.51 s | 152.5 | |
| 5′ | 6.863 (dd 8.0, 0.4) | 6.86 (d 8.2) | 116.09 d | 116.1 |
| 6′ | 7.529 (dd, 8.0, 2.0) | 7.56 (dd, 8.2, 2.2) | 124.37 d | 124.5 |
| 7′ | - | 166.33 s | 166.4 | |
| MeO | 3.550 s | - | 52.33 q | - |
360 MHz for 1H and 90 MHz for 13C in CD3OD
500 MHz for 1H and 125 MHz for 13C in CD3OD, from Hillenbrand et al.42
Figure 2.
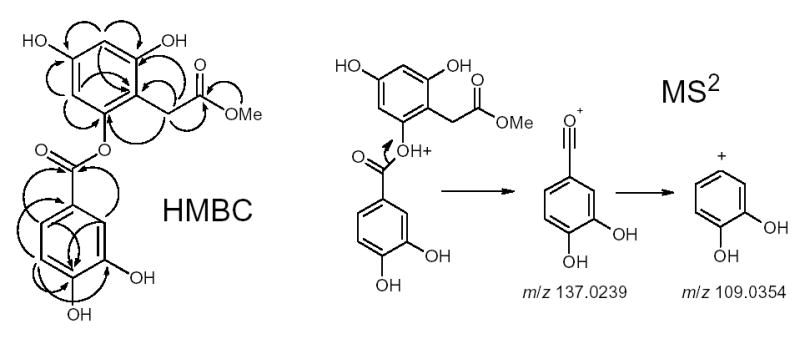
HR-MS fragmentation pattern and HMBC correlations of 2-O-(3,4-dihydroxybenzoyl)-2,4,6-trihydroxyphenylmethylacetate (3).
It should be noted that it is rare to find depsides in organisms that are not lichens,44 although there is evidence that ionizing radiation, such as that used to preserve foods, can break down quercetin into various depsides.45 Whether 3 is in fact a genuine cranberry secondary metabolite, or from some other biological source, related or unrelated to cranberries, deserves further investigation. Considering published evidence,42 however, the methyl ester group in 3 appears unlikely to be due to artifact formation in the presence of methanol/acid.
Experimental Section
General Experimental Procedures
Optical rotations were obtained with a Perkin-Elmer 241 polarimeter (Perkin-Elmer, Inc., MA). 1H and 13C NMR were measured on Bruker Avance 360 and Avance 500 instruments (Bruker, Zürich, Switzerland). Chemical shifts (δ) were expressed in ppm with reference to the deuterated MeOH signals (1H: 3.305, 13C: 49.000 ppm) or D2O (4.8000 ppm). The digital resolution was always better than 0.1 Hz equivalent to 0.0002 ppm (e.g., 32K real datapoints, 8 ppm spectral width for 1H NMR) in the 1H and 1.2 Hz equivalent to 0.008 ppm (32K real data points, 250 ppm spectral width) in the 13C domain. HR-ESI MS data were recorded on a Micromass (Manchester, UK) Q-TOF-2 system. Infrared spectra were recorded from a thin film on a germanium ATR unit (JASCO FT/IR-410, JASCO Inc., MD). Thin-layer chromatography was performed on pre-coated TLC plates (250 μm thickness, KGF Si gel 60 and KGF RP-18 Si gel 60, EM Science, Germany, or 200 μm ALUGRAM® SIL G/UV254, Macherey-Nagel, Germany) with compounds visualized by spraying the dried plates with 5% H2SO4 in EtOH or 2% anisaldehyde and 5% H2SO4 in EtOH, followed by heating. Semipreparative HPLC was carried out on a Waters Delta 600 system with a Waters 996 photodiode array detector, Waters 717 plus autosampler, and Millennium32 Chromatography Manager (Waters Co., MA) on a GROM-Sil 120 ODS-4 HE (Watrex-International, Inc., CA) semipreparative column (5 μm, 300 × 20 mm) with a flow rate of 6 mL/min. Diaion® HP-20 (Supelco Co., PA) and lipophilic Sephadex LH-20 (25–100 μm, Sigma Chemical Co., MO) were used for column chromatography. A High Speed Countercurrent Chromatography system (Pharma-Tech CCC-1000, Pharma-Tech, MD) was also used for fractionation. All solvents were HPLC grade (Fisher Scientific Co., Hanover Park, IL).
Biological Activity
Fractionation was guided by an assay that measured inhibition of adherence of uropathogenic E. coli to human uroepithelial cells; for experimental details see our recent report.40 In brief, p-fimbriated E. coli were grown on CFA agar to promote fimbrial growth. Human T-24 cells were grown to confluence in 96-well microplates. Substances to be tested were briefly incubated with E. coli, and then poured over the T-24 cells, and incubated at 35 °C for one hour. Unadhered bacteria were carefully rinsed away, and new media was added to wells. Adherent bacteria were allowed to multiply for four to six hours, and detected with a microplate reader. Differences in initial quantities of adhered bacteria, compared with solvent controls, were calculated using a standard curve.
Botanical Source
Cranberry juice concentrate was obtained from Ocean Spray, Inc., in two separate lots (lot numbers T073101 and T111802). Because juice does not contain recognizable genetic material or cellular matter, botanical identification was not possible. An HPLC chromatogram for each lot, with peak matching for several cranberry metabolites, is available; see Supporting Information.
Fractionation and Isolation
Briefly, fractionation of cranberry juice consisted of four levels: initially following the general guidelines of Kandil et al.,46 the first column was HP-20 reversed phase resin. HP-20 active fractions were secondarily separated with high-speed countercurrent chromatography (HSCCC, conditions see below), followed, third, by a 4-meter Sephadex LH-20 column, and the final fourth-level fractionation was accomplished on a semipreparative HPLC reverse-phase column (Figure 3). Bacterial antiadherence activity-guided fractionation was performed through the first three levels. The overall method was specifically designed to eliminate benzoic acid early in the fractionation scheme; this method primarily follows bioactivity, and differs from more traditional schemes developed for the targeted isolation of polyphenolic compounds.
Figure 3.
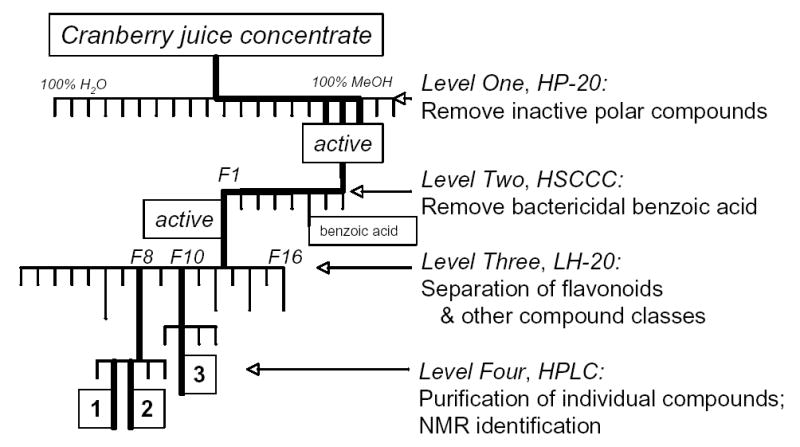
Bioactivity-guided fractionation of cranberry juice, leading to isolation of three compounds (1–3). Bold lines follow activity through the first three levels of fractionation. Longer lines from level three indicate further separation on HPLC.
First Level Fractionation
Two lots of cranberry juice concentrate, 31.8 kg total including H2O content (approximately 6.4 kg dry weight, processed in two runs, approximately 12 kg and 19 kg, respectively), were adjusted on ice from pH 2.4 to approximately pH 5.5 with 28% ammonia (Fisher), and were fractionated over 2.25 kg Dianon® HP-20 (Supelco/Sigma) that had been conditioned with deionized H2O. Juice was allowed to flow through the column, which retained all colored anthocyanins. Effluent of this first fraction contained 95% of the mass (dry weight). The subsequent mobile phase sequence consisted of 100% deionized H2O (10 and 14 L, first and second run, respectively), followed by 20% MeOH in deionized H2O (8 and 9 L, resp.), then 50% MeOH (11 and 19 L, resp.), and finally 100% MeOH (11 and 13 L, resp.). Transitions in MeOH concentration were accomplished with a short gradient of approximately one liter total volume. One-liter fractions were collected throughout and tested for antiadherent activity. Antiadherent fraction yield, from 100% MeOH fractions, was 34.6 g.
Second Level Fractionation
Because the above active HP-20 first level fractions contained large quantities of bactericidal benzoic acid, which interfered with antiadherence assessment, HSCCC separation on a Pharma-Tech CCC-1000 system was next used. 12.9 g of the active fraction from level one was separated using a solvent system of CHCl3-MeOH-H2O 10:7:5, flow rate of 1.5 mL/min, UV detection (T2H mode, upper phase mobile). The resulting fractions were recombined per TLC into 7 fractions. The first of these 7 fractions, collected in 10 runs averaging 62.5 mL solvent each (total 625 mL), with a yield of 5.05 g dry weight, contained antiadherent activity but no benzoic acid.
Third Level Fractionation
The first HSCCC fraction (5.05 g, KD<0.2), containing all visible anthocyanins but no benzoic acid, was then fractionated over a four-meter Sephadex LH-20 column (300 mm × 25 mm ID pre-column; main column 4 m × 10 mm ID, 108 g total LH-20; isocratic 100% MeOH mobile phase) in six injections of approximately 800 mg each (300 fractions at 8 mL each collected for each injection). Fractions obtained were recombined into 16 fractions per TLC.
Fourth Level Fractionation
Fractions 6, 8–12, 14, and 16 from level three were further separated on a semiprep HPLC reversed phase column (Grom, SIL-120 ODS-4 HE, 300 × 20 mm, 5 um) at 5 mL/min, with a mobile phase consisting of a mixture of MeOH (solvent A) and H2O containing 0.1% TFA (solvent B). Compounds 1 and 2 were isolated using a gradient of 50:50 to 76:24 A:B over 35 min. Retention time for 1 was 23.6 min (6.8 mg), and retention time for 2 was 28.4 min (1.9 mg). For the isolation of compound 3, an isocratic method (55:45 A:B) was used. Retention time for 3 was 23.3 min (1.7 mg).
10-p-trans-coumaroyl-1S-dihydromonotropein (1):colorless, amorphous solid; [α]25D −44.4 (c 0.20, MeOH); UV(MeOH/H2O) λmax 215, 230, 310; IR (Ge ATR) νmax 3308, 1684 br, 1634, 1604, 1515, 1169 and 1074 cm−1; 1H NMR and 13C NMR see Tables 1 and 2; HR-ESIMS m/z 537.1629 (calcd for [C25H30O13-H]−, 537.1608)
10-p-cis-coumaroyl-1S-dihydromonotropein (2):colorless, amorphous solid; [α]25D +37.8 (c 0.10, MeOH); UV(MeOH/H2O) λmax 215, 231, 314; IR (Ge ATR) νmax 3308, 1695, 1684, 1558, 1507, 1457, 1188, 1160, and 1071 cm−1; 1H NMR and 13C NMR see Tables 1 and 2; HR-ESIMS m/z 537.1649 (calcd for [C25H30O13-H]−, 537.1608)
2-O-(3,4-dihydroxybenzoyl)-2,4,6-trihydroxyphenylmethylacetate (3):colorless, amorphous solid; UV(MeOH/H2O) λmax 215, 230, 315; IR (Ge ATR) νmax 3308, 1717, 1700, 1684, 1653, 1560, 1295, 1191, and 1138 cm−1; 1H NMR and 13C NMR see Tables 1 and 2; HR-ESIMS m/z 333.0606 (calcd for [C16H14O8-H]−, 333.0611).
Supporting Information
Acknowledgments
Research was supported by an NRSA Fellowship for Allison Turner (F3 AT00623) from the National Center for Complementary and Alternative Medicine (NCCAM), and by grant number P50 AT00155 from the National Center for Complementary and Alternative Medicine (NCCAM), the Office of Dietary Supplements (ODS), the Office for Research on Women’s Health (ORWH), and the National Institute of General Medical Sciences (NIGMS). The contents are solely the responsibility of the authors and do not necessarily represent official views of NCCAM, ODS, ORWH or NIGMS. We thank Ocean Spray Cranberries, Inc. for the cranberry juice concentrate used in this study, and for an unrestricted gift.
Footnotes
Supporting Information. HPLC chromatograms of the two lots of cranberry juice used in this research, including retention time for known cranberry compounds ideain chloride, resveratrol, and benzoic acid, are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bennett J, Brown CM. J Am Pharm Assoc. 2000;40:353–358. doi: 10.1016/s1086-5802(16)31082-8. [DOI] [PubMed] [Google Scholar]
- 2.Blumenthal M, Brinckmann J, Wollschlaeger B. The ABC Clinical Guide to Herbs. 1. American Botanical Council; Austin, Tex: 2003. p. 480. [Google Scholar]
- 3.Bodel PT, Cotran R, Kass EH. J Lab Clin Med. 1959;54:881–888. [PubMed] [Google Scholar]
- 4.Avorn J, Monane M, Gurwitz JH, Glynn RJ, Choodnovskiy I, Lipsitz LA. J Am Med Assoc. 1994;271:751–754. doi: 10.1001/jama.1994.03510340041031. [DOI] [PubMed] [Google Scholar]
- 5.Dignam RR, Ahmed M, Kelly KG, Denman SJ, Zayon M, Kleban M. Ann Long-Term Care. 1998;6:163–167. [Google Scholar]
- 6.Gibson L, Pike L, Kilbourn JP. J Naturopathic Med. 1991;2:45–47. [Google Scholar]
- 7.Haverkorn MJ, Mandigers J. J Am Med Assoc. 1994;272:590. doi: 10.1001/jama.272.8.590a. [DOI] [PubMed] [Google Scholar]
- 8.Howell AB, Foxman B. J Am Med Assoc. 2002;287:3082–3083. doi: 10.1001/jama.287.23.3082. [DOI] [PubMed] [Google Scholar]
- 9.Papas PN, Brusch CA, Ceresia GC. Southwest Med. 1966;47:17–20. [PubMed] [Google Scholar]
- 10.Stothers L. Can J Urol. 2002;9:1558–1562. [PubMed] [Google Scholar]
- 11.Walker EB, Barney DP, Mickelsen JN, Walton RJ, Mickelsen RA., Jr J Fam Pract. 1997;45:167–168. [PubMed] [Google Scholar]
- 12.Foda MM, Middlebrook PF, Gatfield CT, Potvin G, Wells G, Schillinger JF. Can J Urol. 1995;2:98–102. [PubMed] [Google Scholar]
- 13.Schlager TA, Anderson S, Trudell J, Hendley JO. J Pediatr. 1999;135:698–702. doi: 10.1016/s0022-3476(99)70087-9. [DOI] [PubMed] [Google Scholar]
- 14.Kinney AB, Blount M. Nurs Res. 1979;28:287–290. [PubMed] [Google Scholar]
- 15.Schultz A. J Community Health Nurs. 1984;1:159–169. doi: 10.1207/s15327655jchn0103_5. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt DR, Sobota AE. Microbios. 1988;55:173–181. [PubMed] [Google Scholar]
- 17.Sobota AE. J Urol. 1984;131:1013–1016. doi: 10.1016/s0022-5347(17)50751-x. [DOI] [PubMed] [Google Scholar]
- 18.Foo LY, Lu Y, Howell AB, Vorsa N. J Nat Prod. 2000;63:1225–1228. doi: 10.1021/np000128u. [DOI] [PubMed] [Google Scholar]
- 19.Habash MB, Van der Mei HC, Busscher HJ, Reid G. Can J Microbiol. 1999;45:691–694. doi: 10.1139/w99-065. [DOI] [PubMed] [Google Scholar]
- 20.Howell AB, Vorsa N, Der Marderosian A, Foo LY. N Engl J Med. 1998;339:1085–1086. doi: 10.1056/NEJM199810083391516. [DOI] [PubMed] [Google Scholar]
- 21.Ahuja S, Kaack B, Roberts J. J Urol. 1998;159:559–562. doi: 10.1016/s0022-5347(01)63983-1. [DOI] [PubMed] [Google Scholar]
- 22.Zafriri D, Ofek I, Adar R, Pocino M, Sharon N. Antimicrob Agents Chemother. 1989;33:92–98. doi: 10.1128/aac.33.1.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foo LY, Lu Y, Howell AB, Vorsa N. Phytochemistry. 2000;54:173–181. doi: 10.1016/s0031-9422(99)00573-7. [DOI] [PubMed] [Google Scholar]
- 24.Reid G. World J Urol. 1999;17:359–363. doi: 10.1007/s003450050161. [DOI] [PubMed] [Google Scholar]
- 25.Tsang C, Auger C, Mullen W, Bornet A, Rouanet JM, Crozier A, Teissedre PL. Br J Nutr. 2005;94:170–181. doi: 10.1079/bjn20051480. [DOI] [PubMed] [Google Scholar]
- 26.Baba S, Osakabe N, Natsume M, Terao J. Free Radic Biol Med. 2002;33:142–148. doi: 10.1016/s0891-5849(02)00871-7. [DOI] [PubMed] [Google Scholar]
- 27.Deprez S, Brezillon C, Rabot S, Philippe C, Mila I, Lapierre C, Scalbert A. J Nutr. 2000;130:2733–2738. doi: 10.1093/jn/130.11.2733. [DOI] [PubMed] [Google Scholar]
- 28.Deprez S, Mila I, Huneau JF, Tome D, Scalbert A. Antioxid Redox Signal. 2001;3:957–967. doi: 10.1089/152308601317203503. [DOI] [PubMed] [Google Scholar]
- 29.Donovan JL, Manach C, Rios L, Morand C, Scalbert A, Remesy C. Br J Nutr. 2002;87:299–306. doi: 10.1079/bjnbjn2001517. [DOI] [PubMed] [Google Scholar]
- 30.Downs CT, McDonald PM, Brown K, Ward D. J Chem Ecol. 2003;29:845–858. doi: 10.1023/a:1022975531372. [DOI] [PubMed] [Google Scholar]
- 31.Duweler KG, Rohdewald P. Pharmazie. 2000;55:364–368. [PubMed] [Google Scholar]
- 32.Gonthier M-P, Donovan Jennifer L, Texier O, Felgines C, Remesy C, Scalbert A. Free Radical Biol Med. 2003;35:837–844. doi: 10.1016/s0891-5849(03)00394-0. [DOI] [PubMed] [Google Scholar]
- 33.Holt RR, Lazarus SA, Sullards MC, Zhu QY, Schramm DD, Hammerstone JF, Fraga CG, Schmitz HH, Keen CL. Am J Clin Nutr. 2002;76:798–804. doi: 10.1093/ajcn/76.4.798. [DOI] [PubMed] [Google Scholar]
- 34.Jimenez-Ramsey LM, Rogler JC, Housley TL, Butler LG, Elkin RG. J Agric Food Chem. 1994;42:963–967. [Google Scholar]
- 35.Nakamura Y, Tonogai Y. J Agric Food Chem. 2003;51:7215–7225. doi: 10.1021/jf030250+. [DOI] [PubMed] [Google Scholar]
- 36.Spencer JP, Chaudry F, Pannala AS, Srai SK, Debnam E, Rice-Evans C. Biochem Biophys Res Comm. 2000;272:236–241. doi: 10.1006/bbrc.2000.2749. [DOI] [PubMed] [Google Scholar]
- 37.Spencer JPE, Schroeter H, Shenoy B, Srai SKS, Debnam ES, Rice-Evans C. Biochem Biophys Res Comm. 2001;285:588–593. doi: 10.1006/bbrc.2001.5211. [DOI] [PubMed] [Google Scholar]
- 38.Howell AB, Leahy MM, Kurowska E, Guthrie N. FASEB J. 2001;15:A284. [Google Scholar]
- 39.Vattem DA, Jang HD, Levin R, Shetty K. J Food Biochem. 2006;30:98–116. [Google Scholar]
- 40.Turner A, Chen SN, Joike MK, Pendland SL, Pauli GF, Farnsworth NR. J Agric Food Chem. 2005;53:8940–8947. doi: 10.1021/jf052035u. [DOI] [PubMed] [Google Scholar]
- 41.Jensen HD, Krogfelt KA, Cornett C, Hansen SH, Christensen SB. J Agric Food Chem. 2002;50:6871–6874. doi: 10.1021/jf0205110. [DOI] [PubMed] [Google Scholar]
- 42.Reynertson KA, Wallace AM, Adachi S, Gil RR, Yang H, Basile MJ, D'Armiento J, Weinstein IB, Kennelly EJ. J Nat Prod. 2006;69:1228–30. doi: 10.1021/np0600999. While this paper was under review, the discovery of the same compound, named jaboticabin, was published by. [DOI] [PubMed] [Google Scholar]
- 43.Hillenbrand M, Zapp J, Becker H. Planta Med. 2004;70:380–382. doi: 10.1055/s-2004-818956. [DOI] [PubMed] [Google Scholar]
- 44.Miao V, Coeffet-LeGal MF, Brown D, Sinnemann S, Donaldson G, Davies J. Trends Biotechnol. 2001;19:349–355. doi: 10.1016/s0167-7799(01)01700-0. [DOI] [PubMed] [Google Scholar]
- 45.Marfak A, Trouillas P, Allais DP, Champavier Y, Calliste CA, Duroux JL. J Agric Food Chem. 2002;50:4827–4833. doi: 10.1021/jf020165m. [DOI] [PubMed] [Google Scholar]
- 46.Kandil FE, Smith MAL, Rogers RB, Pepin MF, Song LL, Pezzuto JM, Seigler DS. J Agric Food Chem. 2002;50:1063–1069. doi: 10.1021/jf011136z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.