

Abstract
Several approaches have been used in an effort to identify proteins that interact with β-amyloid precursor protein (APP). However, few studies have addressed the identification of proteins associated with APP in brain tissue from patients with Alzheimer’s disease. We report the results of a pilot proteomic study performed on complexes immunoprecipitated with APP in brain samples of patients with Alzheimer’s disease and normal control subjects. The 21 proteins identified could be grouped into five functional classes: molecular chaperones, cytoskeletal and structural proteins, proteins involved in trafficking, adaptors, and enzymes. Among the proteins identified, six had been reported previously as direct, indirect, or genetically inferred APP interactors. The other 15 proteins immunoprecipitated with APP were novel potential partners. We confirmed the APP interaction by Western blotting and coimmunolocalization in brain tissues, for 5 of the 21 interactors. In agreement with previous studies, our results are compatible with an involvement of APP in axonal transport and vesicular trafficking, and with a potential association of APP with cellular protein folding/protein degradation systems.
Alzheimer’s disease (AD), a progressive dementia affecting approximately 10% of people older than 65 years, is characterized by the accumulation of amyloid plaques, neurofibrillary tangles, and the progressive loss of synapses and neurons in the brain.1–3
The major proteinaceous component of amyloid plaques is amyloid-β peptide (Aβ), a variably sized (typically 40–42 residues) peptide derived from the amyloid precursor protein (APP). APP is an integral membrane protein whose transmembrane region contains the carboxyterminal portion of the Aβ peptide.4 Proteolytic processing by α, β, and γ secretases causes the poorly soluble Aβ1–42 (as well as Aβ 1–40), the soluble p3 peptide, and a 99-amino acid carboxyterminal fragment, among other cleavage products.5 The cytosolic region has a caspase recognition site at Asp 664 that generates a cytotoxic 31-amino acid C-terminal fragment, C31.6–9 Although the mechanisms by which APP generates Aβ are becoming increasingly well understood, the physiological function of APP remains unknown. However, APP has long been known to accumulate at nerve terminals.10 Recent studies have pointed to a function for APP in axonal trafficking,11,12 in cellular motility,13,14 in vesicular transport,12 and in the regulation of synaptic function.15 Yeast two-hybrid screens of mouse and human brain libraries have identified various proteins that interact with the intracellular domain of APP,16–22 including the nuclear adaptor protein Fe65, X11, Mint 3 and neuron-specific Mint 1 and 2,23 Dab1 (disabled gene 1),18 JIP-1,17 and IB1 (Jip-1b), which scaffolds APP with JNK.19
Whereas two-hybrid screens and cell culture studies examine protein–protein interactions in settings that often differ considerably from those in which the function of the protein of interest is performed, microarray techniques and two-dimensional gel electrophoresis allow for the profiling of gene and protein expression patterns directly from tissue samples. Several proteomic studies have examined the complete proteome in AD versus nondiseased human tissues.24,25 A study by Yoo and colleagues26 used matrix-associated laser desorption ionization (MALDI) mass spectrometry to identify and quantify chaperone proteins in AD and normal brains. In this study, six chaperone proteins including heat shock 70 kDa protein 1 (HSP70.1), heat shock cognate protein 71 (HSC71), and αB-crystallin, were found to be aberrantly expressed in different regions of the AD brain. In addition, Castegna and colleagues27 identified proteins that are targets for oxidation and nitration in AD brains using a proteomics approach.
Although typically less sensitive than one-dimensional (1D) electrophoresis followed by Western blotting, two-dimensional electrophoresis of immunoprecipitated material followed by mass spectrometry does not require the prediction of candidate interactors. We have used two-dimensional gel electrophoresis combined with mass spectrometry to identify proteins present in complexes coimmunoprecipitated with APP from brain tissues of patients with AD and normal control subjects. The use of antibodies that specifically recognize (1) the extracellular domain of APP28; (2) the 15 C-terminal amino acids of APP,29 and (3) the neoepitope that is generated after C-terminal cleavage of APP at Asp6647 allowed us to explore the interactions of APP that may involve the C-terminal 31-amino acid stretch that contains motifs required for the interaction of APP with several cytosolic proteins.
We have identified 21 proteins in complexes with APP, of which 15 were found to be novel potential interactors. The identities of 5 of these 21 potential APP interactors (2 previously described interactors and 3 of the 15 potential novel interactors) were confirmed by selective immunoprecipitation followed by immunoblotting or immunohistochemistry. The functional grouping of known and novel interactors is consistent with a role of APP in trafficking and vesicular transport and confirms the association of APP with molecular chaperones and motor proteins.
Materials and Methods
Sample Preparation and Coimmunoprecipitations
Human brain extracts were prepared from frozen autopsy material. Hippocampus was provided by the Harvard Brain Bank Tissue Resource Center, which is supported by Public Health Service (PHS) grant number MH/NS 31862. Cortical tissue was provided by the University of California, San Diego Brain Bank. Extracts for coimmunoprecipitation were solubilized in 0.15M NaCl, 0.01M tris(hydroxymethyl)aminomethane (Tris), 1.0% Triton X-100 (Sigma, St. Louis, MO), pH 8.0, containing a cocktail of protease inhibitors (Mini-Complete; Roche, Indianapolis, IN). Extracts were maintained at 4°C. Homogenization of 50mg/ml tissue (wet weight) was performed using an Ultra-Turrax T8 polytron (IKA-Werke GMBH & Co. KG, Staufer, Germany). Extracts were centrifuged for 10 minutes at 14,000 g and pre-cleared with Protein A-Agarose (Pierce, Rockford, IL). Antibody (20μg/ml) was added to 0.2ml extract. Protein A/G Microbeads (Santa Cruz Biotechnology, Santa Cruz, CA) were added, and the mixture was incubated on ice for 30 minutes. MACS separation columns (Miltyeni Biotec, Auburn, CA) were used according to the manufacturer’s specifications. Proteins were eluted from the MACS columns with BioRad Reagent 3 (5M urea, 2M thiourea, 0.01M Tris, 2% CHAPS, and 2% ampholytes; BioRad, Hercules, CA) in preparation for two-dimensional gel electrophoresis.
Antibodies
Polyclonal CT1529 recognizes the C-terminal 15 residues of APP. APPNeo recognizes the neoepitope generated after cleavage of APP695 at Asp664. 22C11 (Chemicon, Temecula, CA) recognizes an epitope on the extracellular domain of APP. Antibodies specific for HSC70, HSP90, αB-crystallin, and N-ethylmaleimide sensitive factor (NSF) were purchased from StressGen (Victoria, British Columbia, Canada). Antibodies specific for dynamin, dynein, kinesin, and myelin basic protein were purchased from Chemicon. Antibodies specific for 14-3-3 were purchased from Santa Cruz, and those specific for Fe65 and Ask-1 were purchased from BioSource (Piscataway, NJ).
Electrophoresis and Imaging
Isoelectric focusing was performed on a BioRad Protean II IEF (isoelectric focusing) unit using 11cm immobilized pH gradient (IPG) strips (3–10, linear; BioRad). Protein mixtures were reduced with tributylphosphine (20μM); IPG strips were rehydrated for 12 hours (50V) and focused with a linear gradient (250–8,000V) for 35,000V–hr. IEF strips were either frozen at −70°C or equilibrated in urea sodium dodecyl sulfate (SDS) for SDS polyacrylamide gel electrophoresis. Equilibration buffer for SDS polyacrylamide gel electrophoresis was 6M urea, 2% SDS, 20% glycerol, 0.01M Tris, pH 8.8. Disulfides were reduced with 2% dithiothreitol (DTT) for 10 minutes, followed by a 10-minute alkylation reaction with 2.5% iodoacetamide. Equilibrated strips were transferred to BioRad Criterion gels (8–16%; BioRad) and overlaid with low-melt agarose. Electrophoresis was performed at 200V for 55 minutes. Gels were fixed with 7% acetic acid/10% methanol, poststained with Sypro Ruby (Molecular Probes, Eugene, OR) and destained with 7% acetic acid/10% methanol. Gels were imaged on a Typhoon 8500 (Amersham Biosciences (Piscataway, NJ)). A transilluminator was used to visualize the proteins, which were excised with a scalpel and stored at −80°C until trypsin digestion and mass spectrometry.
Western Blotting
Ten percent of the R3 eluate was mixed with NuPage (In-vitrogen, La Jolla, CA) sample buffer and reduced with β-mercaptoethanol for 1 hour at room temperature (RT). Electrophoresis was on a 10% NuPage gel using 2-(N-morpholino)-ethanesulfonic acid (MES) buffer. Proteins were transferred to nitrocellulose (0.45μm; BioRad) using NuPage sample buffer. After transfer, membranes were stained with Sypro Ruby (Molecular Probes), imaged, and blocked with 5% milk at 4°C overnight. Antibodies were used at the dilution recommended by the manufacturer. Horseradish peroxidase secondary antibodies were developed with horseradish peroxidase–developing reagent (Amersham Biosciences).
Mass Spectrometry
Excised spots were digested with trypsin using a DigestPro robot (Abimed, Langenfeld, Germany). Peptides were extracted from the gel spots with 5% acetonitrile/2% trifluoracetic acid (TFA). The extract was dried under vacuum, and the peptides were resuspended with sonication in 5% acetonitrile/2% TFA. MALDI matrix solution was prepared by mixing a 2:1:1 solution of recrystallized α-cyano-4-hydroxycinnamic acid (40mg/ml in acetone):nitrocellulose, Transblot (20mg/ml in acetone; BioRad):isopropanol. Matrix solution was mixed 1:1 with the peptide mixture, spotted on MALDI target, and air-dried. The spots were washed once with 5% formic acid, dried briefly with heating, then rinsed with Milli-Q water (Millipore, Billerica, MA) and dried with moderate heat. MALDI data were acquired with an ABI Voyager-DE Pro MALDI-TOF mass spectrometer (ABI, Foster City, CA). Spectra were manually collected (500 shots/spectra) in Reflector mode using instrument software (Voyager Instrument Control Panel version 5.10, Applied Biosystems, Foster City, CA). The data were then analyzed in ABI Data Explorer (v4.0.0.0; ABI) with internal autolytic tryptic peptides (842.5099 and 2211.0968 daltons) as calibrants. Protein identification was performed using Profound Peptide Mapping software v4.10.5 (http://prowl.rockefeller.edu/profound_bin/WebProFound.exe). Manual nanospray was performed on selected samples as necessary. Digests of tryptic peptide were purified on Poros R2 (Perspective Biosystems, Foster City, CA) packed into glass capillaries. Digests were bound to the purification capillaries, washed five times with 5% formic acid, and eluted directly into a Pd-coated nanospray tip with 50% methanol/5% formic acid. Analysis was performed on a Sciex API3000 triple quadrupole mass spectrometer (MDS Sciex, Concord, Canada). Tandem mass spectrometry spectra from selected peptide ions were searched against the NCBInr (National Center for Biotechnology Information) database using the MASCOT algorithm running on an in-house server.30
Immunohistochemistry
Seven-micrometer microtome sections were deparaffinized in xylene, rehydrated in 100, 95, 80, and 70% ethanol, and then washed in 1X Tris-buffered saline (TBS) for 15 minutes at RT. Microwave antigen retrieval was performed in 10mM citrate buffer (pH 6.0) for 5 minutes at 440 watts. Slides were allowed to cool to RT, and then were washed in 1X TBS for 10 minutes. Samples were blocked in 10% normal donkey serum in 1X TBS for 1 hour at RT. Primary antibody or rabbit preimmune IgG (Sigma) diluted to 1μg/ml in 1% bovine serum albumin, 1X TBS was applied to sections. Sections were incubated overnight at 4°C followed by three 20-minute washes in 1X TBS. Alexa 488- and Alexa 555-conjugated secondary antibodies (Molecular Probes) were applied at 1:250 in 1% bovine serum albumin, 1X TBS, and incubated in the dark for 1 hour at RT. Sections then were washed four times in 1X TBS for 30 minutes and incubated in a 1:250 dilution of TOTO3 (Molecular Probes) for 20 minutes. Sections were subject to two 10-minute washes in 1X TBS and mounted in Vectashield mounting medium for fluorescence (Vector Laboratories). Single confocal images or z-stacks of images were acquired using a Nikon Eclipse-800 microscope (Nikon, Melville, NY) and collected using Compix Simple PCI software (Compix Incorporated, Lake Oswego, OR). Deconvolution was done using a maximum likelihood estimation algorithm with Huygens software and processed by the Imaris imaging interphase (Bitplane AG, Zurich, Switzerland). All confocal images then were processed in a SGI Octane R12 computer running Bitplane’s Advanced Imaging Software suite. Analysis of colocalization was done using the Colocalization algorithm in the Imaris Bitplane Suite.
Results
CT15 and APPNeo Immunoprecipitate C-Terminally Intact and C-Terminally Cleaved Forms of Amyloid Precursor Protein
As shown in Figure 1, both CT15 and APPNeo immunoprecipitated APP-immunoreactive material from hippocampal, parietal, and frontal cortices of AD and nondiseased control brain lysates (see Figs 1A, B). The specificity of CT15 and APPNeo reactivity was confirmed by the absence of APPNeo-immunoreactive material in complexes immunoprecipitated with CT15, but not in those immunoprecipitated with 22C11 or with APPNeo (see Fig 1C).
Fig 1.

(A) Amyloid protein precursor (APP) was immunoprecipitated with antibodies specific for C-terminally intact (CT15) and C-terminally cleaved (APPNeo) forms of APP. Material immunoprecipitated from normal and Alzheimer’s disease (AD) brain lysates with CT15 and APPNeo antibodies was resolved in gels and immunoblotted with an antibody that recognizes its extracellular portion (22C11). (B) As in (A), CT15 and APPNeo antibodies. (C) APP was immunoprecipitated from lysates of AD or normal control brains with CT15, APPNeo, and 22C11 antibodies and was immunoblotted with APPNeo. hip = hippocampus; par = parietal cortex.
Identification of Amyloid Precursor Protein Interactors in Human Brains
To determine potential interaction partners of APP in AD and nondiseased brains, we used CT15 and APPNeo in immunoprecipitation assays followed by two-dimensional gel electrophoresis. A representative gel is shown in Figure 2.
Fig 2.

Material present in complexes immunoprecipitated with α-amyloid precursor protein (α-APP) antibodies was separated in two-dimensional gels (a representative example is shown) and stained with Sypro Ruby (Molecular Probes), as described in Materials and Methods. Spots corresponding to unambiguously identified proteins present in complexes with APP-immunoreactive material are highlighted.
Twenty-one proteins that were present in complexes immunoprecipitated with APP were unambiguously identified by mass spectrometry (Table). These proteins could be grouped into five functional classes: molecular chaperones, cytoskeletal proteins, proteins involved in vesicular and axonal trafficking, adaptors, and enzymes. All were present in both AD and control brains. Although three additional spots were observed to be present differentially in AD and control brains, these were present at levels less than the sensitivity of the sequencing system used; therefore, sequence data were not obtained from these three.
Table.
Summary of Proteins Identified as co-immunoprecipitants with Amyloid Precursor Protein
| Protein | Immunoprecipitation | Z/Mowse Score | Percentage of Coverage/No. of Peptides |
|---|---|---|---|
| Trafficking and motor proteins, mechanochemical enzymes | |||
| NEM sensitive factor | AD IP neo, CT15 | 1.27 | 16%/11 |
| Syntaxin binding protein 1, Munc18-1,rbSec1 | AD IP CT15 | 1.47 | 15%/9 |
| Dynamin-1 | AD IP neo, CT15 | 2.12 | 25/18 |
| Dynein | AD IP neo, CT15 | 0.26 | 16%/6 |
| Myosin, nonmuscle | AD IP CT15 | (MS-MS) | |
| Chaperones | |||
| Hsp 90 alpha (HSP 86) | AD IP neo, CT15 | 2.04 | 21%/14 |
| HSC 71 | AD IP neo, CT15 | 2.29 | 43%/24 |
| Crystallin alpha-2 | AD IP neo, CT15 | 2.31/41 | 52%/8 |
| Cyclophilin A/cyclosporin | AD IP neo | 1.74 | 59%/11 |
| Alpha spectrin (alpha-fodrin) | AD IP CT15 | 2.29 | 11%/26 |
| Glial fibrillary acidic protein | AD IP neo | (multi.id) | |
| Cytoskeletal and structural | |||
| Beta tubulin | AD IP neo, CT15 | 1.44/59 | 32%/11 |
| Beta actin | AD IP neo, CT15 | 1.44 | 27%/11 |
| Neurofilament light polypeptide | AD IP neo | ||
| Mylein basic protein | AD IP CT 15 | (Western) | |
| Adaptors | |||
| 14-3-3 protein zeta | AD IP neo | 1.59/92 | 35%/9 |
| Fe65 | AD IP CT15 | (Western) | |
| Enzymes | |||
| Ubiquitin carboxy-term hydrolase isozyme L1 | AD IP neo | 77 (mouse) | |
| Phosphoglycerate mutase A | AD IP neo | 1.78 | 41%/7 |
| Uracil DNA glycosylase | AD IP neo | 1.93 | 23%/6 |
| Ask1 | AD IP neo | (Western) | |
Among the proteins identified, six had been reported previously as direct, indirect, or genetically inferred APP interactors. The remaining 15 proteins were novel potential partners. The pattern of immunoprecipitated proteins was similar for CT15 and APPNeo antibodies except in a few cases in which immunoprecipitation was specific for one of the two antibodies (see the Table). The unambiguous identification of CT15-specific versus APPNeo-specific complexes was complicated by the finding that APPNeo bound its epitope with a much greater affinity than CT15. Thus, no conclusions could be made about the mutual exclusivity of binding, except for those cases in which the candidate proteins were found in CT15-immunoprecipitated, but not in APPNeo-immunoprecipitated, complexes. Consistent with this interpretation, Fe65 was found in complexes with APP-immunoreactive material only when CT15, but not when APPNeo, was used as the immunoprecipitating antibody (see the Table and Fig 2). The specificity of the immunoprecipitation reactions was tested in preparative experiments by peptide competition (data not shown) and by the use of control rabbit IgGs.
Confirmation of the Identities of Amyloid Precursor Protein Interactors
The identity of 2 of 6 previously described APP interactors, and 4 of the 15 novel potential APP interactors, was confirmed by immunoprecipitation followed by Western blotting, or by immunohistochemistry in mouse brains. As described previously for a variety of experimental models,13,14,20,21,31–33 we found that the adaptor protein Fe65 was present in complexes immunoprecipitated from AD brains when CT15, but not when APPNeo, antibodies were used (Fig 3A). Conversely, 14-3-3 (another adaptor protein that has been found in association with neurofibrillary tangles,34 has been proposed to favor the formation of tau fibrillar polymers,35,36 and has been used routinely as a clinical marker for Creutzfeldt–Jakob disease when detected in cerebrospinal fluid37) was found to be present in complexes immunoprecipitated from AD brains with APPNeo but not with CT15 (see Fig 3B).
Fig 3.

(A) Identification of Fe65 in complexes immunoprecipitated with CT15 and APPNeo from lysates of Alzheimer’s disease (AD) brains by one-dimensional electrophoresis and Western blotting. (B) Identification of 14-3-3 in complexes immunoprecipitated with CT15, APPNeo, and 22C11 from lysates of AD brains by one-dimensional electrophoresis and Western blotting. (C) Identification of αB-crystallin in complexes immunoprecipitated with CT15 and APPNeo antibodies from lysates of normal and AD brains by one-dimensional electrophoresis and Western blotting. (D) Sections from brains of human amyloid precursor protein (hAPP) transgenic animals were stained with antibodies to αB-crystallin together with CT15 or APPNeo, followed by Cy3-conjugated anti–rabbit or Cy5-conjugated anti–mouse secondary antibodies. Confocal images were obtained independently for each channel and overlaid as shown. BF = basal forebrain; FC = frontal cortex; Par = parietal cortex.
αB-crystallin
The interaction of Aβ- and αB-crystallin has been documented in in vitro systems38,39 and in a transgenic Caenorhabditis elegans model.40,41 We identified αB-crystallin in complexes immunoprecipitated from AD and nondiseased control brain lysates with both CT15 and APPNeo by immunoprecipitation with CT15 and immunoblotting. As shown in Figure 3C, αB-crystallin immunoreactivity was present in complexes immunoprecipitated both from normal and AD brains, regardless of the region analyzed. As noted earlier, no conclusions could be made about the relative amounts of protein immunoprecipitated with CT15 versus APPNeo, given that the two antibodies showed different affinities for their respective epitopes. The association between APP and αB-crystallin was further documented in brain sections of human APP (hAPP) transgenic mice that carry two familial Alzheimer’s Disease (FAD)-associated mutations (Swedish, 670K→N/671M→L; Indiana, 717V→F).42,43 These transgenic animals were chosen for study because they show moderate levels of hAPP minigene expression (1.5- to 2-fold greater than endogenous mouse APP protein expression levels)42 and represent a well-established mouse model of AD. Sections from hAPP transgenic animals were stained with antibodies recognizing αB-crystallin together with CT15 or APPNeo. As shown in Figure 3D, αB-crystallin immunoreactivity was pronounced in neuronal bodies in a compartment juxtaposed to the nucleus and colocalized with CT15- and APPNeo-immunoreactive material in brains from hAPP transgenic animals. To explore further the association between APP and αB-crystallin, we examined the intracellular distribution of APP-immunoreactive and αB-crystallin–immunoreactive material in cells from hAPP-transgenic and nontransgenic mouse brains. As shown in Figure 4, CT15-immunoreactive material colocalized with αB-crystallin in a compartment juxtaposed to the nucleus of cells both in brains of hAPP transgenic and nontransgenic animals. The extent of colocalization of endogenous C-terminally cleaved mouse APP and αB-crystallin in nontransgenic brains was comparable with that found in brains from hAPP transgenic animals (see Fig 4). The Pearson correlation coefficient of colocalized material in the region of interest considered (c) was used as a measure of the degree of colocalization.44 Quantitation of c for the colocalization of APPNeo- and αB-crystallin–immunoreactive signals yielded similar values in hAPP transgenic and nontransgenic brains, indicating that endogenous C-terminally cleaved mouse APP associates with αB-crystallin in mouse brain tissues, and that this association is not disrupted by the expression of a human FAD-APP transgene. We did not observe any significant increase in the amount of colocalized APPNeo-immunoreactive and αB-crystallin–immunoreactive material in hAPP mouse transgenic (compared with non-transgenic) brains (c = 0.47 and c = 0.57, respectively).
Fig 4.
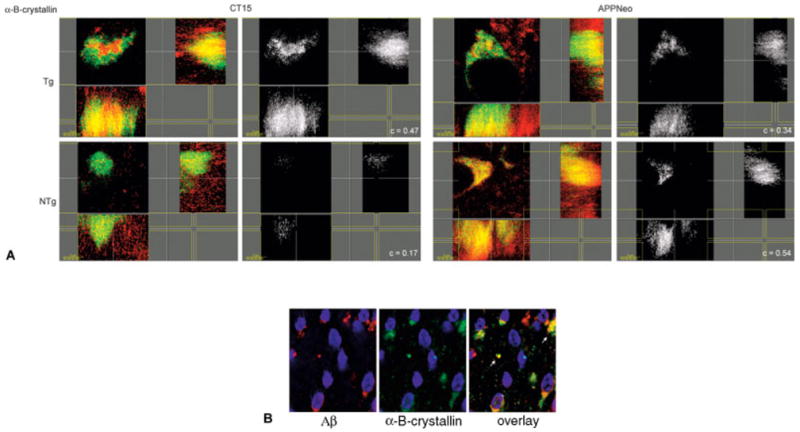
(A) Sections from brains of human amyloid precursor protein (hAPP) transgenic animals were stained with antibodies to αB-crystallin together with CT15 or APPNeo, followed by Cy3-conjugated anti–rabbit or Cy5-conjugated anti–mouse secondary antibodies. z-stacks of confocal images were obtained at high magnification (×3,000 = ×1,000, 3× digital zoom) and processed with Imaris Section, Surpass, and Colocalization applications (Bitplane AG). Pixels displaying colocalization of the signal in both channels were identified using the Colocalization software (Imaris Bitplane, Zurich, Switzerland) and are shown in white. c values corresponding to the Pearson correlation coefficient for the signal values in both channels in the volume of interest were determined. Representative two-dimensional sections across the volumes analyzed are shown. c values for colocalization are shown in the bottom right corner of each panel. (B) Confocal images of sections from brains of hAPP transgenic animals stained with anti-αB-crystallin and anti-Ab (4G8) antibodies and reacted with Cy5-conjugated anti–rabbit or Cy3-conjugated anti–mouse secondary antibodies.
In contrast, a marked increase in C-terminally intact APP (ie, immunoreactive with the CT15 antibody) colocalizing with αB-crystallin was observed in hAPP transgenic compared with nontransgenic mouse brains (c = 0.47 and c = 0.17, respectively), suggesting that the C-terminally intact product of the human APP transgene may associate with αB-crystallin in mouse brains.
Considering that HSP16, the C. elegans ortholog of αB-crystallin, is robustly induced by the expression of Aβ in C. elegans and colocalizes with Aβ deposits, we sought to determine whether αB-crystallin may also associate with Aβ, or with forms of APP containing the Aβ peptide, in vivo. Coimmunolocalization experiments were performed in brains from transgenic mice that accumulate high levels of Aβ early in life (2–3 weeks postnatally). αB-crystallin– and Aβ-immunoreactive signals were found in the perinuclear compartment of all immunoreactive cells, and they were colocalized in deposits in that compartment or in the cytoplasm of cortical neurons in transgenic animals (see Fig 4B). Taken together, our results indicate that αB-crystallin can associate with C-terminally intact and C-terminally cleaved forms of APP in vivo, and that this interaction may involve Aβ- or Aβ-containing forms of APP.
Dynamin
Two recent reports have pointed to a requirement for dynamin function in the modulation of α- and β-secretase cleavage of APP, with retention of the mature form of the protein at the cell surface when endocytosis was blocked by the expression of a dominant negative dynamin mutant.45,46 The impact of endocytosis inhibition in the generation of Aβ peptide is still an unresolved question.46–48 We confirmed the presence of dynamin in complexes immunoprecipitated with anti-APP antibodies from basal forebrain, frontal cortex, and parietal cortex of AD and nondiseased brains. As shown in Figure 5A, dynamin-immunoreactive bands were present in complexes immunoprecipitated with both CT15 and APPNeo from AD and non-AD control brains. An apparent decrease in the amount of dynamin-immunoreactive material immunoprecipitated from frontal and parietal cortices of AD versus non-diseased brains was observed. Dominant negative mutants of dynamin have been reported to significantly reduce the amounts of Aβ peptide generated by proteolytic cleavage of APP,45,46 suggesting that the presence of APP in lipid rafts on the cell surface46 and its recruitment to dynamin-containing endocytic vesicles45 is important for the processes that result in the generation of Aβ.
Fig 5.
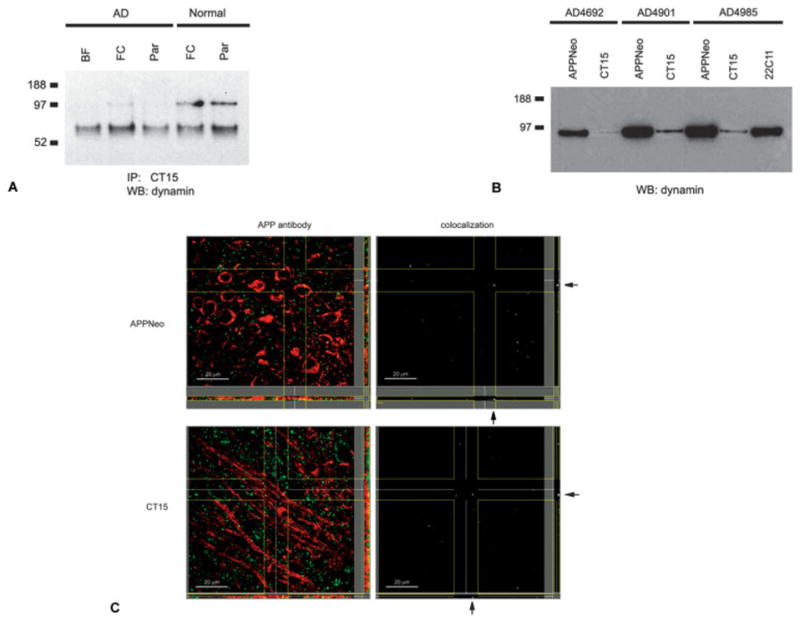
(A) Identification of dynamin in complexes immunoprecipitated with CT15 from lysates of Alzheimer’s disease (AD) and normal control brains by one-dimensional electrophoresis and Western blotting. (B) Identification of dynamin in complexes immunoprecipitated with APPNeo, CT15, and 22C11 from AD brains by one-dimensional electrophoresis and Western blotting. (C) Maximum projection arrangement for z-stacks of confocal images of brain sections of human amyloid precursor protein (hAPP) transgenic mice stained with antibodies specific for dynamin and CT15 or APPNeo. Dynamin immunoreactivity is shown in green; CT15 and APPNeo are shown in red. BF = basal forebrain; FC = frontal cortex; Par = parietal cortex.
Immunohistochemical analyses were performed on brains of FAD-hAPP transgenic and nontransgenic mice. Dynamin immunoreactivity, which was distributed in a punctate pattern throughout the brain but was excluded from fiber tracts, colocalized both with APPNeo and CT15 in a small fraction of those puncta, both in transgenic (see Fig 5C) and nontransgenic mouse brains. Although the channel correlation value for the colocalization of immunoreactive signals at those foci was significant (0.53 and 0.48, respectively), the paucity of double-labeled dynamin-immunoreactive puncta in mouse brains can be interpreted as a negative indication with respect to the interaction of dynamin and APP in vivo. Given the high correlation value for the colocalization of the signals, however, it is possible that the interaction of APP and dynamin in the endocytic compartment is sensitive to disruption by the fixation procedures used.
Dynein
A report indicating a role for APP in axonal trafficking11 demonstrated that APP interacts with kinesin, a plus-end–directed motor and the main representative of one of the three main classes of distinct motor proteins: kinesin, dynein, and myosin.49,50 Interestingly, we detected members of the other two main classes of motor proteins, dynein and myosin, in complexes immunoprecipitated from AD and non-diseased control brain lysates with both CT15 and APPNeo (note that the Z-score for dynein was relatively low [see the Table], which may have been caused by the presence of other proteins within the same isolated gel spot; however, the presence of dynein in APP-immunoprecipitated complexes was confirmed by Western blotting). Immunoprecipitation of C-terminally intact and C-terminally cleaved forms of APP from hAPP transgenic and non-transgenic mouse brains followed by Western blotting confirmed the presence of dynein in APP-based complexes in human and mouse brains (data not shown). Increased amounts of dynein-immunoreactive material were found in complexes immunoprecipitated with C-terminally intact forms of APP, suggesting that the C-terminal 15 amino acids of APP were required for the formation of the complex (or enhanced the interaction). To validate these in vitro results, we performed immunohistochemical analyses on FAD-hAPP transgenic and nontransgenic mouse brains. Our studies showed that the association of APP with dynein in mouse brains was robust and dependent on the C-terminal 15 amino acids of APP (Fig 6). This was in contrast with the pronounced compartmentalization of APPNeo and dynein immunoreactivities in FAD-hAPP transgenic and non-transgenic mouse brains (compare top and bottom panels in Fig 6B). These results suggest that the interaction of both endogenous mouse APP and human APP with dynein occurs preferentially in fiber tracts in the mouse brain, and that the C-terminal 15 amino acids of APP are required for this interaction.
Fig 6.

Snapshots of maximum projections of z-stacks of confocal images processed in Imaris (Bitplane AG) of brain sections of human amyloid precursor protein (hAPP) transgenic mice stained with antibodies specific for dynamin and CT15 or APPNeo as indicated. Dynein immunoreactivity is shown in the green channel; CT15 and APPNeo are shown in the red channel.
Discussion
We have used immunoprecipitation followed by a proteomic-type mass spectrometric analysis to identify molecules that associate with APP in the brains of patients with AD and in control brains from patients without neurological diseases. This approach has the advantage of allowing for the unbiased exploration of all possible interactors (subject to the sensitivity of the immunoprecipitation–mass spectrometry combination) in relevant settings such as diseased and nondiseased human brain tissues at a defined time in disease development. The main disadvantage of the method, which is shared by most biochemical approaches, is the required disruption of cell types and compartments at the time of lysis, potentially allowing for interactions that would not occur in intact, compartmentalized tissues or cells. Notwithstanding the latter caveat, potential bona fide interactors can be identified among the list of candidates by further immunohistochemical analyses, as we demonstrate in this study.
We have unambiguously identified 21 proteins in complexes immunoprecipitated with APP-specific antibodies from human AD brains. These proteins included APP interactors that had been described previously, such as Fe65 and αB-crystallin, indicating that we could effectively identify APP interactors in human brain tissues. Interestingly, all proteins identified were in a few functional groups: proteins involved in axonal transport, vesicle formation, and trafficking (dynamin, NEM [N-ethyl-maleimide]-sensitive factor, Munc18, dynein, myosin); cytoskeletal and structural proteins (spectrin, actin, tubulin, glial fibrillary acidic protein, neurofilament light polypeptide, myelin basic protein); chaperone proteins (αB-crystallin, HSP90, HSC71, cyclophilin A), adaptors (Fe65, 14-3-3), and enzymes (ubiquitin C-terminal hydrolase L-1 [UCH-L1], phosphoglycerate mutase A, uracil DNA glycosylase, ASK1). As noted earlier, one of the limitations of the approach used here is the bias toward proteins of high expression, and thus the failure to identify interactors of lower expression. It is likely that this limitation accounts for our failure to identify some of the known interactors of APP, such as kinesin, which was observed only when Western blotting was used.
Experimental evidence indicating a functional role for several of the identified proteins in the formation of complexes with APP was already available12,13,20–22,32,33,40 and provides additional support for our results. Munc18a recently was found to interact with the N-terminus of X-11, an APP-interacting protein that requires the presence of the YENPTY motif at the C-terminus of APP.51
It has been proposed that HSC70 depends on NEM-sensitive factor activity to release kinesin from trafficking vesicles.52 Our data provide further support for the notion that APP is involved in vesicle trafficking, as we identified a member of the HSC70 family, HSC71, together with NEM-sensitive factor, in complexes with APP in lysates of human brains. Although we did not identify kinesin in complexes immunoprecipitated with APP and resolved by two-dimensional electrophoresis, we were able to detect its presence in complexes immunoprecipitated by APP antibodies and Western blotting (data not shown). Consistent with the notion that APP may be involved in vesicle transport, we detected dynamin, a GTPase that regulates endocytosis, and dynein, a retrograde transport–specific motor protein, in complex with APP in human brain lysates. Confirming the previously described genetic interaction between APP and dynamin,45 we identified this protein in complexes with APP immunoprecipitated from human AD and nondiseased brains. Interestingly, a pronounced decrease in the amount of dynamin in APP-based complexes was observed in frontal and parietal cortices of AD compared with nondiseased brains. These results suggest that APP-mediated retrograde vesicle trafficking may be compromised in the brains of patients with AD.
Our data demonstrate that APP can interact also with a minus-end–directed motor protein, dynein, which also has been genetically linked to a putative function of APP in axonal transport.11 The interaction requires the presence of the C-terminal 15 amino acids of APP in vivo. This observation validates the rationale for the experimental approach chosen and indicates the usefulness of proteomics methods as a first-stage approach to the study of interactions of a protein of interest.
We also have identified several cytoskeletal components in APP-based complexes immunoprecipitated from human brain lysates. Actin is a fundamental constituent of the cytoskeleton and is required for the maintenance of cellular structure and a variety of processes dependent on cytoskeletal integrity, including the recycling of synaptic vesicles.53 Although APP was not present in compartments overlapping with tubulin-containing structures in one of these studies,13 our results suggest that APP may be associated with the microtubule-based cytoskeleton at some point during axonal trafficking.
Confirming previous reports (in the case of Fe6513,21,33) and consistent with the proposed role of the intracellular domain of APP in the assembly of signaling complexes, both Fe65 and 14-3-3, which have been implicated as adaptors in different signal transduction pathways,33,54 were found in APP-containing complexes immunoprecipitated from human brain lysates. Interestingly, Fountoulakis and colleagues55 had shown previously in a two-dimensional-based study that 14-3-3 levels are increased in the AD brain.
Spectrin (α-fodrin) links cortical actin to the plasma membrane.56 Interestingly, a genetic interaction between spectrin and the components of the γ-secretase complex presenilin and nicastrin has been found in Drosophila.57 Presenilin or nicastrin mutations that affect Notch processing were also found to disrupt the spectrin cytoskeleton. The identification of spectrin in APP-based complexes in human brain lysates may point to a physical association between APP and spectrin that may underlie the observed genetic effect. We also found that both glial fibrillary acidic protein and neurofilament light polypeptide are present in complexes immunoprecipitated with APP from human AD brains.
Similar to previous findings in C. elegans40 and in mammalian cell culture systems,58,59 several molecular chaperones were found in complexes with APP in human brain lysates. αB-crystallin is a major constituent of the lens and is abundantly expressed in other tissues, such as in the heart.60 This chaperone has been proposed to interact stoichiometrically with partially assembled amyloid precursors and inhibit their aggregation at the nucleation step.61 HSP16, the C. elegans αB-crystallin homolog, is induced robustly by the expression of Aβ in the worm and colocalizes with Aβ deposits.40 Interestingly, Aβ-associated toxicity in this model could be suppressed by inhibition of a negative regulator of HSP70 function, implying an important role for chaperones in Aβ-induced toxicity. Inhibition of proteasome activity has been shown to increase levels of αB-crystallin62 and facilitate the interaction of HSP73, a member of the HSP70 family of cytosolic chaperones, with APP.63 In this study, C-terminally cleaved forms of endogenous mouse APP were preferentially found in complexes with αB-crystallin in brain tissues when compared with C-terminally intact forms of the molecule.
Further supporting a role for alterations in protein folding/degradation homeostasis in AD, several reports have documented the involvement of the ubiquitin/proteasome system in pathogenic changes associated with the disease.64 We have identified a key activity in the ubiquitin pathway, UCH-L1, in complexes with APP in brain lysates. Interestingly, UCH-L1 had been identified previously as a target of protein oxidation specific to AD brains.27
Finally, ASK1, an apical mitogen-activated protein kinase kinase kinase that specifically activates the stress response through the stress-activated protein kinases JNK and p38 and is also required for sustained activation of the unfolded protein response,65 was found in complexes immunoprecipitated with APP from human brain lysates. Although we did not detect the presence of JIP adaptor proteins or JNK kinases in complexes immunoprecipitated with APP from human brain lysates using two-dimensional gel electrophoresis and mass spectrometric analysis, Western blotting of the immunoprecipitated material did show the presence of JIP-1 and JNK proteins in APP-containing complexes (these data resulted from a separate study and, together with data on the interaction of APP and ASK1, will be reported elsewhere).
In summary, this study offers a look at the spectrum of in vivo interactors with APP, and potentially with proteolytic fragments derived from APP, in AD and in control brains. Our results support previous suggestions that APP function may involve a role in axonal transport and vesicular trafficking, and also support its association with the protein folding/protein degradation machinery.
Acknowledgments
This work was supported by the NIH (National Institute on Aging, AG05131, D.E.B., E.H.K.; AG12282, D.E.B., National Institute of Neurological Disorders and Stroke, NS45093, D.E.B.), the Joseph Drown Foundation to the Buck Institute for Age Research, the Alzheimer’s Association (NIRG-04-1054, V.G.), and a John Douglas French Foundation Fellowship (V.G.).
We thank Dr L. Mucke for the PDAPP(J9) transgenic mice. We thank E. Goodhew Barnett for her support. We also thank B. Calagui for animal husbandry assistance and M. Susag for assistance in the preparation of the manuscript.
References
- 1.Mattson MP. Addendum: pathways towards and away from Alzheimer’s disease. Nature. 2004;431:107. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.National Institute of Mental Health. Clinical trials: Alzheimer’s disease and related disorders. Vol. 2005. Washington, DC: U.S. Department of Health and Human Services; 2005. [Google Scholar]
- 3.The Alzheimer’s Disease Education and Referral (ADEAR) Center. National Institute on Aging progress report on Alzheimer’s disease. Vol. 1998. Silver Spring, MD: U.S. Department of Health and Human Services, National Institutes of Health, National Institute on Aging; 1998. [Google Scholar]
- 4.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 5.Nunan J, Small DH. Proteolytic processing of the amyloid-beta protein precursor of Alzheimer’s disease. Essays Biochem. 2002;38:37–49. doi: 10.1042/bse0380037. [DOI] [PubMed] [Google Scholar]
- 6.Lu DC, Rabizadeh S, Chandra S, et al. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 7.Galvan V, Chen S, Lu D, et al. Caspase cleavage of members of the amyloid precursor family of proteins. J Neurochem. 2002;82:283–294. doi: 10.1046/j.1471-4159.2002.00970.x. [DOI] [PubMed] [Google Scholar]
- 8.Pellegrini L, Passer BJ, Tabaton M, et al. Alternative, non-secretase processing of Alzheimer’s beta-amyloid precursor protein during apoptosis by caspase-6 and -8. J Biol Chem. 1999;274:21011–21016. doi: 10.1074/jbc.274.30.21011. [DOI] [PubMed] [Google Scholar]
- 9.Weidemann A, Paliga K, Durrwang U, et al. Proteolytic processing of the Alzheimer’s disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J Biol Chem. 1999;274:5823–5829. doi: 10.1074/jbc.274.9.5823. [DOI] [PubMed] [Google Scholar]
- 10.Buxbaum JD, Thinakaran G, Koliatsos V, et al. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- 12.Kamal A, Almenar-Queralt A, LeBlanc JF, et al. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 13.Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J Neurosci. 2003;23:5407–5415. doi: 10.1523/JNEUROSCI.23-13-05407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol. 2001;153:1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamenetz F, Tomita T, Hsieh H, et al. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 16.Taru H, Iijima K, Hase M, et al. Interaction of Alzheimer’s beta-amyloid precursor family proteins with scaffold proteins of the JNK signaling cascade. J Biol Chem. 2002;277:20070–20078. doi: 10.1074/jbc.M108372200. [DOI] [PubMed] [Google Scholar]
- 17.Scheinfeld MH, Roncarati R, Vito P, et al. Jun NH2-terminal kinase (JNK) interacting protein 1 (JIP1) binds the cytoplasmic domain of the Alzheimer’s beta-amyloid precursor protein (APP) J Biol Chem. 2002;277:3767–3775. doi: 10.1074/jbc.M108357200. [DOI] [PubMed] [Google Scholar]
- 18.Homayouni R, Rice DS, Sheldon M, Curran T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J Neurosci. 1999;19:7507–7515. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuda S, Yasukawa T, Homma Y, et al. c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer’s amyloid precursor protein with JNK. J Neurosci. 2001;21:6597–6607. doi: 10.1523/JNEUROSCI.21-17-06597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiore F, Zambrano N, Minopoli G, et al. The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of Shc bind the intracellular domain of the Alzheimer’s amyloid precursor protein. J Biol Chem. 1995;270:30853–30856. doi: 10.1074/jbc.270.52.30853. [DOI] [PubMed] [Google Scholar]
- 21.Zambrano N, Buxbaum JD, Minopoli G, et al. Interaction of the phosphotyrosine interaction/phosphotyrosine binding-related domains of Fe65 with wild-type and mutant Alzheimer’s beta-amyloid precursor proteins. J Biol Chem. 1997;272:6399–6405. doi: 10.1074/jbc.272.10.6399. [DOI] [PubMed] [Google Scholar]
- 22.McLoughlin DM, Miller CC. The intracellular cytoplasmic domain of the Alzheimer’s disease amyloid precursor protein interacts with phosphotyrosine-binding domain proteins in the yeast two-hybrid system. FEBS Lett. 1996;397:197–200. doi: 10.1016/s0014-5793(96)01128-3. [DOI] [PubMed] [Google Scholar]
- 23.Biederer T, Cao X, Sudhof TC, Liu X. Regulation of APP-dependent transcription complexes by Mint/X11s: differential functions of Mint isoforms. J Neurosci. 2002;22:7340–7351. doi: 10.1523/JNEUROSCI.22-17-07340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schonberger SJ, Edgar PF, Kydd R, et al. Proteomic analysis of the brain in Alzheimer’s disease: molecular phenotype of a complex disease process. Proteomics. 2001;1:1519–1528. doi: 10.1002/1615-9861(200111)1:12<1519::aid-prot1519>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 25.Tsuji T, Shimohama S. Analysis of the proteomic profiling of brain tissue in Alzheimer’s disease. Dis Markers. 2001;17:247–257. doi: 10.1155/2001/386284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo BC, Kim SH, Cairns N, et al. Deranged expression of molecular chaperones in brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun. 2001;280:249–258. doi: 10.1006/bbrc.2000.4109. [DOI] [PubMed] [Google Scholar]
- 27.Castegna A, Aksenov M, Aksenova M, et al. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura Y, Takeda M, Niigawa H, et al. Amyloid beta-protein precursor deposition in rat hippocampus lesioned by ibotenic acid injection. Neurosci Lett. 1992;136:95–98. doi: 10.1016/0304-3940(92)90656-r. [DOI] [PubMed] [Google Scholar]
- 29.Sisodia SS, Koo EH, Hoffman PN, et al. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci. 1993;13:3136–3142. doi: 10.1523/JNEUROSCI.13-07-03136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 31.Baek SH, Ohgi KA, Rose DW, et al. Exchange of N-CoR core-pressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 2002;110:55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 32.Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyl-transferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 33.Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ. The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001;276:40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- 34.Umahara T, Uchihara T, Tsuchiya K, et al. 14-3-3 proteins and zeta isoform containing neurofibrillary tangles in patients with Alzheimer’s disease. Acta Neuropathol (Berl) 2004;108:279–286. doi: 10.1007/s00401-004-0885-4. [DOI] [PubMed] [Google Scholar]
- 35.Hernandez F, Cuadros R, Avila J. Zeta 14-3-3 protein favours the formation of human tau fibrillar polymers. Neurosci Lett. 2004;357:143–146. doi: 10.1016/j.neulet.2003.12.049. [DOI] [PubMed] [Google Scholar]
- 36.Hashiguchi M, Sobue K, Paudel HK. 14-3-3zeta is an effector of tau protein phosphorylation. J Biol Chem. 2000;275:25247–25254. doi: 10.1074/jbc.M003738200. [DOI] [PubMed] [Google Scholar]
- 37.Choe LH, Green A, Knight RS, et al. Apolipoprotein E and other cerebrospinal fluid proteins differentiate ante mortem variant Creutzfeldt-Jakob disease from ante mortem sporadic Creutzfeldt-Jakob disease. Electrophoresis. 2002;23:2242–2246. doi: 10.1002/1522-2683(200207)23:14<2242::AID-ELPS2242>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 38.Stege GJ, Renkawek K, Overkamp PS, et al. The molecular chaperone alphaB-crystallin enhances amyloid beta neurotoxicity. Biochem Biophys Res Commun. 1999;262:152–156. doi: 10.1006/bbrc.1999.1167. [DOI] [PubMed] [Google Scholar]
- 39.Liang JJ. Interaction between beta-amyloid and lens alphaB-crystallin. FEBS Lett. 2000;484:98–101. doi: 10.1016/s0014-5793(00)02136-0. [DOI] [PubMed] [Google Scholar]
- 40.Fonte V, Kapulkin V, Taft A, et al. Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc Natl Acad Sci U S A. 2002;99:9439–9444. doi: 10.1073/pnas.152313999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Link CD, Taft A, Kapulkin V, et al. Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol Aging. 2003;24:397–413. doi: 10.1016/s0197-4580(02)00224-5. [DOI] [PubMed] [Google Scholar]
- 42.Hsia AY, Masliah E, McConlogue L, et al. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mucke L, Masliah E, Yu GQ, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costes SV, Daelemans D, Cho EH, et al. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chyung JH, Selkoe DJ. Inhibition of receptor-mediated endocytosis demonstrates generation of amyloid beta-protein at the cell surface. J Biol Chem. 2003;278:51035–51043. doi: 10.1074/jbc.M304989200. [DOI] [PubMed] [Google Scholar]
- 46.Ehehalt R, Keller P, Haass C, et al. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parkin ET, Turner AJ, Hooper NM. Amyloid precursor protein, although partially detergent-insoluble in mouse cerebral cortex, behaves as an atypical lipid raft protein. Biochem J. 1999;344:23–30. pt 1. [PMC free article] [PubMed] [Google Scholar]
- 48.Tun H, Marlow L, Pinnix I, et al. Lipid rafts play an important role in A beta biogenesis by regulating the beta-secretase pathway. J Mol Neurosci. 2002;19:31–35. doi: 10.1007/s12031-002-0007-5. [DOI] [PubMed] [Google Scholar]
- 49.Bridgman PC. Myosin-dependent transport in neurons. J Neurobiol. 2004;58:164–174. doi: 10.1002/neu.10320. [DOI] [PubMed] [Google Scholar]
- 50.Shea TB, Flanagan LA. Kinesin, dynein and neurofilament transport. Trends Neurosci. 2001;24:644–648. doi: 10.1016/s0166-2236(00)01919-6. [DOI] [PubMed] [Google Scholar]
- 51.Ho CS, Marinescu V, Steinhilb ML, et al. Synergistic effects of Munc18a and X11 proteins on amyloid precursor protein metabolism. J Biol Chem. 2002;277:27021–27028. doi: 10.1074/jbc.M201823200. [DOI] [PubMed] [Google Scholar]
- 52.Tsai MY, Morfini G, Szebenyi G, Brady ST. Release of kinesin from vesicles by hsc70 and regulation of fast axonal transport. Mol Biol Cell. 2000;11:2161–2173. doi: 10.1091/mbc.11.6.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shupliakov O, Bloom O, Gustafsson JS, et al. Impaired recycling of synaptic vesicles after acute perturbation of the presynaptic actin cytoskeleton. Proc Natl Acad Sci U S A. 2002;99:14476–14481. doi: 10.1073/pnas.212381799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Der Hoeven PC, Van Der Wal JC, Ruurs P, et al. 14-3-3 isotypes facilitate coupling of protein kinase C-zeta to Raf-1: negative regulation by 14-3-3 phosphorylation. Biochem J. 2000;345:297–306. doi: 10.1042/0264-6021:3450297. pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fountoulakis M, Cairns N, Lubec G. Increased levels of 14-3-3 gamma and epsilon proteins in brain of patients with Alzheimer’s disease and Down syndrome. J Neural Transm Suppl. 1999;57:323–335. doi: 10.1007/978-3-7091-6380-1_23. [DOI] [PubMed] [Google Scholar]
- 56.Denker SP, Barber DL. Ion transport proteins anchor and regulate the cytoskeleton. Curr Opin Cell Biol. 2002;14:214–220. doi: 10.1016/s0955-0674(02)00304-6. [DOI] [PubMed] [Google Scholar]
- 57.Lopez-Schier H, St Johnston D. Drosophila nicastrin is essential for the intramembranous cleavage of notch. Dev Cell. 2002;2:79–89. doi: 10.1016/s1534-5807(01)00109-5. [DOI] [PubMed] [Google Scholar]
- 58.Johnson RJ, Xiao G, Shanmugaratnam J, Fine RE. Calreticulin functions as a molecular chaperone for the beta-amyloid precursor protein. Neurobiol Aging. 2001;22:387–395. doi: 10.1016/s0197-4580(00)00247-5. [DOI] [PubMed] [Google Scholar]
- 59.Yang Y, Turner RS, Gaut JR. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem. 1998;273:25552–25555. doi: 10.1074/jbc.273.40.25552. [DOI] [PubMed] [Google Scholar]
- 60.Chiesi M, Longoni S, Limbruno U. Cardiac alpha-crystallin. III. Involvement during heart ischemia. Mol Cell Biochem. 1990;97:129–136. doi: 10.1007/BF00221054. [DOI] [PubMed] [Google Scholar]
- 61.Hatters DM, Lindner RA, Carver JA, Howlett GJ. The molecular chaperone, alpha-crystallin, inhibits amyloid formation by apolipoprotein C-II. J Biol Chem. 2001;276:33755–33761. doi: 10.1074/jbc.M105285200. [DOI] [PubMed] [Google Scholar]
- 62.Ito H, Kamei K, Iwamoto I, et al. Inhibition of proteasomes induces accumulation, phosphorylation, and recruitment of HSP27 and alphaB-crystallin to aggresomes. J Biochem (Tokyo) 2002;131:593–603. doi: 10.1093/oxfordjournals.jbchem.a003139. [DOI] [PubMed] [Google Scholar]
- 63.Kouchi Z, Sorimachi H, Suzuki K, Ishiura S. Proteasome inhibitors induce the association of Alzheimer’s amyloid precursor protein with Hsc73. Biochem Biophys Res Commun. 1999;254:804–810. doi: 10.1006/bbrc.1998.9977. [DOI] [PubMed] [Google Scholar]
- 64.Song S, Kim SY, Hong YM, et al. Essential role of E2–25K/Hip-2 in mediating amyloid-beta neurotoxicity. Mol Cell. 2003;12:553–563. doi: 10.1016/j.molcel.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 65.Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]