

Abstract
Background
Late Na+ current (INaL) in human and dog hearts has been implicated in abnormal repolarization associated with heart failure (HF). HF slows inactivation gating of late Na+ channels, which could contribute to these abnormalities.
Aims
To test how altered gating affects INaL time course, Na+ influx, and action potential (AP) repolarization.
Methods
INaL and AP were measured by patch clamp in left ventricular cardiomyocytes from normal and failing hearts of humans and dogs. Canine HF was induced by coronary microembolization.
Results
INaL decay was slower and INaL density was greater in failing hearts than in normal hearts at 24°C (human hearts: τ=659±16 vs. 529±21 ms; n=16 and 4 hearts, respectively; mean±SEM; p<0.002; dog hearts: 561±13 vs. 420±17 ms; and 0.307±0.014 vs. 0.235±0.019 pA/pF; n=25 and 14 hearts, respectively; p<0.005) and at 37°C this difference tended to increase. These INaL changes resulted in much greater (53.6%) total Na+ influx in failing cardiomyocytes. INaL was sensitive to cadmium but not to cyanide and exhibited low sensitivity to saxitoxin (IC50=62nM) or tetrodotoxin (IC50=1.2μM) tested in dogs. A 50% INaL inhibition by toxins or passing current opposite to INaL, decreased beat-to-beat AP variability and eliminated early afterdepolarizations in failing cardiomyocytes.
Conclusions
Chronic HF leads to larger and slower INaL generated mainly by the cardiac-type Na+ channel isoform, contributing to larger Na+ influx and AP duration variability. Interventions designed to reduce/normalize INaL represent a potential cardioprotective mechanism in HF via reduction of related Na+ and Ca2+ overload and improvement of repolarization.
Keywords: heart failure, late Na+ current inactivation, action potential, early afterdepolarizations
Introduction
Mortality in patients with heart failure (HF) remains high despite recent progress in treatment, with approximately 40% of patients dying suddenly [1]. Ventricular tachycardia and fibrillation have been documented in ~80% of these sudden deaths [2]. While the electrophysiological mechanisms leading to such arrhythmias remain unclear, erratic control of the action potential (AP) in the diseased myocardium is widely believed to be the proximate cause. Although the major cause of AP prolongation observed in HF [1] was previously thought to be downregulation of K+ channels [3], there is a growing body of evidence that it could also be due to alteration of the late Na+ current (INaL) produced by voltage-gated Na+ channels (Nav) that continue to be active during the AP plateau.
The significance of INaL in cardiac arrhythmias was demonstrated directly through the discovery of a genetic mutation in the cardiac Na+ channel isoform (Nav1.5), resulting in abberant inactivation of Na+ current in patients with this congenital defect (LQT3 syndrome) [4, 5]. Further studies showed that wild type Nav1.5 could also produce an intrinsic INaL, which is present in ventricular cardiomyocytes (VCs) of normal and failing human hearts [6, 7]. Because of its slow decay (τ~0.5s), which is almost complete after 2 s, this INaL can be distinguished from INaT, as well as from the persistent Na+ current reported in LQT3 patients.
INaL density is increased in VCs in experimental chronic HF in dogs [8], and INaL is implicated in the increased AP duration and early afterdepolarizations (EADs) observed in both canine and human failing VCs [6, 8]. The increased density of INaL in HF was recently confirmed in VCs from a canine model of pacing-induced HF as well as in failing human hearts [9], and INaL has been suggested as one of the major mechanisms for arrhythmias in HF [1, 10]. Despite the emerging importance of INaL in HF, some important data and conceptual links are still missing:
The molecular and gating mechanisms of the HF-related changes observed in whole-cell INaL are not well defined. A recent single-channel study showed that inactivation gating of individual late Nav is altered in human failing VCs [7] and a new numerical model of Nav operating in different gating modes predicted slower whole-cell INaL decay in HF [11], but this prediction has not been tested experimentally.
Changes in INaL density in HF were previously found only for the portion of INaL remaining after prolonged membrane depolarization (500 ms [8] or 750 ms [9]), much longer than the AP plateau in human and canine VCs. Since the entire time course of INaL has not been compared in normal and failing VCs, the role of INaL in AP abnormalities in HF remains unclear.
Accordingly, we conducted the present study in order to: 1) identify HF-related changes during almost entire time course of INaL in VCs of human and canine hearts, 2) characterize their importance for abnormal repolarization, and 3) delineate principles of cardioprotection in HF based on INaL correction.
Methods
Our investigation of human heart tissue conforms to the principles outlined in the Declaration of Helsinki, and was approved by the Henry Ford Health System Human Rights Committee (Institutional Review Board). The canine study conforms to the Guidelines for Care and Use of Laboratory Animals published by the US National Institutes of Health and was approved by the Institutional Animal Care and Use Committee (IACUC) of the Henry Ford Health System.
We used 4 normal donor human hearts, which for technical reasons were deemed unsuitable for transplantation, and 16 explanted failing hearts (Table 1). To complement our studies in humans, we used a canine model of chronic HF. Briefly, HF was produced in 25 canine hearts ~3 months after multiple sequential coronary artery microsphere embolizations [12]. By 3 months left ventricle (LV) ejection fraction had diminished to ~25–27%, and the dog hearts manifested ventricular arrhythmias and sudden death similar to human HF [13]. Fourteen normal dog hearts served as controls. Cardiomyocytes were enzymatically isolated from the apical LV mid-myocardium as reported previously [6]. We used the mid-myocardial layer to avoid inconsistencies related to the transmural INaL density profile [14, 15]. The yield of viable rod-shaped, Ca2+-tolerant myocytes varied from 10 to 50%.
Table 1.
Demography of human hearts used in the study. Medications were given during preparation for transplantation.
| Patient/heart | Aetiology | Age, y | Sex | τ, ms | Medications |
|---|---|---|---|---|---|
| 1 | ICM | 48 | M | 663 | ACE inhibitor, beta blocker, Digoxin, Furosemide |
| 2 | IDC | 57 | M | 624 | ACE inhibitor, Digoxin, Furosemide |
| 3 | ICM | 57 | M | 665 | Amiodarone, ACE inhibitor, Digoxin, Furosemide |
| 4 | ICM | 39 | M | 723 | ACE inhibitor, Digoxin, Isosorbide dinitrate, Furosemide |
| 5 | IDC | 27 | F | 653 | ACE inhibitor, Digoxin, Furosemide |
| 6 | IDC | 44 | M | 633 | ACE inhibitor, Digoxin, Furosemide, Milrinone |
| 7 | IDC | 45 | M | 688 | ACE inhibitor, Digoxin, Furosemide, Milrinone |
| 8 | ICM | 55 | M | 623 | Amiodarone, ACE inhibitor, Digoxin |
| 9 | ICM | 62 | M | 728 | Digoxin, Isosorbide dinitrate, Furosemide |
| 10 | IDC | 54 | M | 692 | Amiodarone, ACE inhibitor, Digoxin, Furosemide |
| 11 | ICM | 52 | M | 675 | Amiodarone, ACE inhibitor, Digoxin, Furosemide |
| 12 | ICM | 53 | M | 536 | ACE inhibitor, Spironolactone, Digoxin |
| 13 | IDC | 56 | F | 611 | Amiodarone, Diltiazem, Digoxin, Isosorbide dinitrate, Gemfibrozil, Atenolol |
| 14 | ICM | 61 | F | 570 | ACE inhibitor, Furosemide, Milrinone |
| 15 | IDC | 63 | M | 641 | Beta blocker, Digoxin, Furosemide, Losartan |
| 16 | IDC | 39 | F | 820 | ACE inhibitor, Spironolactone, Digoxin |
| 17 | *Normal | N/D | N/D | 528 | |
| 18 | *Normal | N/D | N/D | 572 | |
| 19 | Normal | N/D | N/D | 546 | |
| 20 | Normal | N/D | N/D | 472 |
indicates INaL decay time constant, IDC indicates idiopathic dilated cardiomyopathy; ICM, ischaemic cardiomyopathy.
Some data on the normal hearts have been reported previously [6]. N/D-not disclosed.
INaL was measured using a whole-cell patch-clamp technique [6]. It was assessed by 2-s membrane depolarizations to -30 mV from a holding potential of −140 mV applied with a stimulation frequency of 0.1 Hz. To avoid voltage-clamp problems, we inactivated INaT by a 5-ms pre-pulse to +50 mV (Fig.1A, inset), which substantially diminishes INaT but does not change INaL [6]. The solution contained (in mM): 140 NaCl, 5 CsCl, 1.8 CaCl2, 2 MgCl2, 5 glucose, 0.002 nifedipine, and 5 HEPES-CsOH buffer (pH 7.3). The pipette solution contained (in mM): 5 NaCl, 133 CsCl, 2 MgATP, 20 tetraethylammonium chloride, 10 EGTA, and 5 HEPES-CsOH buffer (pH 7.3). Experiments were performed at room temperature (22–24°C). INaL decay was evaluated by a single exponential fit starting 200 ms after the onset of depolarization [6]. The dose-response curve describing INaL block (B%) by cadmium (Cd2+), tetrodotoxin (TTX), or saxitoxin (STX) was approximated by a one-binding-site model:
Figure 1.
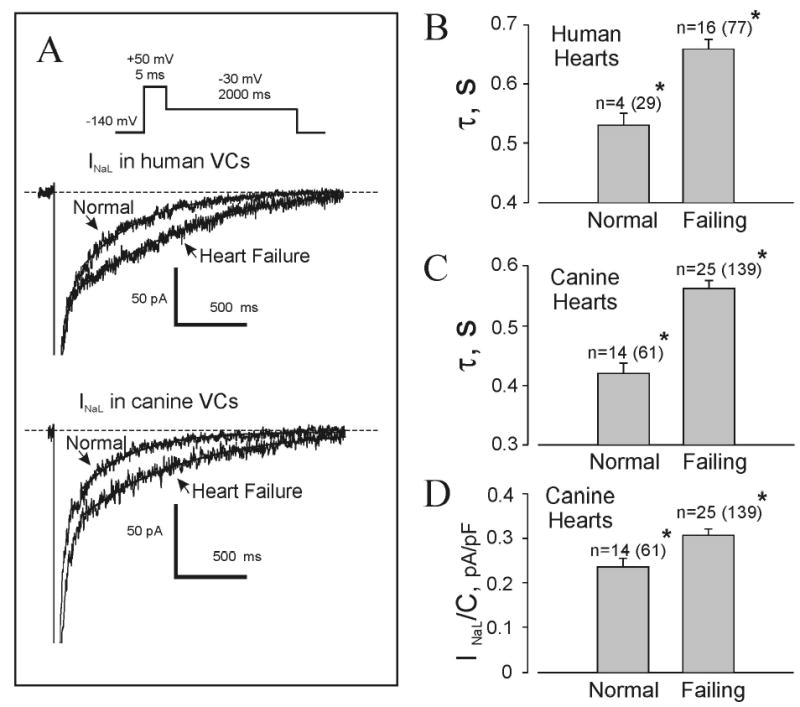
Changes in whole-cell late Na+ current (INaL) in left ventricular cardiomyocytes from normal and failing human and canine hearts. A, representative superimposed whole-cell traces recorded in normal and failing (patient #16, Table 1) human hearts (upper traces) and dog hearts (lower traces) using a voltage-clamp protocol (inset). Current was sent through a low-pass filter (200 Hz) and truncated at -150 pA. Dashed lines represent zero current. Solid lines show exponential fits to the experimental recordings, with decay time constants (τ) of 435 and 929 ms (human) or 351 and 739 ms (canine) for normal and failing myocardium, respectively. B & C, summarized data on τ for human and canine normal and failing hearts. D, average data on INaL density in normal and failing canine hearts. Plots show mean ± SEM; n=number of hearts (with number of tested cells in parenthesis); *p < 0.005, normal vs. failing hearts (ANOVA).
APs were recorded at a pacing frequency of 0.25 Hz and 37°C using an amphotericin B-perforated patch in current-clamp mode [6].
Statistical methods
Multiple comparisons were made with one-way analysis of variance (ANOVA). Deviations from the mean in all measurements are reported in terms of standard error. In some cases (Fig. 4D) Student’s paired t test was applied. A p value < 0.05 was considered significant.
Figure 4.

Chronic HF increases beat-to-beat action potential duration variability (APDV) in canine ventricular myocytes that can be rescued by a reduction of INaL. A : Superimposed consecutive APs recorded by perforated patch clamp in representative normal and failing dog ventricular myocytes at 37°C and 0.25 Hz pacing rate. B: Average data on APDV assessed as SD/mean. The figure shows the mean±SEM for 18 failing hearts (61 cells, 908 APs) and 4 normal hearts (7 cells, 141 APs). P was evaluated by ANOVA. C and D: Beat-to-beat APDV in failing canine ventricular myocytes was decreased by electrical neutralization of the late Na+ current with a delayed external current (200 ms after AP upstroke) at 37°C and 0.25 Hz pacing rate. C: A representative example of a delayed external current effect on the consecutive APs (overlapped traces) in one cell. D: Average APDV data (mean±SEM) assessed as SD/mean in 3 cells. Statistical comparison was made by Student’s paired t test.
The quality of the single-site binding model (Fig. 3A,B) was evaluated by F-test (StatMost 2.5 Software, DataMost, UT), testing the null hypothesis that variability of the experimental data and values predicted by the model are the same. The F-value was determined as the ratio of the larger over the smaller variance and compared with the tabulated F-values for the respective degrees of freedom and a confidence level of 0.95.
Figure 3.

Pharmacological characterization of INaL. A-C: Dose-response curve fitting percent of INaL blockade by TTX (A) and STX (B) in canine failing ventricular cardiomyocytes and by Cd2+ in human VCs (C, patient #3, Table 1) with a one-binding-site model (see Methods). Insets in A&B: superimposed whole-cell traces recorded in failing canine ventricular cardiomyocytes with different concentrations of tetrodotoxin (TTX) and saxitoxin (STX) in the bath (indicated at the traces), respectively. Traces were truncated at –200 pA, Vh= -140mV, Vt= -30mV, low-pass filter 50 Hz The inset in panel C shows superimposed INaL current trace fragments from 200 to 2000 ms before (control) and after application of 1 mM Cd2+. D, summarized data on INaL current density recorded in control and cyanide-treated cardiomyocytes from human failing hearts. The inset shows representative raw INaL traces before (left) and after (right) exposure of cardiomyocytes isolated from a failing heart (patient #2, Table 1) to cyanide for 2 hours. Room temperature was 22–23°C in all experiments.
Results
Chronic HF results in a slower and greater INaL in VCs
Fig. 1A shows superimposed representative raw INaL traces from VCs isolated from normal and failing hearts. Data on the INaL decay constant (τ) are summarized in Fig. 1B,C and shown for individual human hearts in Table 1. INaL was significantly and consistently slower in failing human hearts compared to normal hearts (by ~25%, i.e., τ = 659 ± 16 vs. 529 ± 21 ms; n=16 and 4, respectively; p < 0.002). We also found a similar slowing of INaL decay (~34%) in canine failing cardiomyocytes (561 ± 13 vs. 420 ± 17 ms; n = 25 and 14 hearts, respectively; p < 0.001). The distribution of τ values in individual failing cells was broader, with a slight skew towards higher values compared to normal cells (Supplemental Fig. I.). Significantly, 14 out of 16 failing human hearts had τ > 600 ms, whereas all normal hearts had τ < 600 ms (Table 1). In some patients τ was relatively normal (536 and 570 ms for patients #12 and #14); others showed a dramatic increase, reaching as much as 1.5 times normal (723, 728, and 820 ms for patients #4, #9, and #16). Indeed, in some cells τ was close to 1 s (patient #16).
To describe total INaL decay, in addition to time constants, we also measured an “initial” value of INaL density (INaL/C). However, identification and interpretation of HF-related INaL/C changes in humans proved complicated, as INaL/C varied greatly from patient to patient, probably due to differences in treatment, aetiology, sex, age, etc. (Table 1) [6]. Accordingly, we measured and analyzed HF-related changes in density using our reproducible canine HF model. INaL/C assessed 200 ms after membrane depolarization was significantly greater in the failing canine VCs (0.307±0.014 pA/pF, n=25 hearts vs. 0.235±0.019 pA/pF, n=14 normal hearts; p<0.005) (Fig.1D).
Reconstitution of the INaL time course at a physiological temperature
Using a simple single exponential decay model for INaL time course, we then evaluated the effect of the identified changes in INaL density and inactivation gating on the INaL time course and total Na+ influx into the cells. Based on the τ values and the densities from Fig.1C,D, we calculated the time course of the average INaL (Fig. 2A) and its integral (Fig.2C) for normal and failing canine cells of the same size (200 pF), thus excluding the effect of ventricular myocyte hypertrophy reported previously in this canine chronic HF model [8]. Using Q10 factors, we then calculated the time course of INaL and its integral at a physiological temperature, 37°C (Fig. 2B,D).
Figure 2.

Idealized total INaL time course calculated using an exponential decay function: I(t)=C*d200ms*exp[- (t-200ms)/τ] with mean time constants (τ) and INaL densities, d=INaL/C at 24°C (A) and 37°C (B) taken from Fig.1C,D for normal (solid) and failing canine ventricular cardiomyocytes (dashed line) having the same electric capacitance C=200 pF. The curves for 37°C were calculated using Q10 factors: and with Q10d=1.5 for Nav conductance and Q10τ=2.2 for late Nav gating [11, 38]. The absolute difference between normal and failing cells is shown by the dotted lines. The inset shows the relative difference (per cent change) between normal and failing cells. C and D, total electrical charge transferred by INaL, assessed as an INaL integral at 24°C and 37°C (shown in A and B, respectively).
Important findings are:
The absolute difference between failing and normal cells (dotted lines in Fig.2) was bell-shaped, with the peak (-14.5 pA at 240 ms for 24°C or -24.5 pA at 90 ms for 37°C), occurring well within the AP plateau.
The relative difference between normal and failing cells increased progressively with membrane depolarization, doubling after 330 ms at 37°C (Fig. 2B, inset).
Both absolute and relative differences were much greater at 37°C, thus predicting a greater impact of INaL on membrane repolarization under physiological conditions.
The slower and greater INaL transferred far more Na+ ions into failing cells (Fig.2C,D). The integrals for INaL during 2 s were 48.3 vs. 31.9 pC (51.4% increase) at 24°C and 30.66 vs. 19.96 pC (53.6% increase) at 37°C.
Pharmacological characterization of INaL
TTX, STX and Cd2+ are valuable pharmacological tools to identify the specific Na+ channel isoform underlying a Na+ current. We found that INaL in human VCs was sensitive to Cd2+; dose-response curves in one patient (patient #14, Table 1) revealed that Cd2+ blocked 50% of INaL at 104 μM (Fig. 3C).While it is known that INaL in human VCs exhibits low sensitivity to TTX and STX [6], the dose-response curves of these toxins for INaL in canine VCs has not been reported to our knowledge. We found that INaL of canine failing VCs exhibited a low affinity for TTX and STX (Fig. 3A,B). We also tested whether potassium cyanide (KCN) could modulate INaL in normal and failing human VCs. KCN did not influence INaL in failing human VCs either applied acutely during the patch-clamp experiment (40 min, 1 mM KCN; n = 5 VCs) or after short-term pre-incubation of VCs before the patch-clamp experiment (2 h, 5 mM KCN) (Fig. 3D). KCN was also found to have no significant effect in normal VCs (not shown). In fact, the only remarkable effect of KCN was a significant increase in nonselective leak conductance (from 1.5- to 5- fold).
Partial INaL inhibition restores normal repolarization
We found that VCs from canine and human failing hearts exhibited variable beat-to-beat AP duration, culminating in early afterdepolarizations (EADs) [6, 8](Supplemental Fig.II). Since HF results in a two-fold increase in INaL at the most critical time point of 330 ms (Fig.2B, inset, 37°C), corresponding to the repolarization phase in normal cells, we used 1.5 μM TTX which would block ~50% of INaL [6] and thus render it “normal”. Partial INaL blockade shortened AP duration in failing human VCs [6,8](Supplemental Fig. II.A). Pharmacological correction of INaL restored “healthy” APs in the failing cells: EADs ceased and both AP duration and variability returned to normal [6](Compare Supplemental Fig. II.A, second trace and Fig.II.B). A similar effect was found in canine failing VCs [8, 16, 17](Supplemental Fig. II.C, D) using STX (100 nM), another INaL blocker (Fig.3B). We also tested the importance of INaL for AP duration and variability and EADs directly, electrically neutralizing INaL by current clamp. Injecting a 50-pA current at the AP plateau, 200 ms after the AP upstroke (see Fig. 2B, dashed line for INaL prediction at 37°C), normalized AP duration and variability and abolished EADs (Fig.4).
Discussion
We believe the present study consistently shows for the first time that INaL decay is significantly slower in left ventricular cardiomyocytes taken from patients with chronic HF undergoing heart transplantation. Our complementary study of a reproducible experimental canine model of chronic HF revealed that in addition to this INaL slowing, the density of INaL significantly increased in the failing VCs. We then explored the biophysical and physiological consequences of altered INaL in HF myocytes, since a slower and larger INaL might influence intracellular Na+/Ca2+ balance and the balance of ion currents at the AP plateau, thereby affecting cell contractile performance and AP repolarization, respectively. Finally, we tested pharmacologically which Nav isoform could be responsible for the change in INaL.
Greater INaL in HF VCs: possible link to AP abnormalities
We previously showed a significantly longer AP in HF VCs using a canine chronic HF model [8], in line with data from other experimental HF models as well as from human HF VCs [6, 18]. In the present study, based on data from an extended number of canine hearts, we found that besides increased AP duration, beat-to-beat AP duration variability (assessed as SD/mean) was significantly increased in the failing VCs (Fig. 4A,B). This finding could explain greater variability of refractoriness in patients with HF, in turn believed to be a predictor of arrhythmic events and sudden death [1, 19, 20]. Such variability of AP duration could be at least partly owed to slower and greater INaL in failing VCs as reported in the present study. Computer simulations based on INaL data have confirmed that INaL contributes to the VC AP plateau [14], and late Na+ channel openings were indeed observed on the AP plateau level in human VCs [7]. Accordingly, as INaL increases and becomes slower in HF cells (Figs.1, 2, and supplemental Fig. I), its contribution is expected to be both greater and more persistent, prolonging AP duration and ultimately causing abnormal repolarization, such as EADs and AP duration variability. We found that EADs ceased and both AP duration and variability returned to normal when INaL was partially inhibited with TTX or STX [6, 8](Supplemental Fig.II). We also observed similar effect by injecting current opposite to INaL during the AP plateau (Fig.4C,D). In contrast to Na+ current blockade by toxins or lidocaine (Supplemental Fig. II, see also [6, 8]), this protocol preserved INaT and hence, the INaT -related Na+ contribution to function of the Na+/Ca2+ exchanger (NCX) [21, 22].
These findings indicate that the slower and greater INaL in HF indeed prolongs the AP plateau and contributes to the high incidence of EADs and greater AP duration variability in VCs of HF patients [6](Supplemental Fig. II.A,B) and canine failing hearts [8, 17] (Fig.4A,B) that is in turn, considered a potential mechanism for ventricular arrhythmias in HF [1, 19, 20]. Thus interventions designed to block INaL and/or accelerate INaL decay [17, 23] in patients may have a beneficial effect on spatial [15] and temporal repolarization abnormalities [19, 20]. However, blockade of INaL should be performed with extreme caution: 1) it should be specific for INaL, because simultaneous blockade of INaT (already downregulated in HF [9, 24, 25]) could cause slowed conduction and therefore could be pro-arrhythmic; and 2) it may also shift the Na+/Ca2+ balance so that more Ca2+ would be transferred from the cell by the NCX, thus decreasing SR load and worsening contractile performance.
Greater Na+ influx via INaL in HF VCs: possible effects on Na+/Ca2+ balance and contractility
A mathematical model based on single-channel data in human VCs predicted that although INaL has a much smaller amplitude than INaT, it lasts much longer; thus both currents transfer roughly the same amount of Na+ during membrane depolarization [11], suggesting a substantial INaL contribution to intracellular Na+ homeostasis. In the present study we found that INaL-associated Na+ flux increased by 53.8% in failing cells vs. normal cells of same size (Fig.2D). Moreover, taking into account that INaT is reduced by 30–40% in HF VCs [9, 24, 25], the role of INaL in Na+ homeostasis should be even more substantial in the failing cells, in line with the recent finding that [Na]i was significantly increased in failing paced cardiomyocytes [26]. Accordingly, in addition to AP modulation, INaL in failing VCs would be expected to offset the enhanced function of the upregulated NCX in HF [27], thus preserving intracellular Ca2+ and SR load which in turn could either improve contractile function at low frequencies or lead to diastolic Ca2+ overload and poor myocyte contraction in failing hearts at higher frequencies [16, 17, 21, 28]. Furthermore, the danger of Ca2+ overload is that it leads to spontaneous Ca2+ release and related EADs and delayed afterdepolarizations culminating in life-threatening arrhythmias [1, 10, 22]. In summary, a greater Na+ influx via INaL in failing VCs can be interpreted as an intrinsic digitalis-like effect on Na+/Ca2+ balance with all its known benefits and risks. Inhibition of INaL can reduce those risks and thus represent a potential cardioprotective mechanism.
Molecular basis of the altered INaL in HF
Our previous studies of late Na+ single-channel activity [7] did not reveal any peculiar Nav activity that might be responsible for INaL in failing compared to normal hearts. Furthermore, heterologous expression of Nav1.5 produced late openings and gating modes similar to human VCs [7, 11], indicating that Nav1.5 seems to be the main source of INaL in human VCs. However, this does not exclude the possibility that another voltage-sensitive Nav αsubunit isoform (NaVx) could contribute to INaL.
A difference of 2 amino acids in the Nav pore region accounts for the high affinity of neuronal Nav and low affinity of cardiac Nav for TTX or STX, a feature used to distinguish these Nav isoforms [29]. A highly TTX-sensitive Nav isoform has been suggested as a source of late Nav openings in rat cardiomyocytes [30]. Different transcripts of highly TTX-sensitive brain isoforms (Nav1.1, 1.3, 1.6) have been discovered in the mouse heart and recently in dogs (Nav1.1, 1.2, 1.3) [31, 32], suggesting species-specific expression. These neuronal Nav isoforms were responsible for 10 to 20% of peak INa in myocardial and Purkinje cells, respectively [32]; however, our previous study showed that the total INaL in human VCs had a low affinity for TTX or STX [6]. The only reported mammalian Nav isoform with a low affinity for TTX other than Nav1.5 is Nav1.8 (gene SCN10A) [33]; however, as far as we know, the mRNA encoding Nav1.8 has not been detected in rat hearts [33], failing human heart tissue or isolated LV cardiomyocytes (Undrovinas and Kyle, unpublished data). In the present study, we found that INaL of failing canine hearts had a low affinity for TTX and STX (Fig. 3A,B) similar to human VCs [6]; IC50 was 1.2 vs. 1.53 μM for TTX and 62 vs. 98 nM for STX comparing dog vs. human cells.
The experiments blocking INaL with different concentrations of TTX and STX were crucial to understand the origin INaL. If INaL represented the activity of channels with different pharmacological and gating properties, the extent of blockade would differ at different time points after membrane depolarization. However from our original INaL traces in dogs (see example in Fig.3A,B) and humans (Fig.3A,B from our previous paper [6]), it is clearly seen that the extent of the toxin-induced blockade was about the same during INaL time course, ranging from 200 to 1000 ms. There was no significant “non-inactivated” or “steady-state” Na+ current blocked by the toxins as of 2 s of membrane depolarization. Thus our data suggest that within this time frame INaL is mainly defined by the activity of only one type of Nav so that INaL in canine and human VCs has a common molecular origin (i.e. Nav1.5) and similar molecular mechanisms could be involved in slowing of INaL in HF.
A possible role for other Nav isoforms in INaL might be substantiated by the use of divalent cations. TTX-sensitive Nav isoforms are blocked poorly by divalent cations, whereas TTX-resistant forms of Nav are blocked by micromolar Cd2+ [34]. The present study indicates that in human VCs INaL is sensitive to Cd2+ (Fig. 3C), supporting our supposition that the Nav isoform with low TTX affinity is primarily responsible for INaL in humans [6]. We recently demonstrated that silencing the SCN5A gene responsible for Nav1.5 expression with a siRNA decreased INaL by 75% and reduced AP duration in dogs with chronic HF [35]. Therefore Nav1.5 generates most of the total INaL.
Experimental hypoxia and metabolic inhibitors such as cyanide can reportedly augment the persistent Na+ current in rats [36]. These authors suggested that a different Nav isoform sensitive to both hypoxia and cyanide is responsible for this persistent Na+ current [30, 36]. However, our data show that INaL is not sensitive to cyanide (Fig.3D). These negative results do not support the hypothesis that a novel cyanide-sensitive Nav isoform is involved in INaL generation in both normal and failing human hearts.
A persistent background INa has been reported in rabbit [37] and rat [30] cardiomyocytes at potentials ranging from -120 to 0 mV. However, we did not find any TTX-blockable (25 μM) INa at potentials less than -80 mV in human or canine VCs (not shown), suggesting species-specific differences in background INa, so that the possible role of Nav in the regulation of resting potential suggested for some animals has not been confirmed for humans and dogs. Our previous finding also supports this result that TTX or STX decreased AP duration but did not change resting potential in human [6] or canine VCs [8].
Study Limitations
There are two major limitations for studies that use live human tissue: 1) the great difficulty of obtaining normal human hearts, and 2) patient-to-patient variability due to various treatments, aetiologies, sex, age, etc. It is well known that gene expression (in our case SCN5A) can be treatment-dependent; for example, the beta adrenoreceptor blockers commonly used in patients with HF can alter INa density [24].
Conclusions
Chronic HF alters late Na+ current in VCs; the current increases and becomes slower, thereby carrying a significantly greater Na+ influx and contributing to abnormal repolarization and beat-to-beat AP duration variability. Interventions normalizing INaL can rescue functional AP profile in HF VCs. The mechanism that causes slowing of INaL decay in HF is related to modulation of gating (e.g., late channel gating modes [11]) of the predominant cardiac channel isoform Nav1.5 rather than expression of a different Nav isoform.
Supplementary Material
Acknowledgments
This study was supported in part by grants from the National Heart, Lung and Blood Institute [HL-53819, HL074238 (AI Undrovinas), HL-49090 (HN Sabbah)], and by a grant-in-aid from the American Heart Association (0350472Z; AI Undrovinas).
References
- 1.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 2.Bayes de Luna A, Coumel P, Leclercq JF. Ambulatory sudden cardiac death: mechanisms of production of fatal arrhythmia on the basis of data from 157 cases. Am Heart J. 1989;117:151–159. doi: 10.1016/0002-8703(89)90670-4. [DOI] [PubMed] [Google Scholar]
- 3.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 4.Bennett PB, Yazawa K, Makita N, et al. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 6.Maltsev VA, Sabbah HN, Higgins RSD, et al. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 7.Undrovinas AI, Maltsev VA, Kyle JW, et al. Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–1489. doi: 10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- 8.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: Role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valdivia CR, Chu WW, Pu J, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92:iv1–iv5. doi: 10.1136/hrt.2005.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res. 2006;69:116 – 127. doi: 10.1016/j.cardiores.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabbah HN, Stein PD, Kono T, et al. A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. Am J Physiol. 1991;260:H1379–1384. doi: 10.1152/ajpheart.1991.260.4.H1379. [DOI] [PubMed] [Google Scholar]
- 13.Sabbah HN, Goldberg AD, Schoels W, et al. Spontaneous and inducible ventricular arrhythmias in a canine model of chronic heart failure: relation to haemodynamics and sympathoadrenergic activation. Eur Heart J. 1992;13:1562–1572. doi: 10.1093/oxfordjournals.eurheartj.a060102. [DOI] [PubMed] [Google Scholar]
- 14.Sakmann BF, Spindler AJ, Bryant SM, et al. Distribution of a persistent sodium current across the ventricular wall in guinea pigs. Circ Res. 2000;87:910–914. doi: 10.1161/01.res.87.10.910. [DOI] [PubMed] [Google Scholar]
- 15.Undrovinas AI, Maltsev VA, Silverman NA. Transmural functional expression of Na+channel in normal and failing myocardium. Circulation. 2000;102:II–594. [Google Scholar]
- 16.Maltsev VA, Sabbah HN, Tanimura M, et al. Relationship between action potential, contraction-relaxation pattern, and intracellular Ca2+ transient in cardiomyocytes of dogs with chronic heart failure. Cell Molec Life Sci. 1998;54:597–605. doi: 10.1007/s000180050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Undrovinas AI, Belardinelli L, Undrovinas NA, et al. Ranolazine improves abnormal repolarizatio and contraction in left ventricular myocytes of dog with heart failure by inhibiting late sodium current. J Cardiovasc Electrophys. 2006;17:S1–S9. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaab S, Nuss HB, Chiamvimonvat N, et al. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res. 1996;78:262–273. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- 19.Galinier M, Vialette JC, Fourcade J, et al. QT interval dispersion as a predictor of arrhythmic events in congestive heart failure. Importance of aetiology Eur Heart J. 1998;19:1054–1062. doi: 10.1053/euhj.1997.0865. [DOI] [PubMed] [Google Scholar]
- 20.Boccalandro F, Velasco A, Thomas C, et al. Relations among heart failure severity, left ventricular loading conditions, and repolarization length in advanced heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 2003;92:544–547. doi: 10.1016/s0002-9149(03)00722-7. [DOI] [PubMed] [Google Scholar]
- 21.Hasenfuss G, Schillinger W, Lehnart SE, et al. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–648. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]
- 22.Spencer CI, Sham JS. Effects of Na+/Ca2+ exchange induced by SR Ca2+ release on action potentials and afterdepolarizations in guinea pig ventricular myocytes. Am J Physiol. 2003;285:H2552–2562. doi: 10.1152/ajpheart.00274.2003. Epub 2003 Aug 2521. [DOI] [PubMed] [Google Scholar]
- 23.Maltsev VA, Sabbah HN, Undrovinas AI. Late sodium current is a novel target for amiodarone: Studies in failing human myocardium. J Mol Cell Cardiol. 2001;33:923–932. doi: 10.1006/jmcc.2001.1355. [DOI] [PubMed] [Google Scholar]
- 24.Maltsev VA, Sabbah HN, Undrovinas AI. Down-regulation of sodium current in chronic heart failure: effects of long-term therapy with carvedilol. Cell Mol Life Sci. 2002;59:1561–1568. doi: 10.1007/s00018-002-8529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zicha S, Maltsev VA, Nattel S, et al. Post-transcriptional alterations in the expression of cardiac Na+ channel subunits in chronic heart failure. J Mol Cell Cardiol. 2004;37:91–100. doi: 10.1016/j.yjmcc.2004.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Despa S, Islam MA, Weber CR, et al. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 27.Studer R, Reinecke H, Bilger J, et al. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- 28.Pieske B, Maier LS, Piacentino V, 3rd, et al. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–453. doi: 10.1161/01.cir.0000023042.50192.f4. [DOI] [PubMed] [Google Scholar]
- 29.Heinemann SH, Terlau H, Imoto K. Molecular basis for pharmacological differences between brain and cardiac sodium channels. Pflugers Arch. 1992;422:90–92. doi: 10.1007/BF00381519. [DOI] [PubMed] [Google Scholar]
- 30.Saint DA, Ju YK, Gage PW. A persistent sodium current in rat ventricular myocytes. J Physiol. 1992;453:219–231. doi: 10.1113/jphysiol.1992.sp019225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maier SK, Westenbroek RE, Schenkman KA, et al. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–4078. doi: 10.1073/pnas.261705699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haufe V, Cordeiro JM, Zimmer T, et al. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res. 2005;65:117–127. doi: 10.1016/j.cardiores.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 33.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressedy sensory neurones. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 34.Satin J, Kyle JW, Chen M, et al. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science. 1992;256:1202–1205. doi: 10.1126/science.256.5060.1202. [DOI] [PubMed] [Google Scholar]
- 35.Undrovinas AI, Mishra S, Undrovinas NA.Silencing of SCN5A gene by siRNA descrease late sodium current and action potential duration in ventricular cardiomyocytes from dogs with chronic heart failure Circulation 2005112II–127.s [Google Scholar]
- 36.Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zilberter YI, Starmer CF, Starobin J, et al. Late Na channels in cardiac cells: the physiological role of background Na channels. Biophys J. 1994;67:153–160. doi: 10.1016/S0006-3495(94)80464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagatomo T, Fan Z, Ye B, et al. Temperature dependence of early and late currents in human cardiac wild-type and long Q-T ΔKPQ Na+ channels. Am J Physiol. 1998;275:H2016–H2024. doi: 10.1152/ajpheart.1998.275.6.H2016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.