

Abstract
SV40 chromosomes undergoing transcription operationally defined by the presence of RNA Polymerase II (RNAPII) were immune-selected with antibody to RNAPII and subjected to secondary chromatin immunoprecipitation with antibodies to hyperacetylated or unacetylated H4 or H3. Immune Selection Fragmentation and Immunoprecipitation (ISFIP) was used to determine the hyperacetylation status of histones independent of the location of the RNAPII and ReChromatin Immunoprecipitation (ReChIP) was used to determine their hyperacetylation status when associated with RNAPII. While hyperacetylated H4 and H3 were found in the coding regions regardless of the location of RNAPII, unacetylated H4 and H3 were only found at sites lacking RNAPII. The absence of unacetylated H4 and H3 at sites containing RNAPII was correlated with the specific association of the Histone Acetyl Transferase (HAT) p300 with the RNAPII. In contrast, the presence of unacetylated H4 and H3 at sites lacking RNAPII was shown to result from the action of a histone deacetylase (HDAC) based upon the effects of the inhibitor sodium butyrate. These results suggest that the extent of hyperacetylation of H4 and H3 during transcription alternates between hyperacetylation directed by an RNAPII associated HAT and deacetylation directed by an HDAC at other sites.
Keywords: Simian Virus 40, Histone Hyperacetylation, Transcription, RNA Polymerase II, Histone Acetylase, Histone Deacetylase, Transcription Complex
Introduction
The histones that are organized with eukaryotic DNA to form chromatin undergo diverse forms of post-translational modifications (1). Because these modifications can potentially impact the interactions between the histones and associated DNA or other proteins, there has been great interest in understanding the function of these modifications in eukaryotic biological processes. The covalent addition of acetyl groups to histone tails is one form of modification which has been extensively investigated and has been associated with transcription in a number of different studies (2,3). Moreover, the enzymes responsible for histone acetylation and deacetylation, histone acetyl transferases (HATs) and histone deacetylases (HDACs), respectively, have also been extensively studied (4) and in many cases the HATs and HDACs have been found to be associated with various transcription factors (5).
These studies while clearly establishing a link between histone hyperacetylation and transcription are limited by the fact that invariably they are based upon an association between a difference in the extent of transcription such as the induction of a gene and the changes in the properties of the total cellular chromatin containing that gene. To date there has not yet been an extensive characterization of histone hyperacetylation directly in chromatin obtained in vivo which is specifically undergoing transcription.
We have been using the simian virus 40 (SV40) chromosome as a eukaryotic model to investigate two aspects of chromatin structure, nucleosome phasing and histone hyperacetylation (6–9). The SV40 chromosome is particularly well suited for this function since it utilizes host cell proteins for its transcription and replication and has been extensively studied as a model for both eukaryotic processes (10–13). In the SV40 model system, transcription has been shown to occur in three distinct phases (Figure 1A). Within approximately one hour of infection, the first phase of transcription occurs with the induction of the early region of the SV40 genome. The second phase of transcription occurs at about eight hours post-infection when the virally encoded early protein T-antigen interacts with its cognate binding site in the promoter and down-regulates further early transcription. The third phase of transcription, which occurs either along with or shortly after down-regulation of early transcription, is characterized by a marked increase in transcription from the late side of the genome. (10)
Figure 1A.
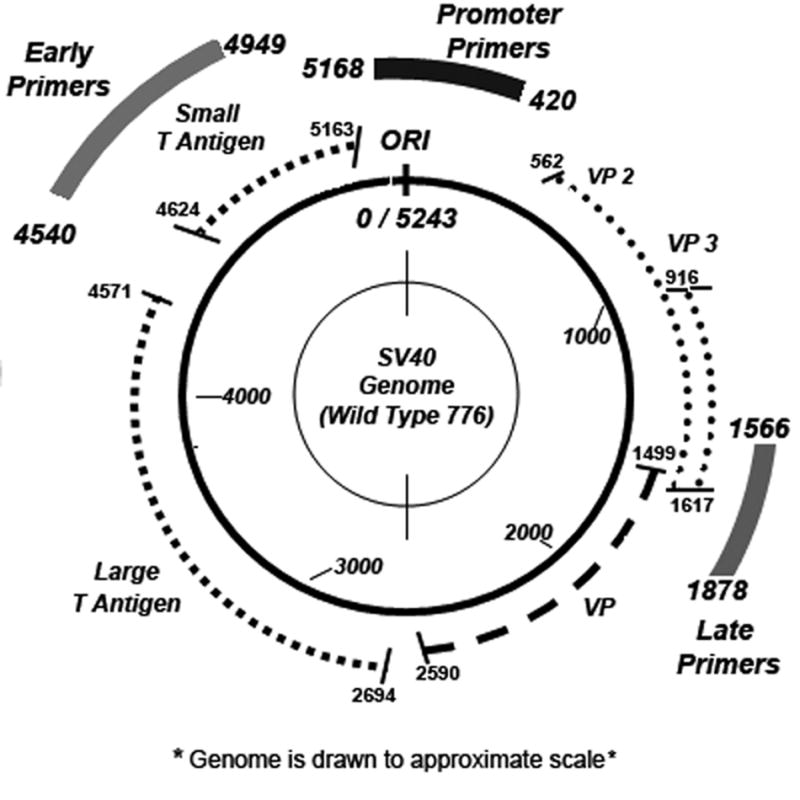
SV40 Genome
In order to obtain transcribing SV40 chromosomes for direct analysis of histone hyperacetylation we reasoned that by using an antibody specific to RNA polymerase II (RNAPII), a protein which is absolutely required for transcription, we would be able to specifically immune select transcribing SV40 chromosomes using chromatin immunoprecipitation (ChIP) procedures (14–16). We expected that the immune selected SV40 chromatin would consist of chromosomes with RNAPII located in the promoter which were undergoing initiation, chromosomes with RNAPII located in coding regions which were undergoing transcriptional elongation, and chromosomes with RNAPII located in either the promoter or coding regions in which the RNAPII was paused. We confirmed that antibody to RNAPII could be used to immune select transcribing SV40 chromosomes using a modified ChIP procedure that we refer to as immune selection and fragmentation (ISF) (6,7). In the ISF procedure the immune selected transcribing SV40 chromosomes were fragmented by sonication into bound chromatin fragments that contained RNAPII and soluble chromatin fragments that lacked RNAPII. Using this strategy we measured the relative occupancy of RNAPII in various regions of the SV40 genome in chromosomes undergoing transcription during the course of an infection and correlated these results to the well characterized early to late shift in SV40 transcription (7). Specifically, we observed that at early times RNAPII was preferentially associated with the promoter and early region of the genome while at late times RNAPII was preferentially associated with the promoter and late region of the genome as expected. At 8 hours post-infection in SV40 chromosomes from wild-type virus, we observed that RNAPII occupancy of the promoter and early region was relatively low consistent with down-regulation, while in a mutant of SV40 (cs1085) in which the T-antigen binding site was lacking and down-regulation could not occur the occupancy of the promoter and early region was very high. Since the occupancy of the SV40 genome during the course of infection correlated directly with the known pattern of transcription, we concluded that we were immune selecting transcribing SV40 chromosomes using antibody to RNAPII(7).
In this publication (7) we also demonstrated the feasibility of directly analyzing chromatin undergoing transcription for the presence of hyperacetylated histones. Because the immune selected chromatin undergoing transcription was fragmented into chromatin fragments that either contained RNAPII or lacked RNAPII, we were able to determine whether a particular hyperacetylated histone was present along with the RNAPII or independent of the RNAPII at any given site in the genome. The strategy that was used for this analysis is outlined in Figure 1B. Histone hyperacetylation in the bound fragments was determined by a Re Chromatin Immunoprecipitation (ReChIP) analysis (17), while the soluble fragments were analyzed by a procedure that we refer to as immune selection fragmentation and immunoprecipitation (ISFIP). The combined procedure is referred to as ISFIP/ReChIP. We used the ISFIP/ReChIP procedure to directly demonstrate that within the early coding region of SV40 chromosomes undergoing early transcription, hyperacetylated H4 and H3 were associated with chromatin fragments that contained RNAPII and also those that lacked RNAPII (7).
Figure 1B.
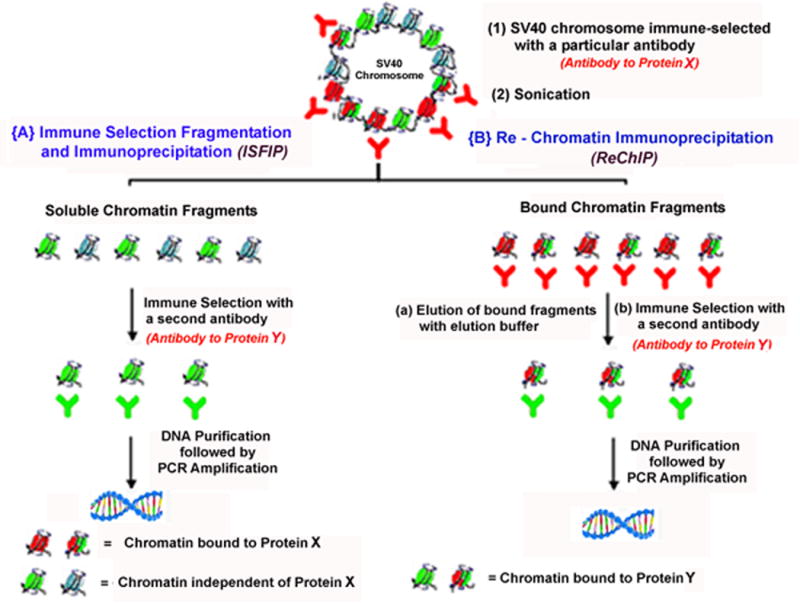
Strategy of Immune Selection Fragmentation followed by Immunoprecipitation (ISFIP) and Re Chromatin Immunoprecipitation (ReChIP)
These ISFIP/ReChIP results indicated that it would be possible to determine the status of histone hyperacetylation or presence of a transcriptional factor at a particular site in the genome of transcribing SV40 chromosomes at any time in the infection. We now describe the organization of the hyperacetylated and unacetylated histones H4 and H3 within the SV40 early and late coding regions during the shift from early to late transcription using the ISFIP/ReChIP procedure. In addition, because the extent of histone hyperacetylation is regulated by the interplay between HATS and HDACs, we describe the organization of the HAT p300 in chromosomes undergoing transcription and the effects of the inhibition of HDACs by sodium butyrate on the presence of hyperacetylated and unacetylated H4 and H3 within the SV40 early and late coding regions.
Results
1. Analysis of histone hyperacetylation on SV40 chromosomes carrying RNAPII during the course of infection
In order to determine the status of histone hyperacetylation in SV40 chromosomes containing RNAPII within the early, late, and promoter regions of the SV40 genome during the course of infection, SV40 chromosomes were isolated at 30 minutes, 8 hours, and 48 hours post-infection and subjected to a ISFIP/ReChIP analysis. At these times one would expect to obtain SV40 chromosomes undergoing induction of early transcription, down-regulation of early transcription and extensive late transcription respectively. Moreover, we have previously shown (7) that the pattern of RNAPII occupancy of the genome is consistent with this expectation. SV40 chromosomes were first immune-selected with antibody to RNAPII, the bound chromatin sonicated, and the bound and soluble fragments then analyzed with antibody to either hyperacetylated H4 or H3. The chromatin fragments generated in this procedure were approximately 200bp to 400 bp in size with the maximum size observed 500bp (7). The status of histone hyperacetylation on the early, late, and promoter regions was determined by PCR amplification of samples with appropriate primer sets (7,6,9). All PCR amplifications were performed within the linear range of amplification (refer to supplementary figure SF9). For the analyses shown, we used SV40 chromatin which had not been formalin fixed prior to isolation because in our previous studies on histone hyperacetylation and RNAPII occupancy in SV40 chromosomes we did not see any differences between fixed and unfixed SV40 chromatin (4, 5, 24). However, each of the experiments described here have been performed with formalin fixed materials at least once and similar results to those obtained with unfixed chromatin were obtained in each case. Typical examples with formalin fixed material are shown in the supplementary data.
Typical examples of this type of analysis are shown in Figure 2. As is apparent from Figure 2, hyperacetylated H4 and H3 were found on the early, late and promoter regions of the genome co-localized with the RNAPII (from ReChIP lanes 6,7,13,14, 20,21) as well as independent of the RNAPII (from ISFIP lanes 4,5,11,12,18,19). In contrast, when the chromatin fragments from the ISFIP were subjected to a ChIP analysis with antibody to RNAPII, no product was generated by PCR amplification (lanes 3, 10, 17) indicating that all of the fragments containing RNAPII remained bound to the protein A agarose during the immune selection step and subsequent chromatin fragmentation.
Figure 2. Simplex PCR analysis of histone hyperacetylation on SV40 chromosomes undergoing transcription.
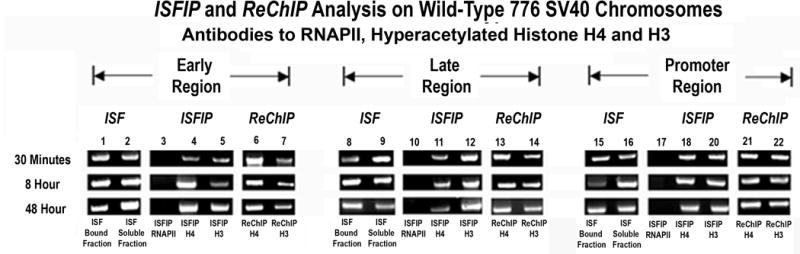
Unfixed SV40 chromosomes were isolated from cells infected with 776 wild type virus for 30 minutes, 8 hours or 48 hours and subjected to an ISFIP/ReChIP analysis with antibodies to hyperacetylated histone H4, histone H3 and RNAPII as described in the materials and methods. The samples were amplified by simplex PCR with primer sets to the early, late and promoter regions. The position of the amplification product from the wild-type 776 DNA is indicated. Lane 1,8,15: bound fraction immunoprecipitated with 10μl of RNAPII antibody (ISF); lane 2,9,16: soluble fraction immunoprecipitated with 10μl of RNAPII antibody (ISF); lane 3,10,17: ChIP with 10 μl of RNAPII antibody (ISFIP); lane 4,11,18: ChIP with 7.5 μl of hyperacetylated histone H4 antibody (ISFIP); lane 5,12,19: ChIP with 10 μl of hyperacetylated histone H3 antibody (ISFIP); lane 6,13,20: ChIP with 7.5 μl of hyperacetylated histone H4 antibody (ReChIP); lane 7,14,21: ChIP with 10 μl of hyperacetylated histone H3 antibody (ReChIP). The PCR products in the ISFIP and ReChIP lanes were amplified from one half of the total amount of DNA obtained from each of the samples. The PCR products in the input lanes were amplified from one fourth of the total amount of DNA present in each of the input samples. Similar results were obtained from at least three separate preparations of SV40 chromosomes for each time point.
2. Analysis of the extent of hyperacetylation of H4 and H3 in chromatin bound to RNAPII and independent of RNAPII
Since hyperacetylated H4 and H3 were present in chromatin associated with RNAPII and independent of RNAPII throughout the SV40 genome, we then determined whether the extent of hyperacetylation of these two histones was similar in the two types of chromatin. Because histone hyperacetylation facilitates transcription by RNAPII (21), we reasoned that either hyperacetylated H4 and H3 would be found to a similar extent throughout the SV40 genome or would be enriched at sites occupied by RNAPII.
The extent of hyperacetylation of H4 and H3 in chromatin bound to RNAPII and independent of RNAPII was determined using two different strategies. In the first strategy we utilized the fact that each nucleosome contains an equal number of copies of histone H4 and H3. If both the H4 and the H3 are hyperacetylated in a nucleosome located at a particular site in chromatin, the same amount of DNA should be present in the immunoprecipitated chromatin associated with this nucleosome using antibody to either hyperacetylated histone. Since the same amount of DNA present in both immunoprecipitates would be the same, one would expect to obtain the same amount of amplification products from both samples following PCR. In contrast, if one of the histones in the nucleosome was not hyperacetylated, one would not expect to obtain any immunoprecipitated DNA or amplification products using antibodies which recognize that form of hyperacetylated histone but would still expect to obtain PCR products using the antibody to the other hyperacetylated histone. For this analysis we subjected the DNA present in the immune precipitates from the ISFIP and ReChIP analyses and the corresponding input chromatin to duplex PCR amplification using primer sets to both the early and late regions. We chose to use duplex PCR because we have previously used this strategy to characterize the preferred location of hyperacetylated H4 and H3 in pooled SV40 chromosomes and RNAPII in transcribing SV40 chromosomes (6,7) and because it allows us to take into account any variations in sample preparations (9). In the duplex procedure we would expect to see identical ratios of amplification products from the early and late regions following immunoprecipitation with antibody to hyperacetylated H4 or H3 as long as the nucleosomes present in those regions contain similar amounts of hyperacetylated H4 or H3. However, if in some of the SV40 chromosomes one of the histones is not hyperacetylated to the same extent as the other histone; we would expect to see the ratio of early to late amplification products to be different.
Examples of this type of analysis are shown in Figure 3. As expected from the results in Figure 2, we observed that PCR amplification products were obtained from both the early and late regions of the genome regardless of whether they were generated by the ISFIP or ReChIP procedure, the time post-infection, or the antibody which was used for the immunoprecipitation. Interestingly, however, we observed a distinct difference between the results obtained from the ISFIP analysis (lanes 1–3) compared to the ReChIP analysis (lanes 4–6). In order to show these differences quantitatively we used densitometry to measure the percentage of the PCR amplification products which were derived from the early region and the late region. In the ISFIP analysis we observed that the ratios of amplification products from immunoprecipitation with antibody to hyperacetylated H4 and H3 were very different at each time point. For example, following immunoprecipitation with antibody to hyperacetylated H4 (lane 2), we consistently observed more amplification product from the early region than from the late region (densitometry 30 minutes 68%/32%, 8 hours 72%/28%, and 48 hours 55%/45%). In contrast, following immunoprecipitation with antibody to hyperacetylated H3 (lane 3), we observed more amplification product from the late region than the early region (densitometry 30 minutes 43%/57%, 8 hours 46%/54%, and 48 hours 43%/57%). In the ReChIP analysis of the same chromosomes we observed that the relative amounts of PCR amplification products from the early and late regions were essentially identical with either antibody to hyperacetylated H4 (Lane 5) or H3 Lane 6) at all three time points (densitometry approximately 53%/47%). In each of these ReChIP analyses there was more product generated from the early region than there was from the late region. These results suggest that the nucleosomes associated with RNAPII (from ReChIP analysis) contain similar amounts of hyperacetylated H4 and H3, while at sites independent of RNAPII (from ISFIP) the hyperacetylation of H4 and H3 in nucleosomes does not occur to the same extent.
Figure 3. Duplex PCR analysis of histone hyperacetylation on SV40 chromosomes undergoing transcription.
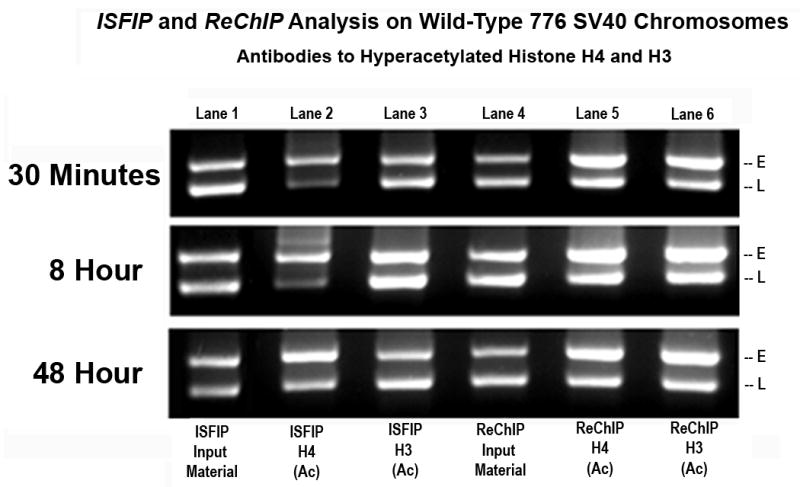
Unfixed SV40 chromosomes were isolated from cells infected with 776 wild type virus for 30 minutes, 8 hours or 48 hours and subjected to an ISFIP/ReChIP analysis as described in materials and methods. The samples were amplified by duplex PCR using primer sets to the early and late regions. E and L indicate the positions of the early and late PCR amplification products, respectively. The position of the amplification product from the wild-type 776 DNA is indicated. Lane 1: ISFIP input fraction; lane 2: ChIP with 7.5 μl of hyperacetylated histone H4 antibody (ISFIP); lane 3: ChIP with 10 μl of hyperacetylated histone H3 antibody (ISFIP); lane 4: ReChIP input fraction; lane 5: ChIP with 7.5 μl of hyperacetylated histone H4 antibody (ReChIP); lane 6: ChIP with 10 all of hyperacetylated histone H3 antibody (ReChIP). The PCR products in the ISFIP and ReChIP lanes were amplified from one half of the total amount of DNA obtained from each of the samples. The PCR products in the input lanes were amplified from one fourth of the total amount of DNA present in each of the input samples. Similar results were obtained from at least three separate preparations of SV40 chromosomes for each time point.
In a second type of analysis we directly determined whether the unacetylated forms of H4 or H3 were present in chromatin fragments associated with RNAPII or independent of RNAPII. For these studies we used antibodies prepared against peptides consisting of the unmodified amino terminal tails of H4 and H3 (obtained from Santa Cruz Biotechnology). The specificity of these antibodies for the unmodified form of H4 and H3 respectively are shown in the Supplementary Data (SF10A & SF10B). We reasoned that if H4 and H3 were hyperacetylated to the same extent in both types of chromatin we would find either no unacetylated histones or similar amounts of unacetylated histones in the two types of chromatin. In contrast if hyperacetylated H4 and H3 were preferentially associated with chromatin bound to RNAPII we would expect to find less unacetylated H4 and H3 in this fraction compared to the chromatin fraction independent of RNAPII. Typical examples of the results from this type of analysis are shown in Figure 4. As shown in Figure 4A, transcribing SV40 chromosomes isolated 30 minutes post-infection were first analyzed for the presence of hyperacetylated or unacetylated H4 and H3 on the early coding region following an ISFIP and ReChIP analyses using antibody to RNAPII for immune selection. Although hyperacetylated H4 (lane 2), unacetylated H4 (lane 3), hyperacetylated H3 (lane 4), and unacetylated H3 (lane 5) were all found in the ISFIP analysis, only hyperacetylated H4 (lane 7) and hyperacetylated H3 (lane 9) were found in the ReChIP analysis. No evidence of unacetylated H4 (lane 8) or unacetylated H3 (lane 10) was observed from the immunoprecipitates obtained from the ReChIP analysis .
Figure 4A. Presence of unacetylated and hyperacetylated histone H3 and H4 on the SV40 early region in chromosomes undergoing early transcription.
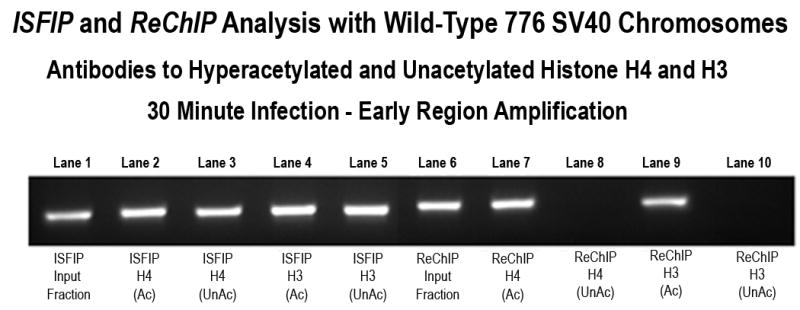
Unfixed SV40 chromosomes were isolated from cells infected with 776 wild type virus for 30 minutes and subjected to an ISFIP/ReChIP analysis with antibodies to unacetylated histone H4 and histone H3 as described in the materials and methods The samples were amplified by simplex PCR with primer sets to the early regions. The position of the amplification product from the wild-type 776 DNA is indicated. Lane 1: ISFIP input fraction; lane 2: ChIP with 7.5 μl of hyperacetylated histone H4 antibody (ISFIP); lane 3: ChIP with 10 μl of unacetylated histone H4 antibody (ISFIP); lane 4: ChIP with 10 μl of hyperacetylated histone H3 antibody (ISFIP); lane 5: ChIP with 10 μl of unacetylated histone H3 antibody (ISFIP); lane 6: ReChIP input fraction; lane 7: ChIP with 7.5 μl of hyperacetylated histone H4 antibody (ReChIP); lane 8: ChIP with 10 μl of unacetylated histone H4 antibody (ReChIP); lane 9: ChIP with 10 μl of hyperacetylated histone H3 antibody (ReChIP); lane 10: ChIP with 10 μl of unacetylated histone H3 antibody (ReChIP). The PCR products in the ISFIP and ReChIP lanes were amplified from one half of the total amount of DNA obtained from each of the samples. The PCR products in the input lanes were amplified from one fourth of the total amount of DNA present in each of the input samples. Similar results were obtained from at least three separate preparations of SV40 chromosomes at 30 minutes post-infection.
This analysis was then extended to include both the early and late coding regions of transcribing SV40 chromosomes preferentially transcribing the early genes (30 minutes post-infection) and the late genes (48 hours post-infection) (Figure 4B). Comparing the results from the ISFIP analysis (lanes 1–3) to the ReChIP analysis (lanes 4–6) in the early and late coding regions, it was clear that unacetylated H4 and H3 were only present in fractions from chromatin from the ISFIP analysis (compare lanes 2 and 3 to 5 and 6). This result confirmed the results from the duplex analysis and indicated that there was a relative increase in the extent of hyperacetylation of H4 and H3 when the histones were associated with RNAPII compared to when the histones were independent of RNAPII.
Figure 4B. Preferential association of unacetylated histones in regions of the SV40 genome lacking RNA Polymerase II in chromosomes undergoing early and late transcription.
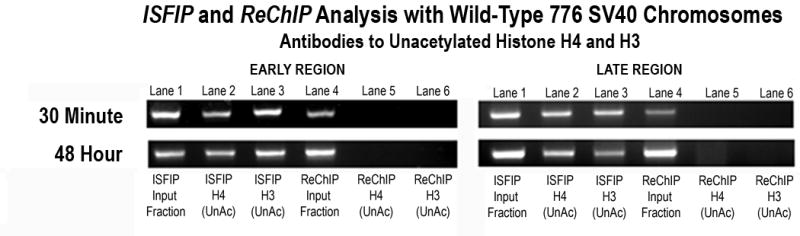
Unfixed SV40 chromosomes were isolated from cells infected with 776 wild type virus for 30 minutes or 48 hours and subjected to an ISFIP/ReChIP analysis with antibodies to hyperacetylated histone H4 and H3 and unacetylated histone H4 and H3 as described in the materials and methods. The samples were amplified with primer sets to the early and late regions. The position of the amplification product from the wild-type 776 DNA is indicated. Lane 1: ISFIP input fraction; lane 2: ChIP with 10 μl of unacetylated histone H4 antibody (ISFIP); lane 3: ChIP with 10 μl of unacetylated histone H3 antibody (ISFIP); lane 4: ReChIP input fraction; lane 5: ChIP with 10 μl of unacetylated histone H4 antibody (ReChIP); lane 6: ChIP with 10 all of unacetylated histone H3 antibody (ReChIP). The PCR products in the ISFIP and ReChIP lanes were amplified from one half of the total amount of DNA obtained from each of the samples. The PCR products in the input lanes were amplified from one fourth of the total amount of DNA present in each of the input samples. Similar results were obtained from at least three separate preparations of SV40 chromosomes for each time point.
Because the previous analyses were performed on SV40 chromosomes in which the occupancy of the early and late regions by RNAPII was relatively high (between 40% and 70%) (7), we next determined whether the presence of unacetylated H4 or H3 in a region of the genome was related to the extent of RNAPII occupancy. This question was addressed by analyzing chromatin from the SV40 mutant cs1085 obtained at 8 hours post-infection for the presence of unacetylated H4 or H3. In this mutant down-regulation of early transcription does not occur and occupancy in the promoter region at this time increases from 30 ± 2% to 93 ± 1%, occupancy in the early region increases from 47 ± 0.5% to 57 ± 2% and occupancy in the late region decreases from 51 ± 1% to 24 ± 2 % (5). As shown in Figure 5, in chromatin from the cs1085 mutant we did not observe any evidence for unacetylated H4 or H3 present in the early region by either ReChIP or ISFIP although significant amounts appeared to be present on the late region. In contrast hyperacetylated H4 and H3 were observed in both regions in the ISFIP/ReChIP analyses. These results suggested that the presence of unacetylated H4 and H3 were inversely related to occupancy by RNAPII and were again consistent with the hypothesis that RNAPII was associated with increased levels of histone hyperacetylation.
Figure 5. Analysis of unacetylated and hyperacetylated histones on mutant cs1085 SV40 chromosomes undergoing down-regulation of early transcription.
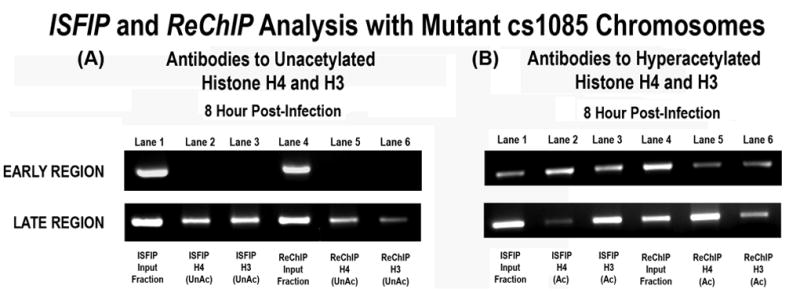
Unfixed SV40 chromosomes were isolated from cells infected with cs1085 mutant virus for 8 hours and subjected to an ISFIP/ReChIP analysis with antibodies to unacetylated histone H4 and histone H3 and hyperacetylated histone H4 and H3 as described in the materials and methods The samples were amplified by simplex PCR with primer sets to the early and late regions. The position of the amplification product from the wild-type 776 DNA is indicated. 5A: Lane 1: ISFIP input fraction; lane 2: ChIP with 10 μl of unacetylated histone H4 antibody (ISFIP); lane 3: ChIP with 10 μl of unacetylated histone H3 antibody (ISFIP); lane 4: ReChIP input fraction; lane 5: ChIP with 10 μl of unacetylated histone H4 antibody (ReChIP); lane 6: ChIP with 10 all of unacetylated histone H3 antibody (ReChIP) ChIP with 10 μl of unacetylated histone H3 antibody (ReChIP). 5B: Lane 1: ISFIP input fraction; lane 2: ChIP with 7.5 μl of hyperacetylated histone H4 antibody (ISFIP); lane 3: ChIP with 10 μl of hyperacetylated histone H3 antibody (ISFIP); lane 4: ReChIP input fraction; lane 5: ChIP with 7.5 μl of hyperacetylated histone H4 antibody (ReChIP); lane 6: ChIP with 10 μl of hyperacetylated histone H3 antibody (ReChIP)
The PCR products in the ISFIP and ReChIP lanes were amplified from one half of the total amount of DNA obtained from each of the samples. The PCR products in the input lanes were amplified from one fourth of the total amount of DNA present in each of the input samples. Similar results were obtained from at least three separate preparations of SV40 chromosomes at 8 hours post-infection.
3. Analysis of the effects of sodium butyrate inhibition of HDACs on the presence of hyperacetylated H4 and H3 in association with and independent of RNAPII
The results which were obtained from the analysis of chromatin fragments bound with RNAPII and independent of RNAPII for the extent of H4 and H3 hyperacetylation using duplex PCR and antibodies to the unacetylated tails of each histone suggested that there was greater hyperacetylation of H4 and H3 in chromatin directly associated with RNAPII than in chromatin which was independent of RNAPII.
Because the extent of histone hyperacetylation in chromatin is thought to be regulated by the relative action of histone acetyl transferases (HATs) and histone deacetylases (HDACs) interacting with the chromatin, there were two possible explanations for the presence of unacetylated histones in regions lacking RNAPII. Either the chromatin was undergoing sequential hyperacetylation and deacetylation, or the chromatin was not undergoing hyperacetylation at all and the lack of acetylation simply reflected the basal unmodified state of the histones. Depending upon the fate of histones during passage of the RNAPII through the chromatin, the hyperacetylation in the former case could be due either to the direct action of a histone acetyl transferase on nucleosomes in front of the RNAPII or alternatively it could be due to the incorporation of hyperacetylated histones generated elsewhere into a reorganized nucleosome following passage of the RNAPII.
In order to distinguish these two possibilities we treated cells prior to and during infection with the HDAC inhibitor sodium butyrate (22). Sodium butyrate has been extensively studied for its ability to inhibit a broad range of HDACs (23) and has been shown previously to affect the extent of histone hyperacetylation in SV40 chromosomes (24). We hypothesized that if the unacetylated histones were present in regions of chromatin actively undergoing hyperacetylation, we would expect to find that following inhibition of HDACs with sodium butyrate histone hyperacetylation in the chromatin fragments independent of RNAPII would appear similar to histone hyperacetylation in the chromatin fragments bound by RNAPII. In contrast if the unacetylated histones were present in chromatin not undergoing hyperacetylation we would not expect to see any effect on the acetylation status of the histones following inhibition by sodium butyrate.
For example, we would expect to see that the ratios of PCR amplification products from the early and late regions in an ISFIP analysis would be the same following immunoprecipitation with antibody to hyperacetylated H4 and H3 as we observed in the ReChIP analysis (Figure 3) instead of being different as seen in an ISFIP analysis of untreated cells (Figure 3). As shown in Figure 6A we observed that the ratio of PCR products from the early and late regions following ISFIP analysis with antibody to hyperacetylated H4 were essentially identical to the ratio to antibody to hyperacetylated H3 in chromosomes obtained 30 minutes post infection (compare lane 2 to lane 3 – 30 minutes) and 48 hours post infection (compare lane 2 to lane 3 – 48 hours). This result was similar to what we observed in the ReChIP analysis (Figure 3) of chromosomes obtained at these times and differed significantly from what was observed in the ISFIP analysis (Figure 3) of chromatin from the same time points.
Figure 6A. Sodium butyrate treatment results in increased hyperacetylation of histones in the coding region of SV40 chromosomes undergoing transcription.
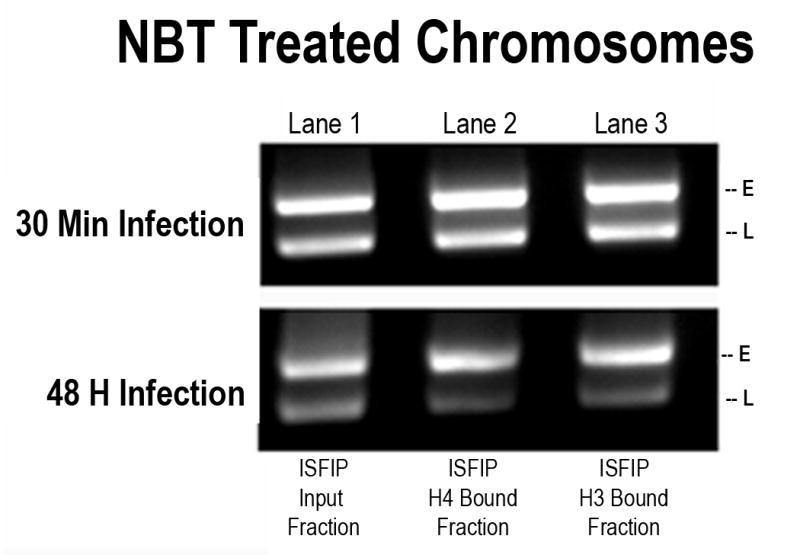
Sodium butyrate treated SV40 chromosomes were isolated from cells infected with 776 wild type virus for 30 minutes or 48 hours and subjected to an ISFIP analysis with antibodies to hyperacetylated histone H4 and H3 as described in the materials and methods. The samples were amplified by duplex PCR using primer sets to the early and late regions. E and L indicate the positions of the early and late PCR amplification products, respectively. The position of the amplification product from the wild-type 776 DNA is indicated. Lane 1: ISFIP input fraction; lane 2: ChIP with 7.5 μl of hyperacetylated histone H4 antibody (ISFIP); lane 3: ChIP with 10 μl of hyperacetylated histone H3 antibody (ISFIP). The PCR products in the ISFIP and ReChIP lanes were amplified from one half of the total amount of DNA obtained from each of the samples. The PCR products in the input lanes were amplified from one fourth of the total amount of DNA present in each of the input samples. Similar results were obtained from at least three separate preparations of SV40 chromosomes for each time point. All analyses were done with separate batches of sodium butyrate treated or untreated chromosomes.
Since the results from the duplex PCR analysis supported the hypothesis that the less acetylated histones in chromatin fragments lacking RNAPII were the result of the action of HATs and HDACs, we then determined whether unacetylated H4 and H3 were still present in the chromatin fragments lacking RNAPII in sodium butyrate treated cells. If as we hypothesized, the unacetylated H4 and H3 seen in the ISFIP analysis were the result of the combination of HAT and HDAC activity, we expected that in the sodium butyrate treated cells there would be little if any unacetylated H4 or H3 following an ISFIP analysis. As shown in Figure 6B this was the result which we obtained for the H4 and H3 located in the late coding region in transcribing SV40 chromosomes isolated late in infection. In agreement with the results shown in Figure 4, unacetylated H4 (lane 2) and unacetylated H3 (lane 3) were found in untreated chromosomes undergoing transcription following an ISFIP analysis but not following a ReChIP analysis (lanes 5 and 6). However, in chromosomes isolated at the same time following treatment with sodium butyrate we observed little if any evidence for the presence of unacetylated H4 (lane 2 and 5) or unacetylated H3 (lane 3 and 6) in the late coding region with either the ISFIP or ReChIP analyses. The absence of unacetylated H4 and H3 in the late coding region of transcribing SV40 chromosomes late in infection following treatment with sodium butyrate indicated that the unacetylated H4 and H3 originally present in untreated transcribing SV40 chromosomes was a result of the action of one or more HDACs. This result is consistent with the hypothesis that within the late region at late times there is coordinate hyperacetylation and deacetylation taking place. The results were not consistent with the hypothesis that the unacetylated histones were present in chromatin not undergoing hyperacetylation in this region of the genome.
Figure 6B. Sodium butyrate treatment results in the reduction of unacetylated histones in the late coding region of SV40 chromosomes undergoing transcription.
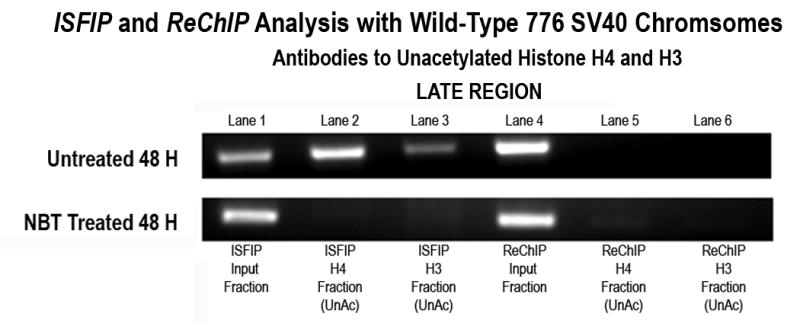
Sodium butyrate and unfixed, untreated SV40 chromosomes were isolated from cells infected with 776 wild type virus for 48 hours and subjected to an ISFIP/ReChIP analysis with antibodies to unacetylated histone H4 and H3 as described in the materials and methods. The samples were amplified by simplex PCR with primer sets to the late region. The position of the amplification product from the wild-type 776 virus is indicated. Lane 1: ISFIP input fraction; lane 2: ChIP with 10 μl of unacetylated histone H4 antibody (ISFIP); lane 3: ChIP with 10 μl of unacetylated histone H3 antibody (ISFIP); lane 4: ReChIP input fraction; lane 5: ChIP with 10 μl of unacetylated histone H4 antibody (ReChIP); lane 6: ChIP with 10 all of unacetylated histone H3 antibody (ReChIP). The PCR products in the ISFIP and ReChIP lanes were amplified from one half of the total amount of DNA obtained from each of the samples. The PCR products in the input lanes were amplified from one fourth of the total amount of DNA present in each of the input samples. Similar results were obtained from at least three separate preparations of SV40 chromosomes at 48 hours post-infection.
4. Organization of p300 on transcribing SV40 chromosomes
The presence of similar levels of hyperacetylated H4 and H3 co-localized with RNAPII in the transcribing regions of SV40 chromosomes, suggested that there might be a histone acetyl transferase (HAT) associated with the RNAPII. While a number of distinct HATs are known, p300 has been previously shown to interact with RNAPII (25), as well as a number of transcriptional factors known to be involved in the regulation of the SV40 promoter such as SP1 (26). For this reason we determined whether p300 was associated with SV40 chromosomes. For this analysis we utilized antibody from Santa Cruz Biotechnology which has been successfully utilized for ChIP analysis previously (27). Specificity of the p300 antibody for its epitope is shown in the Supplementary Data (SF11).
Using this antibody to p300, we then determined the location of p300 on the SV40 genome in transcribing SV40 chromosomes relative to RNAPII using our ISFIP/ReChIP procedure and SV40 chromosomes isolated 30 minutes and 48 hours post-infection. As shown in Figure 7A we observed p300 associated with RNAPII following a ReChIP analysis in the early, late, and promoter regions at each of the times tested (lane 4). However, in the absence of RNAPII (ISFIP analysis) we only observed p300 present in the promoter region (compare lane 2, ISFIP to Lane 4, ReChIP) at each time. We did not observe any p300 associated with either the early or late coding regions following an ISFIP analysis (lane 2).
Figure 7. p300 (HAT) is preferentially associated with RNA Polymerase II in the coding regions of SV40 chromosomes undergoing transcription.
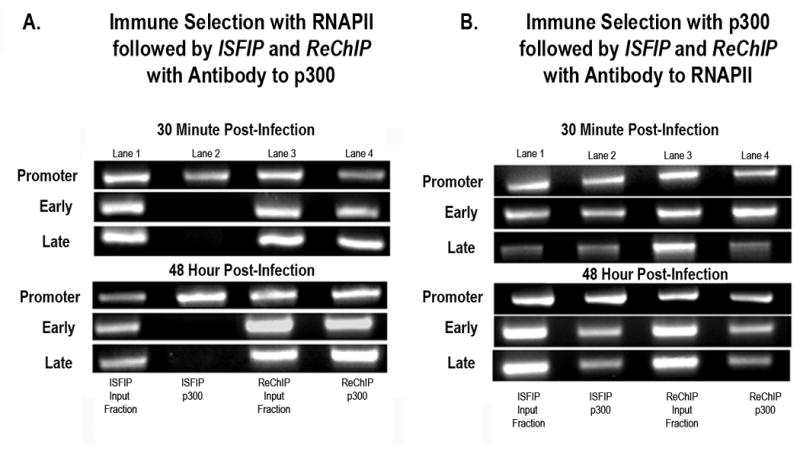
A: Unfixed SV40 chromosomes were isolated from cells infected with 776 wild type virus for 30 minutes or 48 hours and subjected to an ISFIP/ReChIP analysis with antibody to p300 as described in the materials and methods. The samples were amplified by simplex PCR with primer sets to the early, late and promoter regions. The position of the amplification product from the wild-type 776 DNA is indicated. Lane 1: ISFIP input fraction; lane 2: ChIP with 10 μl of p300 antibody (ISFIP); lane 3: ReChIP input fraction; lane 4: ChIP with 10 μl of p300 antibody (ReChIP); B: Unfixed SV40 chromosomes were isolated from cells infected with 776 wild type virus for 30 minutes or 48 hours and subjected to an ISFIP/ReChIP analysis with antibody to RNAPII as described in the materials and methods. The samples were amplified by simplex PCR with primer sets to the early, late and promoter regions. The position of the amplification product from the wild-type 776 DNA is indicated. Lane 1: ISFIP input fraction; lane 2: ChIP with 10 μl of RNAPII antibody (ISFIP); lane 3: ReChIP input fraction; lane 4: ChIP with 10 μl of RNAPII antibody (ReChIP). The PCR products in the ISFIP and ReChIP lanes were amplified from one half of the total amount of DNA obtained from each of the samples. The PCR products in the input lanes were amplified from one fourth of the total amount of DNA present in each of the input samples. Similar results were obtained from at least three separate preparations of SV40 chromosomes for each time point
In a parallel set of experiments we determined whether all of the RNAPII present in SV40 chromosomes also contained co-localized p300. In these analyses we first immune-selected chromosomes for the presence of p300 and then analyzed the chromatin fragments in the ISFIP and ReChIP fractions with antibody to RNAPII. As shown in Figure 7B we observed that RNAPII was present in both the ReChIP and ISFIP fractions (compare lanes 2 and 4) indicating that RNAPII was present in transcribing SV40 chromosomes both associated with p300 and independent of p300 throughout the genome. Taken together these results indicated that a significant fraction but not all of the RNAPII present in the coding regions of transcribing SV40 chromosomes contained associated p300.
Discussion
In order to investigate the status of histone hyperacetylation directly in chromatin undergoing transcription obtained in vivo, we have modified the standard ChIP assay by combining the well known ReChIP procedure (17) to a procedure which we refer to as ISFIP.
This immune combined strategy has two major advantages over many of the other strategies that have been used previously to study histone hyperacetylation during transcription. First, it allows for the direct characterization of relatively uncontaminated chromatin undergoing transcription. During the immune selection step only chromatin undergoing transcription operationally defined by the presence of RNAPII is bound to protein A agarose and any residual contaminating chromatin is removed by extensive washing. Second, because the chromatin undergoing transcription initially immune-selected can be relatively large in size and is subsequently fragmented; it is possible to characterize histone hyperacetylation or the presence of factors at selected sites throughout the initially selected chromatin. In the present study this allowed us to demonstrate that chromatin undergoing transcription contains both hyperacetylated and unacetylated H4 and H3.
Using this strategy we have obtained the first direct in vivo evidence from actively chromatin undergoing transcription that the acetylation of H4 and H3 was dynamic with higher levels of hyperacetylation at sites occupied by RNAPII and lower levels at other sites. Moreover, we have correlated the higher levels at sites occupied with RNAPII with the presence of a co-localized HAT, p300, and the lower levels at other sites with the action of one or more HDACs. We have also observed variations in the extent of hyperacetylation of H4 and H3 in the SV40 promoter, but because of the low resolution of the PCR analysis used and the complexity of the promoter we did not characterize the events occurring in this region to the same extent as the events occurring in the coding region.
The presence of hyperacetylated histones within the coding regions of actively transcribed SV40 chromosomes was not unexpected since hyperacetylated histones have been reported to be associated with transcription by many laboratories (28–30, 2). Since we have previously shown that RNAPII can be found throughout the SV40 genome during infection (7), it was not surprising to see that the hyperacetylated H4 and H3 could also be found throughout the genome.
The direct demonstration that the two hyperacetylated histones were found associated with and independent of RNAPII has not been noted previously except for our most recent publication (7). The observation that more hyperacetylated H4 and H3 were associated with RNAPII than was present in other in other regions of the transcribed chromatin has not been previously reported. However, these results have been predicted (31,30) based upon the observation that RNAPII can be found with piggybacked HATs (5,32). Similarly, the presence of unacetylated forms of H4 and H3 at sites not occupied by RNAPII, which resulted from HDAC activity, has not previously been observed, although their existence has also been suggested.
The observation that hyperacetylated and unacetylated H4 and H3 can be found at the same sites in the coding region of chromatin undergoing transcription depending upon whether RNAPII is present or absent would appear to argue against a model in which these histones in the coding region are labeled according to a “histone code” (33) based on acetylation. Instead the hyperacetylation of the H4 and H3 appears to be a direct result of the passage of the RNAPII through the chromatin. Whether this hyperacetylation serves as a signal for binding of necessary auxiliary transcriptional factors to the chromatin undergoing transcription or to decompact chromatin as has been suggested (31,30,29) remains to be determined. These results do not preclude the possibility that a histone code based on acetylation may function within the SV40 promoter. This possibility is presently under investigation.
The co-localization of the HAT p300 with RNAPII in the coding regions of the transcribing SV40 chromatin suggests that it might be responsible for the observed hyperacetylation of H4 and H3 in these regions. In this regard the p300 may be substituting for the PCAF or “elongator” cofactors, which have previously been shown to bind to RNAPII and are also capable of hyperacetylating H4 and H3 (32,25,34). While this is the first report that p300 is bound to RNAPII in actively chromatin undergoing transcription, p300 has been shown to bind to RNAPII (32,25,34). It is also possible that other HAT activities including PCAF or elongator are associated with the RNAPII in transcribing SV40 chromatin. Since we did not analyze directly for the presence of these activities, we cannot exclude their existence.
We used the inhibitor sodium butyrate to determine whether histone deacetylation was occurring in the regions of actively chromatin undergoing transcription which lacked RNAPII and contained unacetylated H4 and H3. Sodium butyrate has been used for many years because of its ability to inhibit histone deacetylases (22,35). Moreover, many laboratories have shown that the inhibition of histone deacetylases by sodium butyrate results in a significant increase in the level of hyperacetylated histones in chromatin and a corresponding increase in transcription (36,37). This association between the inhibition of histone deacetylation and increased transcription has lead in part to the suggestion that histone hyperacetylation and deacetylation are an integral part of the transcription process. Our results using immune-selected chromatin undergoing transcription from cells treated with sodium butyrate has directly confirmed the hypothesis that inhibition of histone deacetylases results in an increase in the level of histone hyperacetylation in actively chromatin undergoing transcription (30).
Histone deacetylation has been shown previously to occur within the coding region of certain transcribing genes in yeast (38,39). This deacetylation is mediated by histone H3 methylation by Set2 and is thought to suppress the initiation of transcription from sites within the coding region of genes (38,39). Although the same regulatory pathway may be occurring in the SV40 coding regions, we think it is unlikely that it would account for the extensive deacetylation which we observe in actively chromatin undergoing transcription.
In these studies we did not use extensive quantitation in our ChIP analyses except for the duplex analyses as we have previously used (6,7,9) for a number of reasons. First, we have previously shown that the percentages of chromatin fragments containing either hyperacetylated H4, hyperacetylated H3, or RNAPII which are bound or solubilized by the ISF procedure are highly reproducible based upon the quantitation of a large number of samples from multiple preparations of SV40 chromosomes (6,7). Second, we used semi-quantitative duplex PCR analysis to characterize the relative amounts of chromatin containing hyperacetylated H4 and H3. We have used this procedure previously and found it to be a reliable method to measure the relative proportions of chromatin from two different regions of the SV40 genome (7,6) because the procedure allows for the direct comparison of two samples prepared under identical conditions when the sample amounts are limited. Third, the critical observations in this study did not require quantitation. For example, while it might be of interest to know the percentage of the chromatin at a given site which is hyperacetylated or unacetylated, this information does not change the basic observation that both forms of chromatin can be present at the same site in actively chromatin undergoing transcription. Finally, the data presented were highly reproducible. Similar results were obtained for each analysis a minimum of three times using three different preparations of SV40 chromosomes
Based upon the results obtained from these studies we would like to propose the following model for the relationship between transcription and histone hyperacetylation in the coding region of a gene. We emphasize that the model is intended to describe the role of histone hyperacetylation only in the coding region and not in the promoter. We have observed a number of differences between the behavior of hyperacetylated histones, RNAPII, and transcription factors present in the promoter and in the coding region and believe that the role of histone hyperacetylation in the promoter is likely to be different and more complex. As shown in Figure 8, we propose that histone hyperacetylation is a dynamic process characterized by maximal histone hyperacetylation when RNAPII moves into a nucleosome followed by deacetylation as the RNAPII continues translocating through the coding region. In this model RNAPII acquires a HAT such as p300 during the initiation of transcription. This HAT forms a complex with the RNAPII and migrates along with the RNAPII through the coding region. When the RNAPII-p300 complex comes into contact with a nucleosome during active transcription, the p300 ensures that the histones present in the nucleosome are suitably hyperacetylated (Figure 8). Following transcription of the DNA present in the nucleosome and translocation of the complex to the next nucleosome, the hyperacetylated histones present in the original nucleosome may undergo deacetylation. Although we show the HDAC in close proximity to the RNAPII/p300 complex, we do not know at this time whether the HDAC is an integral part of a transcriptisome complex or a separate entity. This model is similar to one previously presented (30) although it differs in that we believe that histone deacetylation may occur along with hyperacetylation in the chromatin undergoing active transcription.
Figure 8.
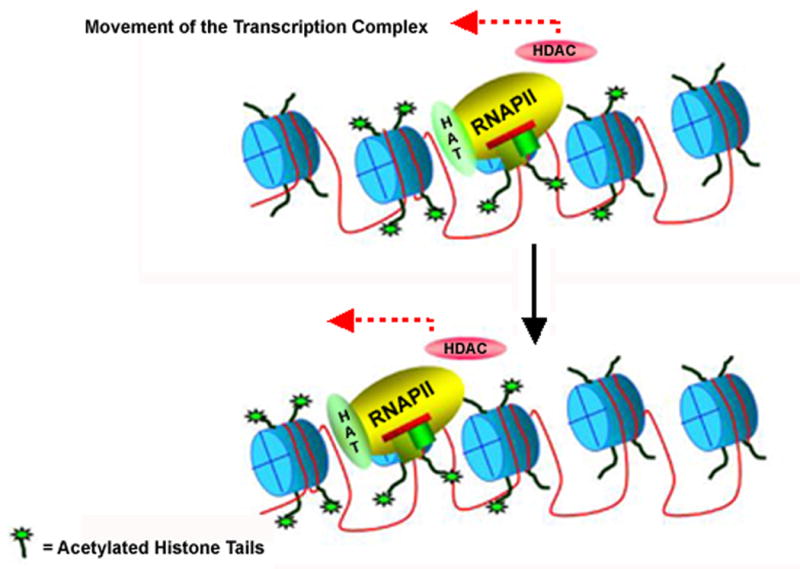
Model for the relationship between transcription and histone hyperacetylation in the coding region of a gene
Materials and Methods
Cells and Viruses
SV40 virus and chromatin were prepared in the BSC-1 cell line of monkey kidney cells (ATCC). The 776 SV40 wild type virus and cs1085 mutant virus was a gift from Dr Daniel Nathans.
Cell Culture and Infections
BSC-1 cells were maintained at 37° C in minimal essential medium containing 10% fetal calf serum, gentamycin, glutamine, and sodium bicarbonate (GIBCO). Sub-confluent monolayer of cells were infected with either the wild-type 776 virus or mutant cs1085 virus as previously described (18). The virus was allowed to adsorb onto the cells for 30 min. The virus was washed off, fresh medium was added and the plates were incubated further for 8 hours and 48 hours at 37° C in an incubator. SV40 chromosomes were harvested at 30 min, 8 hours and 48 hours post-infection respectively. SV40 chromatin was fixed with formalin by adding 0.275 ml of 37% formalin to the cells and incubating the cells with the formalin for 10 min at 37° C in an incubator. The formalin-fixed SV40 chromatin was isolated and prepared the same as the unfixed chromatin described below. Treated cells were grown in the presence of 250 μM of sodium butyrate for 12 hours before infecting with SV40 virus. SV40 chromosomes from treated cells were harvested in a similar manner as described above.
Preparation of SV40 chromosomes
SV40 chromosomes were harvested from infected cells and purified by glycerol gradient centrifugation as previously described (19,20) . Gradient fractions four and five, which contained SV40 chromosomes, were combined for subsequent analysis.
Chromatin Immunoprecipitations followed by Chromatin Fragmentation (ISF)
Sodium butyrate treated or untreated, unfixed SV40 chromosomes were immunoprecipitated with either 10μl antibody to RNA Polymerase II (sc-900; Santa Cruz Biotechnology) or 10 μl antibody to p300 (sc-584, Santa Cruz Biotechnology) using the reagents and protocol supplied by Upstate for the analysis of hyperacetylated H4 with minor modifications as previously described (6,7,9) with the exception of the addition of 175μl of chromatin to the pelleted agarose after 4 hours of incubation with the RNAP II antibody. In the final step of chromatin immunoprecipitation the pelleted agarose was resuspended in 400 μl of TE buffer. 200μl of resuspended agarose was sonicated and prepared for subsequent analysis as previously described (6,7)
Immune Selection Fragmentation followed by a second Immunoprecipitation (ISFIP)
Sodium butyrate treated or untreated, unfixed SV40 chromosomes were immunoprecipitated with antibody to RNAP II or antibody to p300 as described above for the ISF procedure. In the final step of ISF, the soluble fraction obtained after sonication was used as the secondary input sample (200 μl) and immunoprecipitated with antibody to RNAPII, unacetylated histone H4 (sc-8653), unacetylated histone H3 (sc-8657), p300, (Santa Cruz Biotechnology) hyperacetylated histone H4 (17-229), hyperacetylated histone H3 (17-245) (Upstate). The immunoprecipitation was carried out as described previously (6,7,9)
Re Chromatin Immunoprecipitation (Re-ChIP)
ReChIP was performed according to the procedure described by IJpenberg et al (2004) with minor modifications (17). Sodium butyrate treated or untreated, unfixed SV40 chromosomes were immunoprecipitated with antibody to RNAPII or antibody to p300 as described above for the ISF procedure. In the final step of ISF, the bound fraction was eluted twice with 200 μl Immunopure Gentle Ag/Ab Elution Buffer (Pierce). The bound fraction was incubated for 15-minute with elution buffer at room temperature and the eluted chromatin recovered by centrifugation. The eluates were pooled as the secondary input sample (200 μl) and immunoprecipitated with antibody to p300, unacetylated histone H4 and unacetylated histone H3 (Santa Cruz Biotechnology) or hyperacetylated histone H4 or hyperacetylated histone H3 (Upstate). The immunoprecipitation was carried out as described previously (6,7,9)
Preparation of DNA for PCR
Samples were prepared for PCR by phenol/chloroform extraction followed by ethanol precipitation in the presence of paint pellet co-precipitant (Novagen) as previously described (6,7,9). Approximately 100 μl of protein A agarose eluates was purified using phenol/chloroform. The aqueous phase (125μl) was added to a PCR tube that contained 3 μl of pellet paint co-precipitant and 12.5μl of 3 M sodium acetate, pH 5.2 (Novagen). The samples were mixed and 280 μl of 100% ethanol added to each. Following 10-min incubation at room temperature, the samples were centrifuged at 8000 x g for 5 min and the supernatant discarded. The samples were washed with 70% ethanol, vortexed, incubated for 5 min, and then centrifuged at 8000 x g for 5 min. The supernatant was again discarded and the samples were dried in a vacuum
PCR amplifications
DNA was amplified from three different regions of the SV40 genome (the early coding region, the late coding region and the promoter) in a Perkin-Elmer Model 480 thermal cycler using Ampli Taq Gold DNA Polymerase (Applied Biosystems) with primer sets 5’GCTCCCATTCATCAGTTCCA3’ and 5’ CTGACTTTGGAGGCTTCTGG3’ for the amplification of the early region (nt 4540-4949), 5’ CAGTGCAAGTGCCAAAGATC3’ and 5’GCAGTTACCCCAATAACCTC3’ for amplification of the late region (nt 1566-1878) and 5’GCAAAGCTTTTTGCAAAAGCCTAGGCCT3’and 5’CGAACCTTAACGGAGGCCTGGCG3’ for amplification of the promoter region (nt 5168-420). A master mix containing all the required constituents was prepared according to the instructions supplied with the DNA polymerase in advance and kept at −20°C until required. Immediately before use, the master mix was thawed and a volume corresponding to 30 μl for each sample to be amplified was removed to prepare a working mix. The working mix was then prepared by adding the DNA polymerase to the master mix in the ratio of 0.5 μl per 30 μl of master mix. Following thorough mixing, the 30 μl of working mix was added to each previously prepared PCR tube containing a sample of template DNA to be amplified. The tubes were gently vortexed to suspend the pelleted DNA present in the tubes. When suspension was complete, the samples were overlaid with two drops of molecular biology grade mineral oil (Sigma). All previous manipulations were performed in a Nuaire biological safety cabinet Model NU_425-400. The samples were centrifuged for 1 min at 10,000 x g in an Eppendorf micro centrifuge, and the PCR amplifications were hot started by heating the tubes for 2 min and 30 sec at 95° C. The DNA was amplified for 45 cycles with each cycle consisting of annealing at 60° C for early region, 64° C for late region and 70° C for the promoter region for 1 min, DNA synthesis for 1 min at 72° C and denaturation at 95° C for 1 min. Duplex PCR reaction mixes were made by adding early and late reaction mixes in a 1:1 ratio just before PCR amplifications. For the duplex analyses, the DNA was amplified for 45 cycles with each cycle consisting of annealing at 60° C for 1 min, DNA synthesis for 1 min at 72° C and denaturation at 95°C for 1 min.
Analysis of PCR Amplification Products
Following PCR amplification of the DNA samples, the products were separated on 2.4% submerged agarose gels (Sigma) by electrophoresis. (6,7,9,19,20,8) The separated products were visualized by staining with ethidium bromide and electronically photographed using UVP GDS8000 Gel Documentation System (Ultra Violet Products).
Scanning Densitometry
Quantitation of agarose gels was done with Molecular Analyst (Version 1.4) from Bio-Rad. Using Molecular analyst images were obtained by importing those that were captured with UVP GDS8000 Gel Documentation System. On importing the image, quantitation was performed with the Volume Analysis function to determine the percent volume of DNA bands of interest. The Local Background subtraction function was utilized to normalize background noise.
Supplementary Material
Acknowledgments
This work was supported in part by grants from National Institute of Health (1R15GM074811-01A1 to BM), the University of North Dakota SEED program and the University of North Dakota School of Medicine and Allied Health Sciences. We would like to thank Dr. Steve Tronick from Santa Cruz Biotechnology for the generous offer to share the RNAPII, p300 and the unacetylated histone H4 and unacetylated histone H3 antibodies.
Footnotes
Suggestion for Cover Illustration
We would like to suggest using Figure 8: Model for the relationship between transcription and histone hyperacetylation in the coding region of a gene for the cover illustration.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wu J, Grunstein M. 25 years after the nucleosome model: chromatin modifications. Trends Biochem Sci. 2000;25:619–623. doi: 10.1016/s0968-0004(00)01718-7. [DOI] [PubMed] [Google Scholar]
- 2.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 3.Ruiz-Carrillo A, Wangh LJ, Allfrey VG. Processing of newly synthesized histone molecules. Science. 1975;190:117–128. doi: 10.1126/science.1166303. [DOI] [PubMed] [Google Scholar]
- 4.Kingston RE, Narlikar GJ. ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev. 1999;13:2339–2352. doi: 10.1101/gad.13.18.2339. [DOI] [PubMed] [Google Scholar]
- 5.Cho H OG, Sun X Yang XJ, Ogryzko V Lees E, Nakatani Y Reinberg D. A human RNA polymerase II complex containing factors that modify chromatin structure. Mol Cell Biol. 1998;18:5355–5363. doi: 10.1128/mcb.18.9.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balakrishnan L, Milavetz B. Programmed remodeling of hyperacetylated histone H4 and H3 organization on the SV40 genome during lytic infection. Virology. 2005;334:111–123. doi: 10.1016/j.virol.2005.01.025. [DOI] [PubMed] [Google Scholar]
- 7.Balakrishnan L, Milavetz B. Reorganization of RNA polymerase II on the SV40 genome occurs coordinately with the early to late transcriptional switch. Virology. 2006;345:31–43. doi: 10.1016/j.virol.2005.09.039. [DOI] [PubMed] [Google Scholar]
- 8.Milavetz BI. SP1 and AP-1 elements direct chromatin remodeling in SV40 chromosomes during the first 6 hours of infection. Virology. 2002;294:170–179. doi: 10.1006/viro.2001.1308. [DOI] [PubMed] [Google Scholar]
- 9.Milavetz B. Hyperacetylation and differential deacetylation of histones H4 and H3 define two distinct classes of acetylated SV40 chromosomes early in infection. Virology. 2004;319:324–336. doi: 10.1016/j.virol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Fields B, Knipe DM, Howley PM. Fields Virology. 3. Lippincott Raven; 1996. [Google Scholar]
- 11.Hansen U. Methods in Molecular Biology: Chromatin Protocols. Vol. 119. Humana Press; Totowa, N.J: 1999. Transcriptional and structural analyses of isolated SV40 chromatin; pp. 261–290. [DOI] [PubMed] [Google Scholar]
- 12.Randall S, Kelly TJ. The fate of parental nucleosomes during SV40 DNA replication. J Biol Chem. 1992;267:14259–14265. [PubMed] [Google Scholar]
- 13.Tijan R. The biochemistry of transcription and gene regulation. Harvey Lect. 1994–1995;90:19–39. [PubMed] [Google Scholar]
- 14.Alberts AS, Geneste O, Treisman R. Activation of SRF-regulated chromosomal templates by Rho-family GTPases requires a signal that also induces H4 hyperacetylation. Cell. 1998;92:475–487. doi: 10.1016/s0092-8674(00)80941-1. [DOI] [PubMed] [Google Scholar]
- 15.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20:615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 16.Mizzen CA, Allis CD. Linking histone acetylation to transcriptional regulation. Cell Mol Life Sci. 1998;54:6–20. doi: 10.1007/s000180050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.IJpenberg A, Tan NS, Gelman L, Kersten S, Seydoux J, Xu J, Metzger D, Canaple L, Chambon P, Wahli W, et al. In vivo activation of PPAR target genes by RXR homodimers. Embo J. 2004;23:2083–2091. doi: 10.1038/sj.emboj.7600209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kube D, Milavetz B. Generation of a nucleosome-free promoter region in SV40 does not require T-antigen binding to site I. Virology. 1989;172:100–105. doi: 10.1016/0042-6822(89)90111-6. [DOI] [PubMed] [Google Scholar]
- 19.Hermansen R, Sierra MA, Johnson J, Friez M, Milavetz B. Identification of Simian virus 40 promoter DNA sequences capable of conferring restriction endonuclease hypersensitivity. J Virol. 1996;70:3416–3422. doi: 10.1128/jvi.70.6.3416-3422.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friez M, Hermansen R, Milavetz B. Chromatin structure of the simian virus 40 late promoter: a deletional analysis. J Virol. 1999;73:1990–1997. doi: 10.1128/jvi.73.3.1990-1997.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nightingale KP, Wellinger RE, Sogo JM, Becker PB. Histone acetylation facilitates RNA polymerase II transcription of the Drosophila hsp26 gene in chromatin. Embo J. 1998;17:2865–2876. doi: 10.1093/emboj/17.10.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boffa LC, Vidali G, Mann RS, Allfrey VG. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J Biol Chem. 1978;253:3364–3366. [PubMed] [Google Scholar]
- 23.Ayer DE. Histone Deacetylases: Transcription Repression with SINers and NuRDs. Trends Biochem Sci. 1999;9:193–198. doi: 10.1016/s0962-8924(99)01536-6. [DOI] [PubMed] [Google Scholar]
- 24.Roman A. Alteration in the simian virus 40 maturation pathway after butyrate-induced hyperacetylation of histones. J Virol. 1982;44:958–962. doi: 10.1128/jvi.44.3.958-962.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- 26.Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68:1145–1155. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 27.Huang WC CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol. 2005;15:6592–6602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehrenhofer-Murray AE. Chromatin dynamics at DNA replication, transcription and repair. Eur J Biochem. 2004;271:2335–2349. doi: 10.1111/j.1432-1033.2004.04162.x. [DOI] [PubMed] [Google Scholar]
- 29.Bulger M. Hyperacetylated chromatin domains: lessons from heterochromatin. J Biol Chem. 2005;280:21689–21692. doi: 10.1074/jbc.R500004200. [DOI] [PubMed] [Google Scholar]
- 30.Orphanides G, Reinberg D. RNA polymerase II elongation through chromatin. Nature. 2000;407:471–475. doi: 10.1038/35035000. [DOI] [PubMed] [Google Scholar]
- 31.Travers A. Chromatin modification: how to put a HAT on the histones. Curr Biol. 1999;9:R23–25. doi: 10.1016/s0960-9822(99)80037-2. [DOI] [PubMed] [Google Scholar]
- 32.Wittschieben BO,OG, de Bizemont T, Fellows J, Erdjument-Bromage H, Ohba R, Li Y, Allis CD, Tempst P, Svejstrup JQ. A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol Cell. 1999;4:123–128. doi: 10.1016/s1097-2765(00)80194-x. [DOI] [PubMed] [Google Scholar]
- 33.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 34.Otero G, Fellows J, Li Y, de Bizemont T, Dirac AM, Gustafsson CM, Erdjument-Bromage H, Tempst P, Svejstrup JQ. Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol Cell. 1999;3:109–118. doi: 10.1016/s1097-2765(00)80179-3. [DOI] [PubMed] [Google Scholar]
- 35.Riggs MG, Whittaker RG, Neumann JR, Ingram VM. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268:462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 36.Gorman CM, Howard BH, Reeves R. Expression of recombinant plasmids in mammalian cells is enhanced by sodium butyrate. Nucleic Acids Res. 1983;11:7631–7648. doi: 10.1093/nar/11.21.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 39.Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.