

Abstract
Alu elements are the most active and predominant type of short interspersed elements (SINEs) in the human genome. Recently inserted polymorphic (for presence/absence) Alu elements contribute to genome diversity among different human populations, and they are useful genetic markers for population genetic studies. The objective of this study is to identify polymorphic Alu insertions through an in silico comparative genomics approach and to analyze their distribution pattern throughout the human genome. By computationally comparing the public and Celera sequence assemblies of the human genome, we identified a total of 800 polymorphic Alu elements. We used polymerase chain reaction-based assays to screen a randomly selected set of 16 of these 800 Alu insertion polymorphisms using a human diversity panel to demonstrate the efficiency of our approach. Based on sequence analysis of the 800 Alu polymorphisms, we report three new Alu subfamilies, Ya3, Ya4b, and Yb11, with Yb11 being the smallest known Alu subfamily. Analysis of retrotransposition activity revealed Yb11, Ya8, Ya5, Yb9, and Yb8 as the most active Alu subfamilies and the maintenance of a very low level of retrotransposition activity or recent gene conversion events involving S subfamilies. The 800 polymorphic Alu insertions are characterized by the presence of target site duplications (TSDs) and longer than average polyA-tail length. Their pre-integration sites largely follow an extended “NT-AARA” motif. Among chromosomes, the density of Alu insertion polymorphisms is positively correlated with the Alu-site availability and is inversely correlated with the densities of older Alu elements and genes.
Keywords: Retrotransposition, Polymorphism, Bioinformatics, Comparative genomics
1. Introduction
Alu elements are the predominant type of short interspersed elements (SINEs) in the human genome, with over 1 million copies comprising ~10% of the total genome (Houck et al., 1979; Lander et al., 2001; Venter et al., 2001). The origin and amplification of Alu elements are evolutionarily recent events that coincided with the radiation of primates (Batzer and Deininger, 2002; Kapitonov and Jurka, 1996; Quentin, 1988; Shaikh and Deininger, 1996). Alu elements increase in number by retrotransposition, a process involving the insertion of reverse transcribed DNAs of Alu-derived transcripts back into the genome, apparently by hijacking the L1 retrotranspotion machinery (Boeke, 1997; Cost and Boeke, 1998; Dewannieux et al., 2003). Based on a hierarchical series of sequence mutations, Alu elements are classified into three major families designated as J, S, and Y, representing the oldest, intermediate, and youngest Alu sequences, respectively, and each of these families is further divided into one or more levels of subfamilies based on subfamily-specific diagnostic mutations (Batzer et al., 1990, 1996b; Jurka and Smith, 1988). It is estimated that approximately 5000 young Alu elements are specific to humans (Batzer and Deininger, 1991). Among these young Alu elements, ~25% have inserted so recently that they are polymorphic among different human population groups, families, or even individuals with respect to their presence or absence in the genome (Batzer and Deininger, 2002).
Because Alu insertions are unique events that are identical-by-descent or free of homoplasy, they have been useful in genetic mapping and population genetics studies (Batzer et al., 1994; Batzer and Deininger, 1991; Perna et al., 1992; Roy-Engel et al., 2001; Salem et al., 2003, 2005a; Stoneking et al., 1997; Tishkoff et al., 2000). In addition, Alu elements are known to impact several aspects of the genome. For example, Alu insertions provide the evolutionary potential to enhance the coding capacity and versatility of the genome by creating novel proteins via insertion into coding regions or by creating alternatively spliced exons (Lev-Maor et al., 2003; Makalowski et al., 1994; Sorek et al., 2002). De novo Alu insertions can cause genetic diseases by insertion-mediated interruption of gene structures (Deininger and Batzer, 1999; Ganguly et al., 2003; Wallace et al., 1991).
Using various methodologies, over 1000 Alu insertions have been identified as polymorphic among diverse human populations. Earlier studies using genomic library screening with probes/primers specific for young Alu elements contributed to the discovery of a small number of polymorphisms (Arcot et al., 1995; Batzer et al., 1995; Batzer and Deininger, 1991; Roy et al., 1999). With the availability of the human genome sequence, a new and more fruitful approach was developed. Using this strategy, Alu elements belonging to young subfamilies were identified by computational sequence analysis, and oligonucleotide primers were designed based on the flanking regions for polymerase PCR-based assays to ascertain the polymorphism status of these candidates by screening DNA samples from diverse human populations. The first study using such a strategy identified 106 polymorphic Alu insertions out of 475 Ya5 and Yb8 insertions (Carroll et al., 2001). Subsequently, this method has been extensively used to analyze almost all Y subfamilies including Ya (Otieno et al., 2004), Yb (Carter et al., 2004; Roy-Engel et al., 2001), Yc (Roy-Engel et al., 2001; Garber et al., 2005), Yd (Xing et al., 2003), Yg and Yi (Salem et al., 2003), Ye (Salem et al., 2005b) and multiple Y subfamily members on the X chromosome (Callinan et al., 2003). These studies are responsible for the identification of the majority of the known polymorphic Alu insertions.
However, the search for polymorphisms using this strategy has so far been limited to the public version of the human genome sequence. In addition, the selection of candidate polymorphisms is biased towards certain relatively small and young subfamilies for which the numbers of candidates are manageable for PCR assays. Therefore, the currently identified polymorphic elements likely represent a partial list of all potential polymorphic Alu insertions that exist in current human populations. In fact, a very recently study that utilized the human trace genomic sequences representing different human individuals revealed a high proportion of new polymorphic Alu loci (Bennett et al., 2004). In this study, we developed an in silico comparative genomics approach for comparing the public and Celera versions of human genome sequences and identified several hundred new Alu insertion polymorphisms. Our data represents the largest set of polymorphic Alu insertions identified by a single study to date.
2. Materials and methods
2.1. Sources for genomic sequences
The human genomic sequence data used in this study are the public version (Lander et al., 2001) obtained from the UCSC site (April 12, 2003 freeze or hg15) at http://genome.ucsc.edu and the Celera version from the Celera Discovery System (August 2003 version) through private database subscription (http://cds.celera.com, Venter et al., 2001). The Celera sequences represent unconnected scaffolds grouped by chromosome. We also retrieved the Celera whole genome shotgun assembly (WGSA) sequences from GenBank (accessions AADD01000001-AADD01211493, Istrail et al., 2004). All sequences in fasta format were downloaded onto our local bioinformatics server for analyses.
2.2. In silico identification of Alu insertion polymorphisms
To identify polymorphic Alu insertions between the two human genome sequences (public human genome sequence, PHGS and the Celera human genome sequence, CHGS), we developed a strategy as illustrated in Fig. 1. Briefly, all Alu elements in both genome sequences plus 100 bp flanking sequences on both sides were identified by querying the genomic sequences with the Alu consensus sequences using a locally installed basic local alignment search tool (BLAST) program (Altschul et al., 1997). Each of these sequences from PHGS was used to query the corresponding chromosome in CHGS. If a perfect or close to perfect match at full length (Alu element plus flanking sequences with length ≥98% and identity ≥98%) is found, the Alu insertion is considered to be shared between the two genomes and is excluded from further analysis. Otherwise, if the best match is limited to only the Alu or flanking regions, indicating that there is no full-length match, the Alu insertion is considered to be a candidate for being polymorphic and its sequence is subject to another search.
Fig. 1.
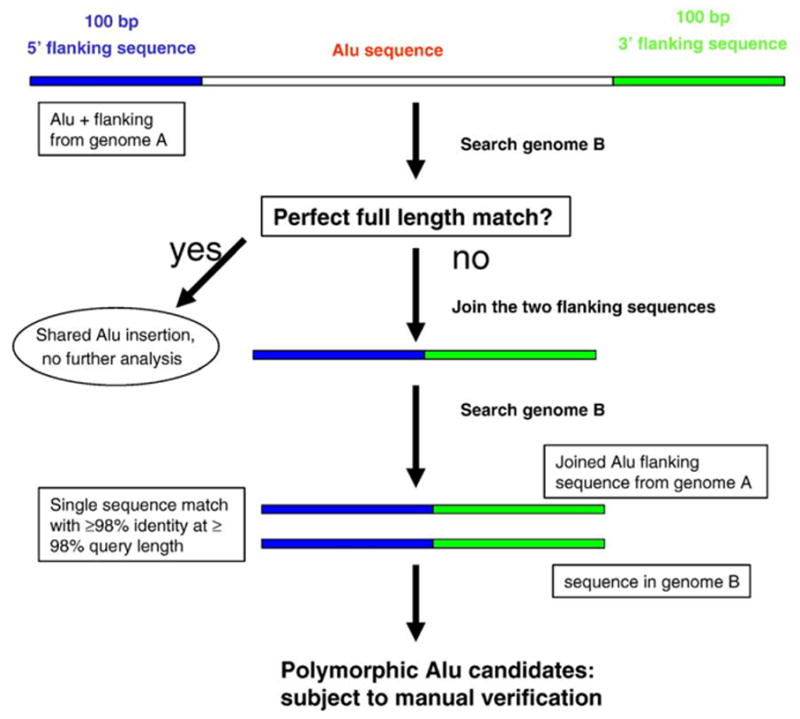
Strategy for detection of Alu insertion polymorphisms using computational comparative genomics. The sequence of an Alu plus 100 bp flanking sequences on both ends from genome A is used to query genome B. If no perfect full-length match is found, then the two flanking sequences are joined (along with the removal of one copy of the TSDs) to query genome B again. If only a single perfect full-length match is found, then this Alu insertion in genome A is considered to be absent from genome B and thus polymorphic.
In the second search, the two flanking sequences of the Alu element are joined and used to query CHGS. If we find only one perfect or close-to-perfect match, then this Alu is considered to be absent in CHGS, i.e. it is polymorphic between the two genomes. Thus, we were able to identify Alu loci that are present in the PHGS, but absent from the CHGS. The procedure is then repeated by exchanging the positions of the two genomes to identify Alu elements that are present in CHGS but absent from PHGS. All polymorphic loci identified through this automatic computer procedure were subjected to manual verification. For an Alu insertion to be considered polymorphic, we required both the existence of a unique perfect match to the joined flanking sequence (with the removal of one copy of the target site duplication) and the absence of the Alu element from the other genome at that specific locus.
2.3. Updated assignment of Alu subfamilies and analysis of Alu sequences
Accurate subfamily assignment for severely truncated Alu elements is not possible. Therefore, we only included elements that have 50 bp or longer non-polyA sequences from the J, S, and Y subfamilies. A total of 1050448 Alu elements identified from the PHGS were used as the starting point for all analyses. Since we noticed that the Alu subfamily assignments annotated in the University of California at Santa Cruz (UCSC) data set based on RepeatMasker do not cover many newer Y Alu lineages, we re-classified the Y subfamilies based on an updated set of consensus sequences for Yg6, Yh9, Yi6, Yj, Ye, Yf1, and Yx. The subfamilies Ya3, Ya4b, and Yb11 were newly characterized in this report. For each Alu sequence, the subfamily assignment was made based on the consensus that gave the highest unique BLAST bit score. For cases in which multiple consensus sequences gave the same best score, the assignment was made based on the consensus sequence representing an older and more general subfamily, such as AluY. To ensure the accuracy of the assignments, we included only the Alu elements that were at least 200 bp long (excluding the polyA-tail) unless they contain sufficient number of subfamily-specific diagnostic mutations, and all remaining identifiable Y elements were placed in the general Y subfamily. This assignment process was performed with the use of a set of PERL scripts. A PERL-based program was also developed to systematically identify the exact start and end positions of Alu elements, as well as the positions of polyA-tails and target site duplications (TSDs). The TSDs are identified as the longest sequence repeats with one copy ending before the start of the Alu and the other copy starting from the 3′ end of the polyA-tail. All of the PERL scripts are available from the authors upon request.
2.4. Experimental verification of Alu insertion polymorphisms
To evaluate the specificity of our in silico method, we randomly selected 16 Alu elements from the 65 polymorphisms identified for chromosome 6, one of which is identical to a published polymorphic insertion, Ya5NBC54 (Watkins et al., 2003). The 16 Alu loci were screened for their presence/absence (Alu+ vs. Alu−) using a panel of 95 individuals originating from Africa (Biaka Pygmies from Cameroon, Burunge from Tanzania), the Middle East, Europe (Northern Europe, Russia), Asia (Chinese, Japanese, Southeast Asia) and the Americas (Mexican Indian, Mayan). Samples from Africans were collected by S.A.T. with informed consent. All other samples were obtained from the Coriell Institute Human Diversity Panels. We designed oligonucleotide primers for PCR of each locus in Alu flanking sequences such that that the amplified product ranged from 100 to 200 bp for Alu− alleles and 400 to 600 bp for Alu+ alleles. Alu loci were genotyped by amplification of 50 ng of genomic DNA in a standard 35-cycle, three-step PCR. The genotypes (+/+, +/−, and −/−) for each locus were recorded and used to calculate the Alu allele frequency and observed heterozygosity within each population. Supplemental Table S1 lists the oligonucleotide primers, annealing temperatures and amplicon fragment sizes for each locus.
2.5. Supplemental data
Online supplemental data is available on our web sites at http://falcon.roswellpark.org/publication/Liang/pAlus/ or http://batzerlab.lsu.edu under publications.
3. Results
3.1. Identification of Alu insertion polymorphisms
By comparing the sequences of PHGS and CHGS, we identified 850 potentially polymorphic Alu loci. Of these, 800 insertions satisfied our criteria of polymorphism. Five hundred sixty-four were identified in PHGS (absent in CHGS) and the remaining 236 were found in CHGS (absent in PHGS). Fig. 2 shows two examples of newly identified polymorphic Alu insertions. From the 236 polymorphic insertions identified in CHGS, we identified 190 from the Celera whole genome shotgun assembly (WGSA) sequences that were obtained exclusively using the Celera proprietary whole genome shotgun sequences (Istrail et al., 2004). The remaining 46 CHGS polymorphic elements were likely from the public human BAC sequences that may have not been used for the assembly of PHGS. To determine how many of the 800 polymorphic Alu elements had been previously identified and published, we compiled a database containing 1051 non-redundant polymorphic Alu insertions (http://falcon.roswellpark.org:9090; Wang et al., in press) from the over 1500 polymorphisms reported in the literature (Batzer et al., 1994, 1995; Bennett et al., 2004; Callinan et al., 2003; Carroll et al., 2001; Carter et al., 2004; Garber et al., 2005; Mamedov et al., 2005; Otieno et al., 2004; Ray et al., 2005; Roy-Engel et al., 2001; Watkins et al., 2003; Xing et al., 2003). A comparison of the genomic locations of our 800 polymorphic Alu insertions with the list revealed that only 266 of the 800 Alu elements correspond with previously reported Alu repeats, suggesting that the remaining 534 insertions represent newly identified polymorphisms. Among the 236 polymorphic insertions identified from CHGS, only 16 (less than 7%) are on the list, indicating that a substantial number of new Alu polymorphisms can be identified from a new genome sequence.
Fig. 2.

Representative examples of newly identified Alu insertion polymorphisms. In this figure, we show the sequences of two new Alu insertion polymorphisms identified in this study using computational comparative genomics. (Panel A) A Yb8 Alu element present in the CHGS but absent from the PHGS. (Panel B) A Ya5 Alu present in the PHGS, but absent from the CHGS (Panel B). The first fasta sequence in each panel shows an Alu sequence (lower case) plus its flanking sequences on both sides (highlights in upper case). The second sequence in each panel represents the pre-integration site that perfectly matches to the joined flanking sequences of the Alu insertions with one copy of the TSDs (double underlined) removed. The chromosome number, genome version, base position in the chromosome (scaffold for Celera), and the Alu subfamilies are indicated in the definition line of each fasta sequence.
3.2. Characterization of three new Alu subfamilies: Ya3, Ya4b, and Yb11
Detailed sequence alignment analysis of the 800 polymorphic loci revealed several distinct classes of Alu. These include two new AluYa subfamilies and one new Yb subfamilies. Following the nomenclature proposed by Batzer et al. (1996a), we designated them as Ya3, Ya4b, and Yb11. Among the 800 polymorphic insertions identified in this study, the copy numbers for Ya3, Ya4b, and Yb11 elements were 7, 22, and 6, respectively. We identified a total of 2904, 313, and 16 elements, respectively, in the PHGS. This makes the Yb11 subfamily the smallest known Alu subfamily (Table 1). As shown in Fig. 3, Ya3 elements lack the first two of the five Ya5 diagnostic mutations. Ya4b Alu elements lack a diagnostic mutation of Ya5 subfamily elements at a position different from that of the Ya4 elements. Yb11 Alu elements contain all diagnostic mutations of Yb9, plus an additional mutation from “G” to “A” at the last base of the insertion sequence, “CAGTCCG”, which is specific to Yb7-9 subfamilies. In addition, Yb11 Alu elements contain a one-base insertion of “T” not shared by Alu elements from other subfamilies (Fig. 3). Thus, the Yb11 subfamily likely originated from the Yb9 subfamily by first gaining the “G” to “A” mutation and then the “T” insertion. This scenario is supported given that we were able to identify intermediates containing the “G” to “A” mutation without the “T” insertion, but not any intermediates in the opposite situation. A sequence alignment of all Yb11 elements is available in supplemental materials (Supplementary Fig. S2). The availability of these new subfamilies provides us with additional subjects for studying Alu amplification processes and for tracking the evolutionary history of different subfamilies.
Table 1.
Distribution of Alu insertion polymorphisms by subfamily
| Alu subfamily | All insertions a | pAlub | pAlu/1k all elements c |
|---|---|---|---|
| AluJb | 95606 | 0 | 0.0 |
| AluJo | 165935 | 0 | 0.0 |
| AluSc | 45464 | 6 | 0.1 |
| AluSg | 111877 | 6 | 0.1 |
| AluSg1 | 12069 | 2 | 0.2 |
| AluSp | 60021 | 5 | 0.1 |
| AluSq | 130047 | 2 | 0.0 |
| AluSx | 274116 | 6 | 0.0 |
| AluY | 110814 | 53 | 0.5 |
| AluYa1 | 2221 | 10 | 4.5 |
| AluYa3 | 2904 | 6 | 2.1 |
| AluYa4 | 1016 | 39 | 38.4 |
| AluYa4b | 313 | 22 | 70.3 |
| AluYa5 | 2887 | 266 | 92.1 |
| AluYa8 | 58 | 6 | 103.4 |
| AluYb3a1 | 13608 | 10 | 0.7 |
| AluYb3a2 | 1705 | 5 | 2.9 |
| AluYb7 | 277 | 10 | 36.1 |
| AluYb8 | 2296 | 171 | 74.5 |
| AluYb9 | 197 | 22 | 116.8 |
| AluYb11 | 16 | 6 | 375.0 |
| AluYc1 | 4173 | 50 | 12.0 |
| AluYc2 | 3357 | 10 | 3.0 |
| AluYd2 | 2001 | 5 | 2.5 |
| AluYd3 | 643 | 3 | 4.7 |
| AluYd8 | 181 | 7 | 38.7 |
| AluYe4-6 | 1189 | 29 | 24.4 |
| AluYf1 | 3106 | 4 | 1.3 |
| AluYg6 | 775 | 21 | 27.1 |
| AluYh9 | 188 | 4 | 21.3 |
| AluYi6 | 1038 | 8 | 7.7 |
| AluYj | 138 | 4 | 29.0 |
| AluYx | 214 | 1 | 4.7 |
| All AluY | 155313 | 773 | 5.0 |
| Total | 1050448 | 800 | 0.8 |
Based on UCSC April 2003 (hg15) assembly and only Alu elements that have 50 bp or longer non-polyA-tail sequences were included.
pAlu: polymorphic Alu elements.
The number of polymorphic Alu elements in 1000 of all Alu repeats. Some minor groups are merged with their closely related major groups.
Fig. 3.

Multiple alignment of Alu consensus sequences. The figure shows a comparison of consensus sequences for six Y lineage Alu subfamilies including those newly identified in this report. Grey residues indicate the diagnostic mutational differences between each of the new subfamilies and their closely related subfamilies. Dots represent identical bases.
3.3. Subfamily-specific levels of Alu insertion polymorphism
We used the ratio of polymorphic elements to all members in a given subfamily to provide a measurement of Alu insertion activity. As shown in Table 1, the majority (96.8%) of the 800 polymorphic elements belong to the Y lineage, while the remaining 3.2% belong to the older S subfamilies. No polymorphisms belonging to the oldest subfamilies (J) were identified. Among the different Y subfamilies, Yb11, Ya8, Ya5, Yb9, Yb8, and Ya4b represent the most active subfamilies, arranged in order from the most to least active. Of all these very active subfamilies, Ya5 and Yb8 have generated the largest numbers of polymorphic insertions, and together they contribute more than 50% (418) of the 800 polymorphic loci identified in the study. The remaining Y subfamilies, Ya4, Yb7, Yc1, Yd8, Ye, Yg6, Yh9, Yi6, and Yj, show intermediate levels of insertion polymorphism, while the rest of the subfamilies show low or very low levels of Alu insertion activity.
3.4. Sequence architecture of recently integrated Alu repeats
To characterize the sequence features in the Alu integration sites, we extracted the flanking sequences from each side of the first nick sites of the L1 endonuclease (EN) (the dinucleotide between the first base of the 5′ TSD and the base in front) and surveyed their base composition. As shown in Fig. 4, the immediate regions flanking first nick sites (positions −2 to 4) are very distinct from the flanking regions, and they roughly follow a previously identified sequence motif, “TT/AAAA” (Cost and Boeke, 1998; Jurka, 1997). The flanking regions are relatively high in A/T content (as high as 70%), with the A/T content showing a gradual decrease in both directions after 15 bp from the first nick site. Table 2 shows the frequency of all observed Alu insertion sites. Interestingly, all motifs that use the “NT-AARA” pattern, except for the “GT-AAAA” site, have much higher site usage (≥0.90 insert per 105 sites) than the rest, and they also represent the most frequently utilized motifs by the 800 polymorphic Alu insertions. By contrast, sequences that differ from the “NT-AARA” motif all show much lower site usage, despite the fact that some of them, such as “AA-AAAA” and “TA-AAAA”, have relatively high occurrences among the 800 integration sites. We observed a high occurrence of “G” replacing “A” at the second to last position of the “TT/AAAA” motif for an unknown reason, but it is consistent with previous observation by Jurka (1997). Therefore, we propose to extend the Alu integration site to “NT-AARA”. Our data demonstrate that the “TpA” di-nucleotide at the first nick site is preferred (~50%) among the 16 possible di-nucleotides (data not shown), agreeing with previously reported results (Cost and Boeke, 1998). To examine whether or not the preference for integration sites differs among Alu subfamilies, we collected all elements containing identifiable TSDs from the PHGS. Similar patterns of preferred integration sites across different subfamilies were obtained.
Fig. 4.
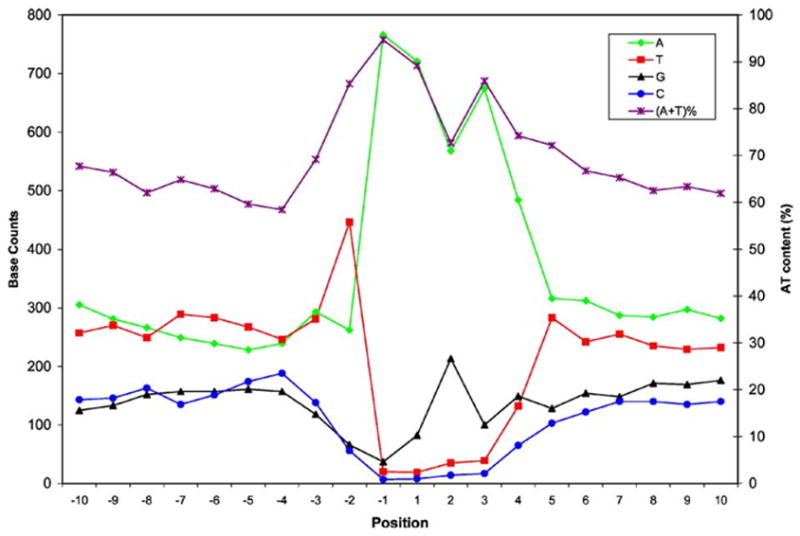
Nucleotide composition of the Alu insertion polymorphism pre-integration sites. Fifteen bp sequences flanking each side of the first nick sites of the 800 polymorphic Alu insertion sites were extracted and the base composition (# of counts on the first Y-axis), as well as the A+T composition (percentage on the second Y-axis) were surveyed for each base position within the 30 bp regions flanking the first nick sites. Base positions − 15 to − 1 represent the 15 bp sequence before the first nick site, while the sequence from positions 1 to 15 represent the genomic sequence starting from the first nick site in the genomic sequence of the Alu− allele.
Table 2.
Motif frequency for the first nick sites for the Alu insertion polymorphisms
| Count a | Motif (number of sites in genome b/site usage c) |
|---|---|
| 78 | ttAAAA (6844770/1.14) |
| 61 | atAAAA (6115727/0.97) |
| 37 | ctAAAA (3427545/1.08) |
| 36 | atAAGA (2040242/1.76) |
| 35 | ttAAGA (2248938/1.55) |
| 32 | aaAAAA (8006008/0.40) taAAAA (7446893/0.43) |
| 23 | gtAAGA (1266992/1.82) |
| 20 | ttAAAG (2636960/0.75) caAAAA(5905656/0.34) |
| 19 | aaAAGA (5553248/0.34) |
| 18 | gtAAAA (2525766/0.58) |
| 17 | (2940505/0.58) ctAAGA (1418507/1.20) |
| 13 | caAAGA (2875842/0.45) |
| 12 | gaAAAA (5451601/0.22) |
| 11 | aaAGAA (6061956/0.18) |
| 10 | taAAAG (3079886/0.32) atAAAG (2801071/0.36) atAGAA (2476795/0.40) tgAAAA (4505577/0.22) |
| 9 | gaAAGA (3257715/0.28) ctAGAA (1925300/0.47)
agAAAA (6851406/0.13) |
| 8 | ctAAAG (1465413/0.48) aaAAAG (5450737/0.15) |
| 7 | taAGAA (2719394/0.26) gaAGAA (3200992/0.22)
atGAAA (3793799/0.18) |
| 6 | ccAAAA (2952060/0.20) tgAAGA (2585632/0.23)
ttAGAA (2603330/0.23) |
| 5 | gaAAAG gtAAAG acAAAA tcAAAA atAATA ttGAAA
gtAGAA |
| 4 | tcAGAA gcAAAG ctAAAT aaAAAT ttAAAT acAAGA
agAAGA tcAAAG tgAAAG ttTAAA caAAAG |
| 3 |
ggAAAA tgAGAA ggAAAG gtAATA ccAAGA atAAAT ggAGAA atAACA tcAAGA ctGAAA aaAATG acAAAG ttAATA acAGAA |
| 2 | aaAAGT gcAAAA gaGAAA gtTAAA taAAAT caAATA
cgAAAA aaATAT ttAACA aaGTTA aaTAAA aaGAAA ggGAAA atTAAA ggAAGA |
| 1 | taAAGC atACAA taAAGG aaATTG cgCTTT ttGGGA
aaAGTT agAGAA aaTGAT acCTTC aaAAAC agGAGA gaGCCC taAAAC acTAAA gtGAAA caAGAA atAAAC tcAATA agAATA aaATGA ttAAAC aaGTCA agGAAA atAGGA atAGGC gcAGAA tgAGCA tcGAAT aaCCAC caAATC gtAAGG aaAACT acAAGC aaAGAG agCTGT agTTGT aaGCAG caGAAA gaAAAC ccAAAT tgGGGG ctAATA aaGGTC atTAGA ctTAAA taTTTA agATTC atAGAT gtAAAC aaAATA ttATAA ttTAGA taAATA aaAATT aaATCA caAAGG agAAAG ccAGAA taGAAA taAATT agAAAT aaATCT aaTTGG gcAAGA ttCAAA atTAAT aaGTGC aaCACA aaAAGC atGCCT ggCCTA agATGT tgTATT aaACAT |
Occurrence of each motif among the 800 polymorphic Alu loci.
The occurrence of the motif in the human genome based on UCSC hg15, with both strands considered. The second and third bases in the motif represent the first nick site by EN. For motif “aaAAAA”, the count in the genome does not include all possibility by shifting 1 bp each time in a run of “A”. Instead, in the case of “A” runs, the count refers to the number of possible shifts by 6-bp each time. The eight sites following the “NT-AARA” motif are underlined.
Site usage represents the ratio of observed occurrence in every 1×105 sites. Site counts and site usage are only shown for sites with more than 5 occurrences among the 800 polymorphic Alu loci.
We compared the average polyA-tail length for all Alu elements with that of the 800 polymorphic elements from each subfamily. The average lengths of polyA-tails for polymorphic insertions are significantly longer than those of all insertions (p < 0.00001), and they positively correlate with the ratio of polymorphic Alu insertions in subfamilies (data not shown), which is in agreement with the previous observation (Roy-Engel et al., 2002).
3.5. Distribution of Alu insertion polymorphisms throughout the human genome
We examined the distribution pattern of the 800 polymorphic insertions with regard to the distribution of all Alu elements. As shown in Table 3, Fig. 5 and Supplementary Fig. S1, the density of polymorphic Alu insertions among human chromosomes varies significantly (p < 0.05), ranging from 0.0 to 0.5 polymorphic insertions/Mb with a genome-wide average of 0.3 polymorphic Alu/Mb. The ratio of polymorphic Alu insertions among all chromosomal insertions also varies, but not significantly (P=0.19), across chromosomes, ranging from 0.1 to 1.9 per 1000 elements. The density of all elements by chromosome is highly correlated with the gene density (Pearson correlation r=0.95 with P < 10−20) and is inversely correlated with the availability of Alu integration sites (r = −0.91, P < 10−33). In contrast, the density of polymorphic Alu loci is inversely related to the densities of fixed elements (r= −0.56, P < 10−8) and of genes (r= − 0.55, P < 10−10), but positively correlated with the density of Alu integration sites (r=0.75, P < 10−20). For example, chromosomes 4, 5, 6, and 13 have lower densities for all Alu elements and genes, but higher densities of new Alu elements and Alu integration sites, while chromosomes 17, 19, 22 have higher densities of all Alu elements and genes but lower densities of polymorphic insertions and Alu integration sites. As an exception to the above trend, the Y-chromosome has the lowest density of all Alu insertions as well as polymorphic Alu insertions (Table 3, Fig. 5).
Table 3.
Distribution of Alu insertion polymorphisms by chromosome
| Chr | Length (Mb) a | All Alub | All_Alu (MB) | pAlu | pAlu (MB) | pAlu/1kAluc | Gene d(Mb) | Alu site (50 kb) e |
|---|---|---|---|---|---|---|---|---|
| 1 | 218.7 | 89555 | 409 | 59 | 0.3 | 0.7 | 14.8 | 429 |
| 2 | 237.0 | 75764 | 320 | 56 | 0.2 | 0.8 | 11.2 | 449 |
| 3 | 193.6 | 60234 | 311 | 53 | 0.3 | 1.0 | 9.8 | 458 |
| 4 | 186.6 | 49248 | 264 | 63 | 0.3 | 1.4 | 8.7 | 497 |
| 5 | 177.5 | 52334 | 295 | 60 | 0.3 | 1.2 | 10.0 | 460 |
| 6 | 166.9 | 52629 | 315 | 65 | 0.4 | 1.3 | 14.0 | 486 |
| 7 | 154.5 | 63416 | 410 | 47 | 0.3 | 0.8 | 12.2 | 442 |
| 8 | 141.7 | 44030 | 311 | 46 | 0.3 | 1.1 | 10.4 | 446 |
| 9 | 115.2 | 42922 | 373 | 28 | 0.2 | 0.7 | 12.7 | 428 |
| 10 | 130.7 | 50584 | 387 | 40 | 0.3 | 0.9 | 11.2 | 415 |
| 11 | 130.7 | 43679 | 334 | 40 | 0.3 | 1.0 | 15.5 | 409 |
| 12 | 129.3 | 52875 | 409 | 23 | 0.2 | 0.5 | 12.9 | 435 |
| 13 | 95.5 | 26172 | 274 | 46 | 0.5 | 1.9 | 8.6 | 494 |
| 14 | 87.2 | 32580 | 374 | 23 | 0.3 | 0.8 | 13.9 | 434 |
| 15 | 81.1 | 34861 | 430 | 28 | 0.3 | 0.9 | 14.9 | 404 |
| 16 | 79.9 | 46393 | 581 | 16 | 0.2 | 0.4 | 16.6 | 343 |
| 17 | 77.5 | 51868 | 669 | 24 | 0.3 | 0.5 | 21.8 | 338 |
| 18 | 74.5 | 22072 | 296 | 29 | 0.4 | 1.4 | 9.0 | 457 |
| 19 | 55.8 | 52324 | 938 | 7 | 0.1 | 0.1 | 31.6 | 279 |
| 20 | 59.4 | 26163 | 440 | 10 | 0.2 | 0.4 | 16.1 | 354 |
| 21 | 33.9 | 11221 | 331 | 6 | 0.2 | 0.6 | 13.0 | 442 |
| 22 | 34.4 | 21922 | 637 | 3 | 0.1 | 0.1 | 24.5 | 284 |
| X | 147.7 | 41691 | 282 | 27 | 0.2 | 0.7 | 9.7 | 454 |
| Y | 22.8 | 5911 | 259 | 1 | 0.0 | 0.2 | 9.7 | 443 |
| G f | 2832.1 | 1050448 | 371 | 800 | 0.3 | 0.8 | 12.8 | 435 |
Length of sequenced region based on UCSC human genome hg15.
Alu elements over 50 bp in length (excluding polyA-tail) from J, S, and Y families.
Ratio of polymorphic Alu insertions is expressed as the number of polymorphic insertions in every 1000 of all Alu insertions.
Only protein coding genes based on annotations in GenBank human genome Build 33 were included.
Density of Alu integration sites based on the “NT-AARA” motif.
Genome with all chromosomes together.
Fig. 5.

Genome-wide distribution of human Alu insertion polymorphisms. The 800 newly reported Alu insertion polymorphisms (blue) and all Alu elements from UCSC hg15 assembly (red) were plotted on the human chromosomal ideogram based on their physical locations. The “all Alu” track in red represents the number of Alu elements per 250 kb genomic region. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.6. Verification of polymorphic insertions
To provide an assessment of the specificity for our method, we used PCR to screen for polymorphism of 16 Alu loci randomly selected from the 65 potentially polymorphic loci on chromosome 6 using a diverse human DNA panel. All 16 Alu loci were polymorphic for presence/absence among 95 individuals from 10 globally diverse populations (Supplementary Table S2), indicating that our method is effective in detecting polymorphic insertions. Our data show that 9 of these 16 Alu loci reached the maximum level of heterozygosity (0.5) in one or more population groups, making them very useful as genetic markers for population genetics studies.
4. Discussion
4.1. In silico comparative genomics approach to identify Alu insertion polymorphisms
In this study, we took advantage of the availability of two different assemblies of the human genome sequence, representing approximately two genomes plus partial genome sequences from additional ethnically diverse individuals (Istrail et al., 2004; Lander et al., 2001; Venter et al., 2001). The ascertainment of the polymorphic status of the each Alu element is based on in silico detection of presence and absence of an Alu sequence in the corresponding genomic regions of the two genome sequences. Since the two genome sequences were generated using different sequencing and assembly strategies and the regions containing Alu sequences are difficult to assemble, we may expect that some Alu insertion differences may simply represent the discrepancies and errors produced during sequence assembly. To reduce the number of these types of false positives, we used stringent criteria to detect polymorphisms. For an Alu locus to be considered polymorphic, we required the element to be absent from the orthologous genomic region of the other genome. Further, we also required that only one copy of perfectly matched pre-integration sites (Alu− loci) exist in the other genome (Fig. 1). The observation that all of the 16 Alu loci tested are indeed polymorphic and that none of our potentially polymorphic loci have been classified as fixed for presence by previous studies suggests our method is effective. In addition, the observation that most of the polymorphic elements we identified contain TSDs also suggests that these elements were recently inserted into the human genome.
4.2. Retrotransposition activity of Alu subfamilies
Until very recently (Bennett et al., 2004), the majority of previously known polymorphic Alu insertion loci were members of the closely related Y subfamilies, with the majority from the Ya5 and Yb8 subfamilies(Batzer et al., 1990; Roy et al., 2000). A comprehensive assessment of the Alu insertion activity for all subfamilies has not been previously possible due to the limited number of polymorphic insertions available and the fact that most of these polymorphic insertions were obtained through a biased subfamily-specific selective screening. Since our method imposes no a priori sequence bias to identify polymorphic insertions from particular subfamilies, our results provide one of the first large and unbiased sets of data suitable for assessment of Alu insertion activities for all Alu subfamilies. Our data confirm the previous observation that Ya5 and Yb8 are very active Alu subfamilies within the human lineage and also recover additional Alu subfamilies, Yb11, Ya8, and Yb9, with appreciable retrotransposition activity. Our data also provides the largest set (a total of 27) of polymorphic elements from the older S subfamilies (Table 1) reported to date, supporting the hypothesis that some older Alu subfamilies may still be mobilizing at very low levels (Bennett et al., 2004; Johanning et al., 2003). However, we cannot rule out the possibility that some or all of these polymorphisms represent recent gene conversion events that have replaced younger subfamily members with sequence from older S type elements. An interesting observation is that, in the case of Ya, Yb, and Yd subfamilies, the activity level seems to correlate very well with their relative ages. For example, among all Yb subfamilies including Yb7, Yb8, Yb9, and Yb11 arranged in order from the oldest to the youngest, based on the increasing number of Yb-specific diagnostic mutations, the ratio of polymorphic insertions (per 1000 elements) shows a gradual increase from the lowest in Yb7 (36.1) to the highest in Yb11 (375).
We noticed that there are significant differences between this study and previous studies with regard to the sizes of certain Y subfamilies, such as the Ya3, Ya4, and Yb3, that are older and less uniform in sequence, while our assignments for the well-studied young subfamilies, such as Ya5, Ya8, Yb7 to Yb9, are in good agreement to previously reported size estimates (Carroll et al., 2001; Carter et al., 2004; Otieno et al., 2004; Salem et al., 2005b; Xing et al., 2003). We believe that these discrepancies are mainly due to the differences in the methods used for subfamily size estimation. In many of the previous studies, the subfamily sizes were obtained using exact match of subfamily-specific sequence motifs, while we assigned subfamily membership based on the best sequence identity to the subfamily consensus sequences. Thus, we identified many elements belonging to intermediate Alu subfamilies in the same series, such as Ya4 and Ya5, or containing certain random mutations that would preclude them from detection by an exact sequence match.
Our conservative identification of 800 polymorphic Alu insertions, plus those reported previously and not included in our list, brings the total number of non-redundant polymorphic Alu loci identified to date to ~1600, with the ratio of polymorphic Alu insertions relative to all Alu insertions in the human genome being approximately 0.16%.
The number of polymorphic loci that can be identified by our method largely depends on the genetic diversity represented by the genome sequences used in the comparison. In accordance with the Human Research Subject Protection Act of 1997, it is not possible to find out the exact ethnic background of the individual donors from whom the public and Celera genome sequences were derived. The fact that some of the previously reported polymorphic Alu elements were observed as shared between PHGS and CHGS suggests that the genome diversity represented by the two versions of genome sequences is limited. This also partially explains the lower ratios of polymorphic Alu insertions for the Ya5 and Yb8 subfamilies obtained from our analysis (9.2% and 7.5%, respectively) in comparison with results of (Carroll et al., 2001) (25% and 20%, respectively). Nevertheless, because we were able to identify 236 polymorphic insertions from the CHGS and because the majority of these loci (95%) are newly identified, we believe that a large number of polymorphic Alu insertions may be identified when additional genomic sequence from diverse ethnic groups becomes available. Therefore, we expect the actual total number of polymorphic Alu loci among human populations to be significantly higher than the number identified to date.
In conclusion, this study demonstrates that our in silico comparative genomic analysis represents an efficient approach for the identification of polymorphic Alu insertions. As additional complete human genome sequences representing different ethnic groups become available, we should be able to identify more polymorphisms. This approach can also be easily adapted for the analyses of other types of mobile elements, such as L1s, and for similar comparative analyses between human and non-human primates as reported by (Hedges et al., 2004). The identification of lineage-specific Alu elements and L1 elements will allow us to reconstruct the history of primate evolution and to understand the contributions of these mobile elements to the biological and physiological differences among the primate species, especially between human and non-human primates.
Supplementary Material
Acknowledgments
This research is in part supported by a Roswell Park Cancer Institute (RPCI) development fund (P.L.), by National Institutes of Health grants, CA101515 (P.L.), CA16056 (RPCI), and GM59290 (M.A.B.), National Science Foundation grants, BCS-9905396 (S.A.T.), BCS-0218338 (M.A.B.) and EPS-0346411 (M.A.B.), by Burroughs Wellcome Fund and David and Lucile Packard Career Awards to S.A.T., and by the Louisiana Board of Regents Millennium Trust Health Excellence Fund HEF (2000–05)-01 (M.A.B.), and the State of Louisiana Board of Regents Support Fund (M.A.B.).
Abbreviations
- PCR
polymerase chain reaction
- SINE
short interspersed element
- WGSA
whole genome shotgun assembly
- TSD
target site duplication
- PHGS
public version of human genome sequences
- CHGS
Celera human genome sequence
- BLAST
basic local alignment search tool
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.gene.2005.09.031.
References
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcot SS, Fontius JJ, Deininger PL, Batzer MA. Identification and analysis of a ‘young’ polymorphic Alu element. Biochim Biophys Acta. 1995;1263:99–102. doi: 10.1016/0167-4781(95)00080-z. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. A human-specific subfamily of Alu sequences. Genomics. 1991;9:481–487. doi: 10.1016/0888-7543(91)90414-a. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev, Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- Batzer MA, et al. Structure and variability of recently inserted Alu family members. Nucleic Acids Res. 1990;18:6793–6798. doi: 10.1093/nar/18.23.6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, et al. African origin of human-specific polymorphic Alu insertions. Proc Natl Acad Sci U S A. 1994;91:12288–12292. doi: 10.1073/pnas.91.25.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, et al. Dispersion and insertion polymorphism in two small subfamilies of recently amplified human Alu repeats. J Mol Biol. 1995;247:418–427. doi: 10.1006/jmbi.1994.0150. [DOI] [PubMed] [Google Scholar]
- Batzer MA, et al. Genetic variation of recent Alu insertions in human populations. J Mol Evol. 1996a;42:22–29. doi: 10.1007/BF00163207. [DOI] [PubMed] [Google Scholar]
- Batzer MA, et al. Standardized nomenclature for Alu repeats. J Mol Evol. 1996b;42:3–6. doi: 10.1007/BF00163204. [DOI] [PubMed] [Google Scholar]
- Bennett EA, Coleman LE, Tsui C, Pittard WS, Devine SE. Natural genetic variation caused by transposable elements in humans. Genetics. 2004;168:933–951. doi: 10.1534/genetics.104.031757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD. LINEs and Alus–the polyA connection. Nat Genet. 1997;16:6–7. doi: 10.1038/ng0597-6. [DOI] [PubMed] [Google Scholar]
- Callinan PA, et al. Comprehensive analysis of Alu-associated diversity on the human sex chromosomes. Gene. 2003;317:103–110. doi: 10.1016/s0378-1119(03)00662-0. [DOI] [PubMed] [Google Scholar]
- Carroll ML, et al. Large-scale analysis of the Alu Ya5 and Yb8 subfamilies and their contribution to human genomic diversity. J Mol Biol. 2001;311:17–40. doi: 10.1006/jmbi.2001.4847. [DOI] [PubMed] [Google Scholar]
- Carter AB, et al. Genome-wide analysis of the human Alu Yb-lineage. Hum Genomics. 2004;1:167–178. doi: 10.1186/1479-7364-1-3-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost GJ, Boeke JD. Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure. Biochemistry. 1998;37:18081–18093. doi: 10.1021/bi981858s. [DOI] [PubMed] [Google Scholar]
- Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- Dewannieux M, Esnault C, Heidmann T. LINE-mediated retro-transposition of marked Alu sequences. Nat Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- Ganguly A, Dunbar T, Chen P, Godmilow L, Ganguly T. Exon skipping caused by an intronic insertion of a young Alu Yb9 element leads to severe hemophilia A. Hum Genet. 2003;113:348–352. doi: 10.1007/s00439-003-0986-5. [DOI] [PubMed] [Google Scholar]
- Garber RK, Hedge DJ, Herke SW, Hazard NW, Batzer MA. The Alu Yc1 subfamily: sorting the wheat from the chaff. Cytogenet Genome Res. 2005;110:537–542. doi: 10.1159/000084986. [DOI] [PubMed] [Google Scholar]
- Hedges DJ, Callinan PA, Cordaux R, Xing J, Barnes E, Batzer MA. Differential Alu mobilization and polymorphism among the human and chimpanzee lineages. Genome Res. 2004;14:1068–1075. doi: 10.1101/gr.2530404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck CM, Rinehart FP, Schmid CW. A ubiquitous family of repeated DNA sequences in the human genome. J Mol Biol. 1979;132:289–306. doi: 10.1016/0022-2836(79)90261-4. [DOI] [PubMed] [Google Scholar]
- Istrail S, et al. Whole-genome shotgun assembly and comparison of human genome assemblies. Proc Natl Acad Sci U S A. 2004;101:1916–1921. doi: 10.1073/pnas.0307971100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanning K, et al. Potential for retroposition by old Alu subfamilies. J Mol Evol. 2003;56:658–664. doi: 10.1007/s00239-002-2433-y. [DOI] [PubMed] [Google Scholar]
- Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci U S A. 1997;94:1872–1877. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Smith T. A fundamental division in the Alu family of repeated sequences. Proc Natl Acad Sci U S A. 1988;85:4775–4778. doi: 10.1073/pnas.85.13.4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov V, Jurka J. The age of Alu subfamilies. J Mol Evol. 1996;42:59–65. doi: 10.1007/BF00163212. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lev-Maor G, Sorek R, Shomron N, Ast G. The birth of an alternatively spliced exon: 3′ splice-site selection in Alu exons. Science. 2003;300:1288–1291. doi: 10.1126/science.1082588. [DOI] [PubMed] [Google Scholar]
- Makalowski W, Mitchell GA, Labuda D. Alu sequences in the coding regions of mRNA: a source of protein variability. Trends Genet. 1994;10:188–193. doi: 10.1016/0168-9525(94)90254-2. [DOI] [PubMed] [Google Scholar]
- Mamedov IZ, Arzumanyan ES, Amosova AL, Lebedev YB, Sverdlov ED. Whole-genome experimental identification of insertion/deletion polymorphisms of interspersed repeats by a new general approach. Nucleic Acids Res. 2005;33:e16. doi: 10.1093/nar/gni018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otieno AC, et al. Analysis of the human Alu Ya-lineage. J Mol Biol. 2004;342:109–118. doi: 10.1016/j.jmb.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Perna NT, Batzer MA, Deininger PL, Stoneking M. Alu insertion polymorphism: a new type of marker for human population studies. Hum Biol. 1992;64:641–648. [PubMed] [Google Scholar]
- Quentin Y. The Alu family developed through successive waves of fixation closely connected with primate lineage history. J Mol Evol. 1988;27:194–202. doi: 10.1007/BF02100074. [DOI] [PubMed] [Google Scholar]
- Ray DA, et al. Inference of human geographic origins using Alu insertion polymorphisms. Forensic Sci. 2005;153:117–124. doi: 10.1016/j.forsciint.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Roy AM, et al. Recently integrated human Alu repeats: finding needles in the haystack. Genetica. 1999;107:149–161. [PubMed] [Google Scholar]
- Roy AM, et al. Potential gene conversion and source genes for recently integrated Alu elements. Genome Res. 2000;10:1485–1495. doi: 10.1101/gr.152300. [DOI] [PubMed] [Google Scholar]
- Roy-Engel AM, et al. Alu insertion polymorphisms for the study of human genomic diversity. Genetics. 2001;159:279–290. doi: 10.1093/genetics/159.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy-Engel AM, et al. Active Alu element “A-tails”: size does matter. Genome Res. 2002;12:1333–1344. doi: 10.1101/gr.384802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem AH, Kilroy GE, Watkins WS, Jorde LB, Batzer MA. Recently integrated Alu elements and human genomic diversity. Mol Biol Evol. 2003;20:1349–1361. doi: 10.1093/molbev/msg150. [DOI] [PubMed] [Google Scholar]
- Salem AH, Ray DA, Batzer MA. Identity by descent and DNA sequence variation of human SINE and LINE elements. Cytogenet Genome Res. 2005a;108:63–72. doi: 10.1159/000080803. [DOI] [PubMed] [Google Scholar]
- Salem AH, Ray DA, Hedges DJ, Jurka J, Batzer MA. Analysis of the human Alu Ye lineage. BMC Evol Biol. 2005b;5:18. doi: 10.1186/1471-2148-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TH, Deininger PL. The role and amplification of the HS Alu subfamily founder gene. J Mol Evol. 1996;42:15–21. doi: 10.1007/BF00163206. [DOI] [PubMed] [Google Scholar]
- Sorek R, Ast G, Graur D. Alu-containing exons are alternatively spliced. Genome Res. 2002;12:1060–1067. doi: 10.1101/gr.229302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoneking M, et al. Alu insertion polymorphisms and human evolution: evidence for a larger population size in Africa. Genome Res. 1997;7:1061–1071. doi: 10.1101/gr.7.11.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, et al. Short tandem-repeat polymorphism/Alu haplotype variation at the PLAT locus: implications for modern human origins. Am J Hum Genet. 2000;67:901–925. doi: 10.1086/303068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Wallace MR, Andersen LB, Saulino AM, Gregory PE, Glover TW, Collins FS. A de novo Alu insertion results in neurofibromatosis type 1. Nature. 1991;353:864–866. doi: 10.1038/353864a0. [DOI] [PubMed] [Google Scholar]
- Wang J, Song L, Grover D, Azrak S, Batzer MA, Liang P. dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Human Mutat. doi: 10.1002/humu.20307. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins WS, et al. Genetic variation among world populations: inferences from 100 Alu insertion polymorphisms. Genome Res. 2003;13:1607–1618. doi: 10.1101/gr.894603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, et al. Comprehensive analysis of two Alu Yd subfamilies. J Mol Evol. 2003;57 (Suppl 1):S76–S89. doi: 10.1007/s00239-003-0009-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.