

Abstract
The neurotoxicity of high levels of methylmercury (MeHg) is well established both in humans and experimental animals. Astrocytes accumulate MeHg and play a prominent role in mediating MeHg toxicity in the central nervous system (CNS). Although the precise mechanisms of MeHg neurotoxicity are ill-defined, oxidative stress and altered mitochondrial and cell membrane permeability appear to be critical factors in its pathogenesis. The present study examined the effects of MeHg treatment on oxidative injury, mitochondrial inner membrane potential, glutamine uptake and expression of glutamine transporters in primary astrocyte cultures. MeHg caused a significant increase in F2-isoprostanes (F2-IsoPs), lipid peroxidation biomarkers of oxidative damage, in astrocyte cultures treated with 5 or 10 μ M MeHg for 1 or 6 hours. Consistent with this observation, MeHg induced a concentration-dependant reduction in the inner mitochondrial membrane potential (ΔΨm), as assessed by the potentiometric dye, tetramethylrhodamine ethyl ester (TMRE). Our results demonstrate that ΔΨm is a very sensitive endpoint for MeHg toxicity, since significant reductions were observed after only 1 h exposure to concentrations of MeHg as low as 1 μ M. MeHg pretreatment (1, 5 and 10 μ M) for 30 min also inhibited the net uptake of glutamine (3H-glutamine) measured at 1 min and 5 min. Expression of the mRNA coding the glutamine transporters, SNAT3/SN1 and ASCT2, was inhibited only at the highest (10 μ M) MeHg concentration, suggesting that the reduction in glutamine uptake observed after 30 min treatment with lower concentrations of MeHg (1 and 5 μ M) was not due to inhibition of transcription. Taken together, these studies demonstrate that MeHg exposure is associated with increased mitochondrial membrane permeability, alterations in glutamine/glutamate cycling, increased ROS formation and consequent oxidative injury. Ultimately, MeHg initiates multiple additive or synergistic disruptive mechanisms that lead to cellular dysfunction and cell death.
1. Introduction
Methylmercury (MeHg) is an organic form of mercury with well established neurotoxicity both in humans and experimental models [25,27]. Although the mechanisms of MeHg-induced neurotoxicity remain unclear, it is intriguing that a compound that readily reacts with sulfhydryl groups shows high organ specificity. MeHg can easily cross the blood-brain and placental barriers and cause central nervous system (CNS) damage in both the adult and developing brain [26,41]. While MeHg can directly cause damage to neurons, numerous studies have established a prominent role for astrocytes in mediating MeHg neurotoxicity [23,36]. The evidence includes observations that MeHg preferentially accumulate in astrocytes [5,18,34] and inhibits uptake systems for glutamate and cysteine transport, both of which will compromise glutathione (GSH) synthesis and redox status in astrocytes [2,16,30,61,62,63]. Furthermore, MeHg causes the activation of cytosolic phospholipase A2 (cPLA2), leading to arachidonic acid release and further inhibition of glutamate transporters and neuronal dysfunction [6,8].
Reactive oxygen species (ROS) generation has been linked to MeHg- induced toxicity both in vivo and in vitro. For example, cultured neurons [53] and glia [64] exposed to MeHg and brain synaptosomes prepared from animals injected with MeHg [1], demonstrate increased ROS production. In addition, an increase in ROS has been observed in mitochondria isolated from MeHg-injected rat brains, isolated rat brain mitochondria exposed to MeHg in vitro [54] and mitochondria from Hg- and glutamate-exposed astrocytes and neurons [14,32]. Evidence suggests that MeHg exposure causes production of ROS, depletion of ATP, excessive accumulation of calcium (Ca2+) and a decrease in mitochondrial membrane potential in mitochondria from the nervous [43] and immune [39,67] systems.
Excessive ROS production, leading to a decrease of mitochondrial membrane potential may also induce the oxidation of membrane polyunsaturated fatty acids, yielding a multitude of lipid peroxidation products. One such family of products is the F2-isoprostanes (F2-IsoPs), prostaglandin-like molecules produced by free radical-mediated peroxidation of arachidonic acid [52]. The measurement of F2-IsoPs has emerged as the most accurate and reliable indicator of oxidative stress in vivo [57]. F2-IsoPs levels are elevated in many tissues exposed to inflammation [45,46], excitotoxicity [47] and anticholinesterase toxicity [48], as well as in diseased regions of the brain in patients who died from advanced Alzheimer’s disease [49]. However, biomarkers of free radical damage in the astrocyte model of MeHg neurotoxicity have not yet been evaluated.
Astrocytes play a primary neuromodulatory role in glutamate-glutamine homeostasis. Glutamine (Gln) is an important amino acid that plays a pivotal role in neuronglia interactions, particularly in the turnover of the transmitter pool of glutamate (Glu) and γ -aminobutyric acid (GABA) [12,29,33], the principal CNS excitatory and inhibitory neurotransmitters respectively [24,28]. After being released during neurotransmission, glutamate is taken up largely by astrocytes. In the astrocytic compartment of the mammalian brain, glutamate is converted into glutamine via a highly active glutamine synthetase (GS) pathway and subsequently released into the intracellular space. Neighboring glutamatergic and GABAergic neurons use glial glutamine as a precursor for neurotransmitter synthesis as a part of the glutamate-glutamine cycle [11,59,60,68]. Studies in recent years have provided evidence that carrier-mediated Gln transport between astrocytes and neurons is a key factor in the glutamate/glutamine cycle. The molecular bases of Gln passage across the astrocytic membrane and neuronal plasma membranes have been investigated extensively over the last few years [12,20,44]. Gln efflux from astrocytes appears to be mediated by sodium-coupled neutral amino acid transporter 3 (SNAT3, SN1), a system N amino acid transporter that is localized to perisynaptic astrocytes and specifically accepts only glutamine, histidine and asparagine [13,19,21]. Gln uptake into neurons is mediated by sodium-dependent transporters of the system A family, two of which, SNAT1 (GlnT, SAT1, ATA1, SA2) and SNAT2 (SAT2, ATA2), are thought to be capable of affecting Glu and/or GABA recycling, and, thereby, synaptic function. Another notable sodium-dependent, glutamine-accepting amino acid transporter in the CNS is ASCT2, which operates in an exchange mode. ASCT2 is more abundantly expressed in astrocytes than in neurons in culture [15,31]. A previous study from our laboratory has shown that exposure of astrocytes to acrylamide affects astrocytic glutamine uptake and expression of mRNA coding for Gln transporters [73] at concentrations comparable to those producing acute toxicity [7,9].
Given these earlier observations , the present study was carried out to examine the effects of MeHg treatment on oxidative injury (F2-IsoPs), mitochondrial inner membrane potential (ΔΨm) and both Gln uptake and expression of SNAT3, ASCT2 and SNAT1 mRNA in primary astrocyte cultures.
2. Materials and methods
2.1. Materials
L-[G-3H]Glutamine (specific activity: 49.0 Ci/mmol) was purchased from Amersham Biosciences (Piscataway, NJ). Methylmercuric chloride (MeHgCl) was purchased from ICN Biomedicals (Costa Mesa, CA). Minimal essential medium (MEM) with Earle's salts, heat-inactivated horse serum, penicillin, streptomycin and tetramethyl rodamine ethyl ester (TMRE) were purchased from Invitrogen (Carlsbad, CA).
2.2. Primary astrocytes culture
Astrocytic cultures from cerebral cortices of newborn (1-day-old) Sprague–Dawley rats were established as previously described [4]. Briefly, rat pups were decapitated, and the cerebral cortices were removed. After carefully removal of meninges, the cerebral cortices were digested with bacterial neutral protease (dispase, Invitrogen), and astrocytes were recovered by the repeated removal of dissociated cells from brain tissues. Twenty-four hours after the initial plating in 6- and 12-well plates, the media were changed to preserve the adhering astrocytes and to remove the neurons and oligodendrocytes. The cultures were maintained at 37°C in a 95% air/5% CO2 incubator for 3 – 4 weeks in minimal essential medium (MEM) with Earle's salts supplemented with 10% heat-inactivated horse serum, 100 U/ml penicillin and 100 μg/ml streptomycin. The media were changed twice per week. These monolayer, surface-adhering cultures were >95% positive for the astrocytic marker, glial fibrillary acidic protein (GFAP).
2.3. F2-isoprostanes Assay
Total F2-IsoPs were measured in primary astrocyte cultures using gas chromatography/mass spectrometry (GC/MS) with selective ion monitoring [52]. Briefly, cells were resuspended in 0.5 ml methanol containing 0.005% butylated hydroxytoluene, sonicated and then subjected to chemical saponification using 15% KOH to hydrolyze bound F2-IsoPs. The cell lysates were adjusted to pH 3, followed by the addition of 0.1 ng of 4H2-labeled 15-F2α -IsoP internal standard. F2-IsoPs were then purified by C18 and silica Sep-Pak extraction and by thin layer chromatography. They were then analyzed as pentafluorobenzyl ester, a trimethylsilyl ether derivative, via gas chromatography, negative ion chemical ionization mass spectrometry.
2.4. Measurement of mitochondrial membrane potential (ΔΨm)
The ΔΨm was measured with the fluorescent dye TMRE [56]. Following MeHg treatments (1, 5 or 10 μ M) for 1 or 6 hr, the culture medium was removed, and the cells were loaded with TMRE at a final concentration of 50 nM in HEPES buffer for 20 min at 37 ºC in a 5% CO2 incubator. At the end of treatment, cells were rinsed with PBS and examined with a Zeiss inverted fluorescent microscope (Zeiss Axiovert S100, Carl Zeiss MicroImaging Inc.) equipped with a cooled digital camera (Photometrics CoolSNAP, Roper Scientific Photometrics, Tucson, AZ). Images of various fields in each plate were captured at 10× magnification with the digital camera. Fluorescent intensities were calculated in 8 randomly selected fields per experiment and were analyzed using the NIH software (Scion Incorporation, Frederick, MD). In each image field, the total number of pixels was quantified on a gray scale (0–255 counts), and the mean pixel value in each image field was obtained and expressed as mean ± S.E.M. The fluorescent intensities were expressed as percent fluorescence change over control. Each experiment was performed in duplicate plates and repeated three times using different astrocyte isolations.
2.5. Analysis of 3H-glutamine uptake in astrocyte cultures
The 3H-glutamine uptake assay was carried out as described previously [2]. Astrocytes were studied at 3 weeks when the monolayers were confluent. Cells in 6-well plates were washed three times with 2 ml of fresh sodium-HEPES buffer consisting of: 122 mM NaCl, 3.3 mM KCl, 0.4 mM MgSO4, 1.3 mM CaCl2, 1.2 mM KH2PO4, 10 mM glucose, and 25 mM HEPES (N-2-hydroxy-ethylpiperazine N’-2-ethansulfonic acid) adjusted to pH 7.4 with 10 M NaOH. Cells were pretreated in Na-HEPES buffer only or with Na-HEPES buffer containing MeHg (1, 5, or 10 μM) for 30 min in a 37 °C, in a 95% air/5% CO2 incubator. Cells were then washed three times with 2 ml of Na-HEPES buffer, and, afterwards, rinsed with 1 ml of pre-warmed buffer containing 1 μCi/ml L-[G-3H]-glutamine in the presence of unlabelled glutamine. A final concentration of 50 μM was added to each well, and glutamine uptake was measured at 1 min and 5 min at room temperature. At each time point, the reactions were terminated by aspirating the buffer from the well, followed by 5 washes with 2 ml of ice-cold mannitol buffer [290 mM mannitol, 10 mM Tris-nitrate, 0.5 mM Ca(NO3)2, pH adjusted to 7.4 with KOH]. At the end of the experiment, cells were lysed in 2 ml of 1M NaOH. An aliquot of 25 μl was used for protein determination with the BCA protein assay (Pierce, Rockford, IL). An aliquot of 750 μl was used for radioactivity measurement. Uptake of glutamine was expressed as cpm/mg protein.
2.6. RT-PCR
Total RNA from cell cultures was extracted using Trizol (Invitrogen). RNA (2 μg) was transcribed using Superscript II (Invitrogen). The primers were obtained from the Laboratory of DNA Sequencing, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw. Each cDNA (2 μl) was amplified by PCR using the primers for the rat amino acid transporters, SNAT3, SNAT1 and ASCT2. The expression was quantitatively related to the gene coding for the constitutive protein, GAPDH. The sequence of primers and the lengths of the products are outlined in Table 1. After 30 cycles of amplification (1 min at 94°C, 1 min at 59°C for SNAT3, 55°C for ASCT2 and SNAT1, and 1 min at 72°C using Biometra thermocycler), the PCR products were recorded using the Nucleovision system (Nucleotech), and densitometric analysis was carried out using the GelExpert 4.0 program.
Table 1.
Primer sequences used for RT-PCR analysis
| Name | Primer (5’– 3’) | Position | Size (bp) | GenBank number |
|---|---|---|---|---|
| Rat | ||||
| GAPDH | F tgaaggtcggagtcaacggatttgg
R catgtaggccatgaggtccaccac |
80–104
1062–1038 |
998 | BC013310 |
| SN1(SNAT3) | F aacatcggagccatgtccag
R aaggtgaggtagccgaagag |
585–557
1159–1140 |
578 | NM006841 |
| ASCT2 | F gccagtccacggccaagatc
R gcctggtcgtgttcgctata |
1769–1750
1086–1102 |
687 | BC080242 |
| ATA1(SNAT1) | F cccattgtcactgctgagaa
R tccctgatagtggggacaaa |
1015–1034
1695–1676 |
681 | NM181090 |
2.7. Statistical analysis
Measurements of 3H-Glutamine uptake, F2-IsoPs levels and mitochondrial membrane potential were conducted in duplicate or triplicate wells/experiment, and the means from three to four experiments were used for statistical analysis. The data were analyzed by one-way analysis of variance (ANOVA), followed by Bonferroni's multiple comparison test with statistical significance set at P<0.05. All analyses were carried out using Graph Pad Prism 4.02 for Windows (Graph Pad Software, San Diego, CA, USA).
3. Results
3.1. Effects of MeHg on the F2 – IsoPs formation in cultured astrocytes
We tested the ability of MeHg to induce oxidative stress in primary astrocytes by measuring levels of F2-IsoPs, a lipid peroxidation biomarker of oxidative injury. Primary astrocytes exposed to 5 μ M or 10 μ M MeHg for 1 or 6 hours showed significant increases in F2-isoPs levels (p<0.05) (Fig. 1). The highest increases in F2-IsoPs levels were detected after 6 hours with 5 μ M MeHg exposure. Concentrations >5 μM did not increase the effect of MeHg on F2-IsoPs, suggesting a maximal effect at his concentration (5 μM). The lowest MeHg concentration (1 μ M) did not cause a significant increase in markers of oxidative stress with F2-IsoPs levels indistinguishable from controls.
Figure 1.
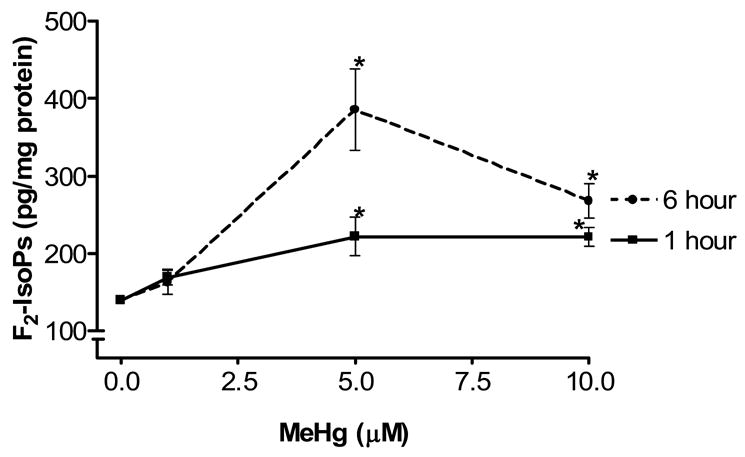
Effect of MeHg on F2-IsoPs formation in cultured astrocytes. Rat primary astrocyte cultures were incubated at 37 °C in the absence or presence of MeHg (1, 5, and 10 μM) and F2-IsoPs levels quantified at 1 and 6 hr, respectively. Data represent the mean ± S.E. from three independent experiments. * p<0.05 versus control by one-way ANOVA followed by Bonferroni multiple comparison tests.
3.2. Effects of MeHg on the ΔΨm in cultured astrocytes
To determine whether mitochondrial dysfunction contributes to MeHg-induced neurotoxicity, we treated astrocytes with MeHg (1, 5 or 10 μ M) for 1 hour (Fig. 2A) and 6 hours (Fig. 2C) and measured the ΔΨm by TMRE fluorescence. Treatments with MeHg for 1 hr (Fig. 2B) and 6 hr (Fig. 2D) resulted in significant dissipation of the ΔΨm, as demonstrated by decreased mitochondrial TMRE fluorescence. Quantification of TMRE fluorescence intensities revealed that all investigated MeHg concentrations induced significant dissipation of the ΔΨm.
Figure 2.
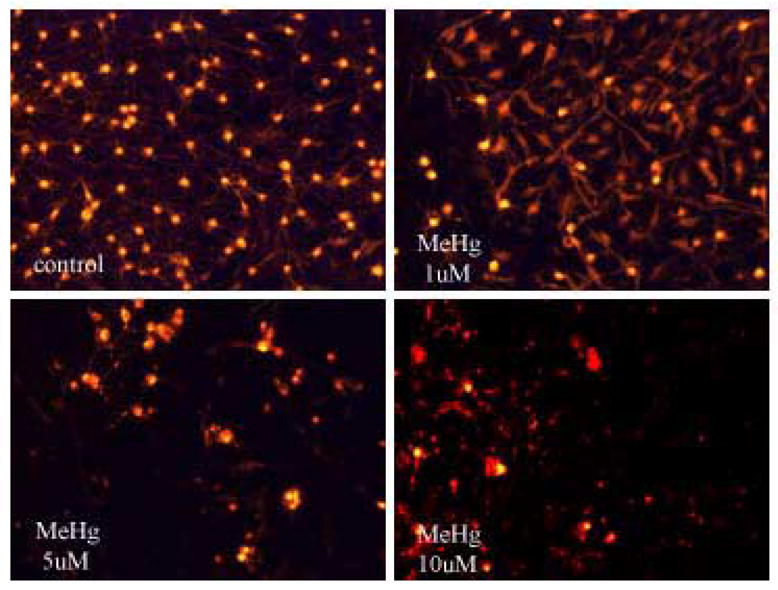
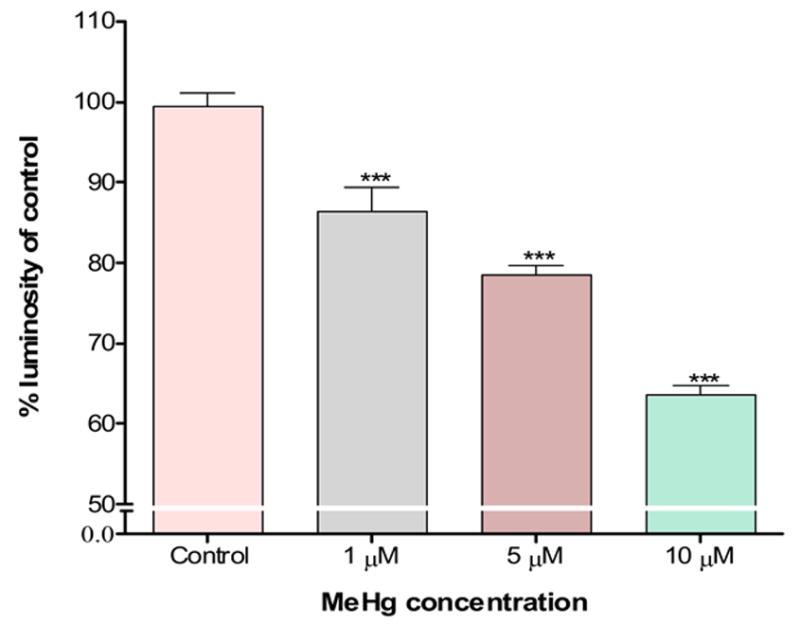
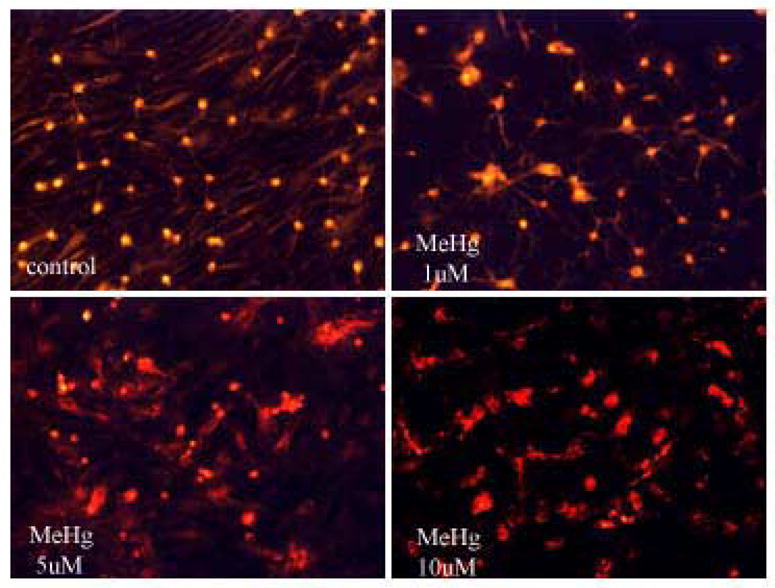
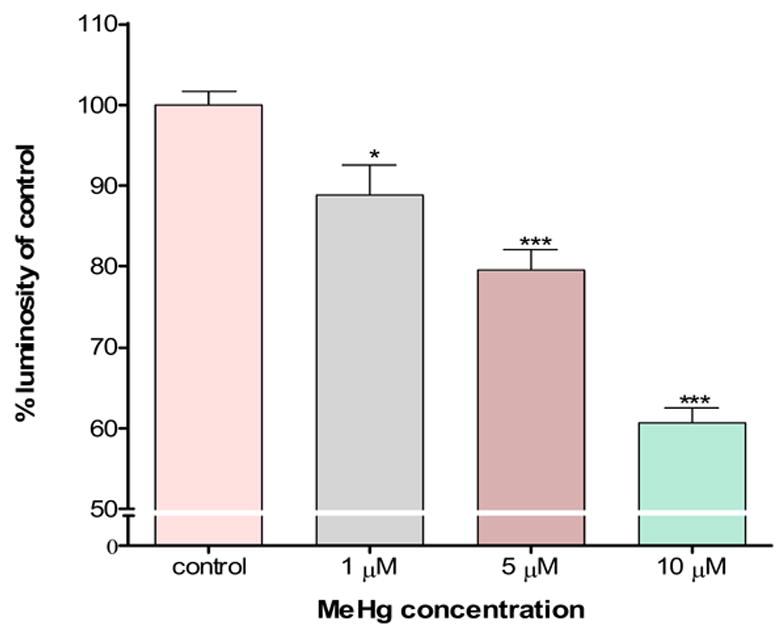
Quantitation of TMRE fluorescent intensities. Cultured astrocytes exposed to MeHg at various concentrations (0, 1, 5, and 10 μM) for 1 hr (A and B) and 6 hr (C and D) and the fluorescent images quantified as described in Section 2. Values are expressed as mean ± S.E.M. of 24 random fields in each group. * p< 0.05, *** p< 0.001 versus control; Δ Δ Δp< 0.001 versus 10μM. note - (2A is image after 1h exposure; 2C is image after 6 h exposure).
3.3. Glutamine uptake inhibition by MeHg in cultured astrocytes
Compared with control uptake rates (100%), pretreatment of astrocytes for 30 min with MeHg inhibited the net uptake of glutamine at 1 min and 5 min in a concentration-dependent manner (Fig. 3). All MeHg treatments (1, 5, or 10 μM) significantly (p<0.001) inhibited astrocytic uptake at both time points. Furthermore, 10 μ M MeHg treatment induced significantly higher inhibition of glutamine uptake at 5 min compared to 1 or 5 μM treatments (p<0.001 and p< 0.05), respectively. No significant differences in glutamine uptake were seen between 1 and 5 min exposures when astrocytes were treated with 1, 5, or 10 μM MeHg.
Figure 3.
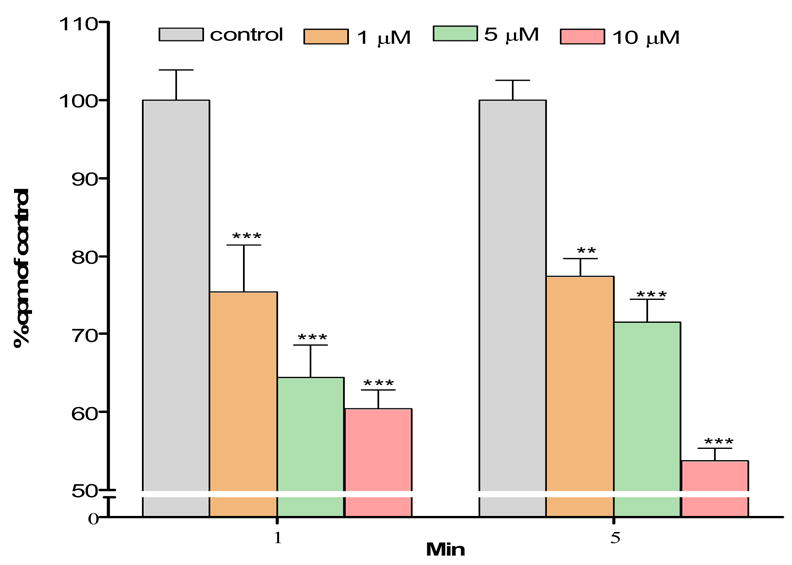
Effects of MeHg on glutamine uptake in astrocytes. Rat primary astrocyte cultures were incubated for 30 min at 37 °C in the absence or presence of MeHg (1, 5, and 10 μM) and the net uptake of glutamine (3H-glutamine) was quantified at 1 and 5 min, respectively. MeHg exposure induced a concentration-dependent decrease in glutamine uptake (*** p<0.001 versus control; Δp<0.05; Δ Δ Δp<0.001 versus 10 μM MeHg, n=6–10; mean ± S.E.M.).
3.4. Influence of MeHg on expression of SNAT3, ASCT2, and SNAT1 mRNA
To explore molecular mechanisms associated with the effects of MeHg on glutamine uptake, we measured the astrocytic amino acid transporter mRNAs by RT-PCR. Figure 4 shows the effects of MeHg treatment on the mRNA expression of SNAT3, ASCT2, and SNAT1. The bars display the ratios of SNAT3, ASCT2 and SNAT1 to GAPDH, a constitutive marker. MeHg treatment at 10 μM significantly (p<0.05) reduced the mRNA expression of SNAT3 and ASCT2, but not of SNAT1 (Figures 4 A and B). The mRNA expression levels of all three amino acid transporters were not significantly different from controls (p>0.05) when primary astrocytes cultures were exposed to 1 or 5 μM MeHg.
Figure 4.
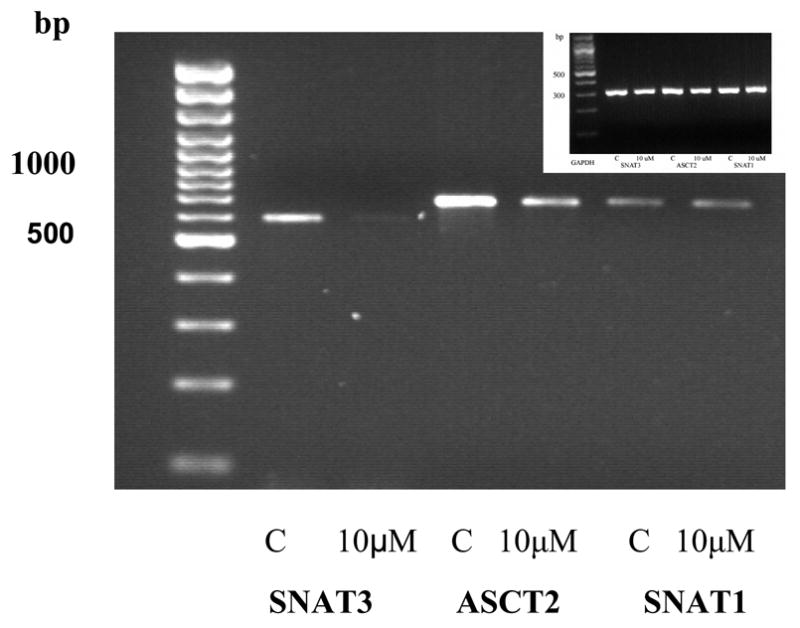
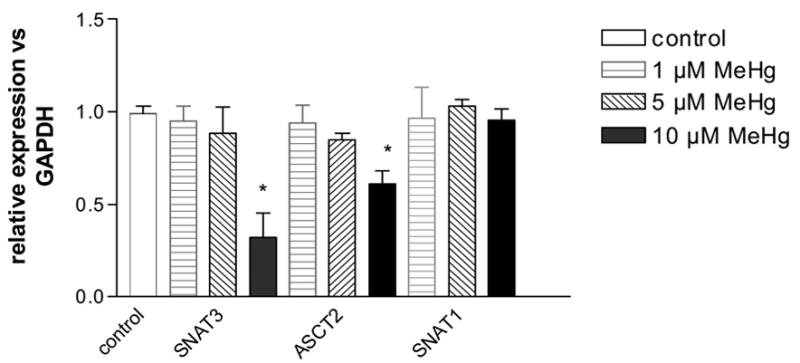
Representative agarose gel electrophoresis of RT PCR products for the expression of SNAT3, ASCT2, SNAT1 mRNA (A). Expression of mRNA for SNAT3, ASCT2 and SNAT1 after treatment with MeHg (1 μM, 5 μM, 10 μM). Results are mean ± SD of three independent isolations.*<0.05 vs. control (Mann -Whitney test) (B). Inset: GAPDH expression.
4. Discussion
The present study established that MeHg exposure in astocytes leads to lipid peroxidation and the induction of mitochondrial permeability transition. To our knowledge, this is the first study to investigate MeHg-induced neurotoxicity in astrocytes by employing biomarkers of oxidative damage (F2-IsoPs) and fluorescent dye (TMRE) for measurement of ΔΨm. These damaging cellular changes initiated by MeHg will gradually lead to the dysfunction of astrocytes and contribute to their inability to maintain optimal control over the extracellular milieu, eventually leading to neuronal death.
In vivo and in vitro biochemical studies employing neuronal cultures and mixed neuronal/glial cultures as well as recent studies with primary astrocyte cultures have shown increased ROS formation with MeHg exposure [1,35,53,63,69,74]. Mitochondria are believed to be major targets of MeHg-induced toxicity [43]. Specifically, highly enriched Hg concentrations were found in mitochondrial fractions with the lowest Hg concentration found in the cytosol of livers from Hg exposed animals [22]. Experiments with human hepatic cell lines also showed that almost half of the metal accumulates in the mitochondria where it causes ultrastructural alterations, as observed by transmission electron microscopy [40]. Most of the bioenergetic experiments with Hg report the uncoupling of oxidative phosphorylation [72], inhibition of ATP synthesis [10], impairment of the respiratory chain [58] and depletion of intracellular ATP and ADP [55]. A study using a selective probe for mitochondrial reactive oxygen intermediates as well as other probes demonstrated a significant MeHg-induced increase in intracellular superoxide anion, hydrogen peroxide and hydroxyl radicals, indicating that the mitochondrial electron transport chain is an early, primary site for ROS formation [2,65,66,74]. Additionally, MeHg exposure disrupts Ca2+ regulation in mitochondria derived from rat brains by decreasing Ca2+ uptake and inducing Ca2+ release [42]. Alterations in Ca2+ homeostasis can also lead to depolarization of the inner mitochondrial membrane and formation of ROS. Our results are consistent with studies showing that MeHg induces increases in ROS. Furthermore, our results extend this knowledge by showing that the lipid peroxidation biomarkers of oxidative injury, F2-IsoPs, are increased in MeHg exposed astocytes.
Several methods exist to determine free radical-mediated damage to cells. While most of these function well in vitro, important limitations arise in living systems where extensive, highly active enzymatic pathways have evolved to metabolize many of the commonly measured products, such as 4-hydroxynonenal [50]. The measurement of F2-IsoPs is a method that has been extensively replicated as an efficient means of quantifying free radical damage in in vivo models associated with other neurodegenerative diseases, including Alzheimer’s disease [49,51], inflammation [45,46], excitotoxicity [47] and anticholinesterase toxicity [48]. In our astrocyte model, levels of F2-isoPs were significantly elevated at 1 or 6 hours following 5 or 10 μ M MeHg exposures (Figure 1).
Another consequence of increased oxidative stress is the induction of the mitochondrial permeability transition (MPT), a Ca2+-dependent process characterized by the opening of the permeability transition pore (PTP) in the inner mitochondrial membrane. This cause increased permeability to protons, ions and other solutes ≤ 1500 Da [75], leading to a collapse of the mitochondrial inner membrane potential (ΔΨm). Loss of the ΔΨm results in colloid osmotic swelling of the mitochondrial matrix, movement of metabolites across the inner membrane, defective oxidative phosphorylation, cessation of ATP synthesis and further generation of ROS. Our experiments demonstrated a concentration-dependent, deleterious effect of MeHg on mitochondrial ΔΨm in cultured astrocytes. Our results also demonstrated that ΔΨm is a very sensitive endpoint for MeHg toxicity, since significant reductions were observed after only 1 hour of exposure to MeHg at concentrations as low as 1 μ M (Figure 2). These results agree with an earlier study by Limke and Atchison [43] that reported that mitochondria in primary cultures of rat cerebellar granule cells depolarize irreversibly after 50 min exposure to 0.5 μ M MeHg. The mechanism by which MeHg induces the MPT in astrocytes is not completely understood, nor is the sequence of events that is associated with this effect. It is generally believed that increased [Ca2+]i trigger ROS formation, and increased oxidative stress, in turn, is generally considered a major factor for MPT induction [17,38] and mitochondrial depolarization. However, the temporal sequence of events reported here with changes in membrane potential preceding oxidative stress suggest that changes in ionic gradients may be the earliest effects of MeHg. This is consistent with early reports from our laboratory showing that MeHg increases cellular permeability to ions such as Na+ (and K+), and that an increase in Na+ permeability via Na+/H+ exchange, offsetting K+ loss, is the primary mechanism in its inhibition of regulatory volume decrease in astrocytes [71].
Active, carrier-mediated efflux of Gln from astrocytes is a critical step in the Gln/Glu cycle, and, as such, a principal event in the neuromodulatory activity of these cells. Results from our study have shown that MeHg pretreatment for 30 min inhibited the net uptake of Gln at both 1 min and 5 min exposures. Under the present experimental conditions, uptake reflected active transport without directional distinction . Inhibition of Gln uptake in MeHg-treated astrocytes is therefore likely to reflect the decreased ability of cells to outwardly transport Gln. The two predominant Gln-transporting proteins of astrocytes are SNAT3(SN1) and ASCT2. Both are capable of mediating inward and outward transport of Gln, with the direction depending on the Gln and/or pH gradients [19]. The relative contribution of ASCT2- and SNAT3/SN1-mediated transport to the overall effect of MeHg remains to be established. Of note, inhibition of the expression of mRNAs coding for SNAT3/SN1 and ASCT2 was only observed at the highest MeHg concentration employed, indicating that, after 30 min treatment, inhibition of uptake was not due to inhibition of transcription. However, decreased expression of both transporters may become a cause for impaired Gln transport following prolonged exposure to MeHg, or, at later time points following a brief exposure. Experiments testing this hypothesis are currently being conducted.
Our results demonstrate that MeHg exposure is associated with changes in membrane permeability and glutamine/glutamate cycling increases in ROS formation and consequent lipid peroxidation. Furthermore, lipid peroxidation increases mitochondrial and cellular permeability alterations, involving GSH [37,70] and calcium depletion [70]. These outcomes work together to create a continuous cycle where acceleration of the mitochondrial chain induces oxidative stress, lipid peroxidation and depletion of antioxidant defenses, which, in turn, diminish membrane permeability and accelerate the respiratory chain, thus generating more ROS. Ultimately, the additive or synergistic mechanisms of cellular disruption caused by MeHg lead to cellular dysfunction and cell death.
Acknowledgments
This study was supported by Public Health Service Grant ES07331 from the National Institute of Health (to MA).
Abbreviations
- ΔΨm
mitochondrial inner membrane potential
- GABA
γ-aminobutyric acid
- Gln
glutamine
- Glu
glutamate
- MeHg
Methylmercury
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ali SF, LeBel CP, Bondy SC. Reactive oxygen species formation as a biomarker of methylmercury and trimethyltin neurotoxicity. Neurotoxocology. 1992;13:637 – 648. [PubMed] [Google Scholar]
- 2.Allen JW, El-Oqayli H, Aschner M, Syversen T, Sonnewald U. Methylmercury has a selective effect on mitochondria in cultured astrocytes in the presence of [U-13C]glutamate. Brain Res. 2001;908:149 –154. doi: 10.1016/s0006-8993(01)02628-2. [DOI] [PubMed] [Google Scholar]
- 3.Aschner M, Du YL, Gannon M, Kimelberg HK. Methylmercury-induced alterations in excitatory amino acid transport in rat primary astrocyte cultures. Brain Res. 1993;602:181 – 186. doi: 10.1016/0006-8993(93)90680-l. [DOI] [PubMed] [Google Scholar]
- 4.Aschner M, Mullaney KJ, Wagoner D, Lash LH, Kimelberg HK. Intracellular glutathione (GSH) levels modulate mercuric chloride (MC)- and methylmercuric chloride (MeHgCl)-induced amino acid release from neonatal rat primary astrocyte cultures. Brain Res. 1994;664:133 – 140. doi: 10.1016/0006-8993(94)91963-1. [DOI] [PubMed] [Google Scholar]
- 5.Aschner M. Astrocytes as modulators of mercury-induced neurotoxicity. Neurotoxicology. 1996;17:663 – 669. [PubMed] [Google Scholar]
- 6.Aschner M. Astrocytic swelling, phospholipase A2, glutathione and glutamate: interactions in methylmercury induced neurotoxicity. Cell Mol Biol. 2000;46:843 – 854. [PubMed] [Google Scholar]
- 7.Aschner M, Cao CC, Wu Q, Friedman MA. The acute effects of acrylamide on astrocyte functions. Ann NY Acad Sci. 2003;993:296–304. doi: 10.1111/j.1749-6632.2003.tb07537.x. discussion 345 – 349. [DOI] [PubMed] [Google Scholar]
- 8.Aschner M, Syversen T. Methylmercury: recent advances in the understanding of its neurotoxicity. Ther Drug Monit. 2005;27:278 – 283. doi: 10.1097/01.ftd.0000160275.85450.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aschner M, Wu Q, Friedman MA. Effects of acrylamide on primary neonatal rat astrocyte functions. Ann NY Acad Sci. 2005;1053:444 – 454. doi: 10.1111/j.1749-6632.2005.tb00053.x. [DOI] [PubMed] [Google Scholar]
- 10.Atchison WD, Harem MF. Mechanisms of methylmercury-induced neurotoxicity. FASEB J. 1994;8:622 – 629. doi: 10.1096/fasebj.8.9.7516300. [DOI] [PubMed] [Google Scholar]
- 11.Benjamin AM, Quastel JH. Metabolism of amino acids and ammonia in rat brain cortex slices in vitro: a possible role of ammonia in brain function. J Neurochem. 1975;25:197 – 206. doi: 10.1111/j.1471-4159.1975.tb06953.x. [DOI] [PubMed] [Google Scholar]
- 12.Bode BP. Recent molecular advances in mammalian glutamine transport. J Nutr. 2001;131:2475S – 2485S. doi: 10.1093/jn/131.9.2475S. [DOI] [PubMed] [Google Scholar]
- 13.Boulland JL, Osen KK, Levy LM, Danbolt NC, Edwards RH, Storm-Mathisen J, Chaudhry FA. Cell-specific expression of the glutamine transporter SN1 suggests differences in dependence on the glutamine cycle. Eur J Neurosci. 2002;15:1615 – 1631. doi: 10.1046/j.1460-9568.2002.01995.x. [DOI] [PubMed] [Google Scholar]
- 14.Brawer JR, McCarthy GF, Gornitsky M, Frankel D, Mehindate K, Schipper HM. Mercuric chloride induces a stress response in cultured astrocytes characterized by mitochondrial uptake of iron. Neurotoxicology. 1998;19:767 – 776. [PubMed] [Google Scholar]
- 15.Broer A, Brookes N, Ganapathy V, Dimmer KS, Wagner CA, Lang F, Broer S. The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J Neurochem. 1999;73:2184 – 2194. [PubMed] [Google Scholar]
- 16.Brookes N, Kristt DA. Inhibition of amino acid transport and protein synthesis by HgCl2 and methylmercury in astrocytes: selectivity and reversibility. J Neurochem. 1989;53:1228 – 1237. doi: 10.1111/j.1471-4159.1989.tb07419.x. [DOI] [PubMed] [Google Scholar]
- 17.Castilho RF, Kowaltowski AJ, Meinicke AR, Bechara EJ, Vercesi AE. Permeabilization of the inner mitochondrial membrane by Ca2+ ions is stimulated by t-butyl hydroperoxide and mediated by reactive oxygen species generated by mitochondria. Free Radic Biol Med. 1995;18:479 – 486. doi: 10.1016/0891-5849(94)00166-h. [DOI] [PubMed] [Google Scholar]
- 18.Charleston JS, Body RL, Bolender RP, Mottet NK, Vahter ME, Burbacher TM. Changes in the number of astrocytes and microglia in the thalamus of the monkey Macaca fascicularis following long-term sub clinical methylmercury exposure. Neurotoxicology. 1996;17:127 – 138. [PubMed] [Google Scholar]
- 19.Chaudry FA, Reimer RJ, Krizaj I, Barber D, Storm-Mathisen J, Copenhagen DR, Edwards RH. Molecular analysis of system N suggests novel physiological roles in nitrogen metabolism and synaptic transmission. Cell. 1999;99:769 –780. doi: 10.1016/s0092-8674(00)81674-8. [DOI] [PubMed] [Google Scholar]
- 20.Chaudry FA, Reimer RJ, Edwards RH. The glutamine commute: take the N line and transfer to the A. J Cell Biol. 2002a;157:349 – 355. doi: 10.1083/jcb.200201070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaudry FA, Schmitz D, Reimer RJ, Larsson P, Gray AT, Nicoll R, Kavanaugh M, Edwards RH. Glutamine uptake by neurons: interaction of protons with system A transporters. J Neurosci. 2002b;22:62 – 72. doi: 10.1523/JNEUROSCI.22-01-00062.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C, Qu L, Zhao J, Liu S, Deng G, Li B, Zhang P, Chai Z. Accumulation of mercury, selenium and their binding proteins in porcine kidney and liver from mercury-exposed areas with the investigation of their redox response. Sci Total Environ. 2006;366:627 – 637. doi: 10.1016/j.scitotenv.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Swanson RA. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J Neurochem. 2003;84:1332 – 1339. doi: 10.1046/j.1471-4159.2003.01630.x. [DOI] [PubMed] [Google Scholar]
- 24.Cherubini E, Conti F. Generating diversity at GABAergic synapses. Trends Neurosci. 2001;24:155 – 162. doi: 10.1016/s0166-2236(00)01724-0. [DOI] [PubMed] [Google Scholar]
- 25.Choi BH. The effects of methylmercury on the developing brain. Prog Neurobiol. 1989;32:447 – 470. doi: 10.1016/0301-0082(89)90018-x. [DOI] [PubMed] [Google Scholar]
- 26.Clarkson TW. The toxicology of mercury. Crit Rev Clin Lab Sci. 1997;34:369 – 403. doi: 10.3109/10408369708998098. [DOI] [PubMed] [Google Scholar]
- 27.Clarkson TW, Magos L, Myers GJ. The toxicology of mercury-current exposures and clinical manifestation. N Engl J Med. 2003;349:1731 – 1737. doi: 10.1056/NEJMra022471. [DOI] [PubMed] [Google Scholar]
- 28.Conti F, Weinberg RJ. Shaping at glutamatergic synapses. Trends Neurosci. 1999;22:451 –458. doi: 10.1016/s0166-2236(99)01445-9. [DOI] [PubMed] [Google Scholar]
- 29.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1 – 105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 30.Dave V, Mullaney KJ, Goderie S, Kimelberg HK, Aschner M. Astrocytes as mediators of methylmercury neurotoxicity: effects on D-aspartate and serotonin uptake. Dev Neurosci. 1994;16:222 – 231. doi: 10.1159/000112110. [DOI] [PubMed] [Google Scholar]
- 31.Dolinska M, Zablocka B, Sonnewald U, Albrecht J. Glutamine uptake and expression of mRNA's of glutamine transporting proteins in mouse cerebellar and cerebral cortical astrocytes and neurons. Neurochem Int. 2004;44:75 – 81. doi: 10.1016/s0197-0186(03)00123-2. [DOI] [PubMed] [Google Scholar]
- 32.Dugan LL, Sensy SL, Canzoniero LM, Hardran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW. Mitochondrial production reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J Neurosci. 1995;15:6377 – 6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erecinska M, Silver IA. Metabolism and role of glutamate in mammalian brain. Prog Neurobiol. 1990;35:245 – 296. doi: 10.1016/0301-0082(90)90013-7. [DOI] [PubMed] [Google Scholar]
- 34.Garman RH, Weiss B, Evans HL. Alkylmercurial encephalopathy in the monkey (Saimiri sciureus and Macaca arctoides): a histopathologic and autoradiographic study. Acta Neuropathol (Berl) 1975;32:61 – 74. doi: 10.1007/BF00686067. [DOI] [PubMed] [Google Scholar]
- 35.Gasso S, Cristofol RM, Selema G, Rosa R, Rodriguez-Farre E, Sanfeliu C. Antioxidant compounds and Ca2+ pathway blockers differentially protect against methylmercury and mercuric chloride neurotoxicity. J Neurosci Res. 2001;66:135 – 145. doi: 10.1002/jnr.1205. [DOI] [PubMed] [Google Scholar]
- 36.Gotz ME, Koutsilieri E, Riederer P, Ceccatelli S, Dare E. Methylmercury induces neurite degeneration in primary culture of mouse dopaminergic mesencephalic cells. J Neural Transm. 2002;109:597 – 605. doi: 10.1007/s007020200049. [DOI] [PubMed] [Google Scholar]
- 37.Gstraunthaler G, Pfaller W, Kotanko P. Glutathione depletion and in vitro lipid peroxidation in mercury or maleate induced acute renal failure. Biochem Pharmacol. 1983;32:2969 – 2972. doi: 10.1016/0006-2952(83)90404-5. [DOI] [PubMed] [Google Scholar]
- 38.Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem. 1997;272:3346 – 3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- 39.InSug O, Datar S, Koch CJ, Shapiro IM, Shenker BJ. Mercuric compounds inhibit human monocyte function by inducing apoptosis: evidence for formation of reactive oxygen species, development of mitochondrial membrane permeability transition and loss of reductive reserve. Toxicology. 1997;124:211 – 224. doi: 10.1016/s0300-483x(97)00153-4. [DOI] [PubMed] [Google Scholar]
- 40.Konigsberg M, Lopez-Diazguerrero NE, Bucio L, Gutierrez-Ruiz MC. Uncoupling effect of mercuric chloride on mitochondria isolated from a hepatic cell line. J Appl Toxicol. 2001;21:323 – 329. doi: 10.1002/jat.763. [DOI] [PubMed] [Google Scholar]
- 41.Lapham LW, Cernichiari E, Cox C, Myers GJ, Baggs RB, Brewer R, Shamlaye CF, Davidson PW, Clarkson TW. An analysis of autopsy brain tissue from infants prenatally exposed to methymercury. Neurotoxicology. 1995;16:689 – 704. [PubMed] [Google Scholar]
- 42.Levesque PC, Atchison WD. Disruption of brain mitochondrial calcium sequestration by Methylmercury. J Pharmacol Exp Ther. 1991;256:236 – 242. [PubMed] [Google Scholar]
- 43.Limke TL, Atchison WD. Acute exposure to methylmercury opens the mitochondrial permeability transition pore in rat cerebellar granule cells. Toxicol Appl Pharmacol. 2002;178:52 – 61. doi: 10.1006/taap.2001.9327. [DOI] [PubMed] [Google Scholar]
- 44.Mackenzie B, Erickson JD. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch. 2004;447:784 – 795. doi: 10.1007/s00424-003-1117-9. [DOI] [PubMed] [Google Scholar]
- 45.Milatovic D, Zaja-Milatovic S, Montine KS, Horner PJ, Montine TJ. Pharmacologic suppression of neuronal oxidative damage and dendritic degeneration following direct activation of glial innate immunity in mouse cerebrum. J Neurochem. 2003;87:1518 – 1526. doi: 10.1046/j.1471-4159.2003.02120.x. [DOI] [PubMed] [Google Scholar]
- 46.Milatovic D, Zaja-Milatovic S, Montine KS, Shie FS, Montine TJ. Neuronal oxidative damage and dendritic degeneration following activation of CD14-dependent innate immune response in vivo. J Neuroinflammation. 2004;1:20 – 26. doi: 10.1186/1742-2094-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milatovic D, VanRollins M, Li K, Montine KS, Montine TJ. Suppression of murine cerebral F2-isoprostanes and F4-neuroprostanes from excitotoxicity and innate immune response in vivo by alpha- or gamma-tocopherol. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;827:88–93. doi: 10.1016/j.jchromb.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 48.Milatovic D, Gupta RC, Aschner M. Anticholinesterase toxicity and oxidative stress. Scientific World Journal. 2006;6:295 – 310. doi: 10.1100/tsw.2006.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Montine TJ, Beal MF, Cudkowicz ME, O'Donnell H, Margolin RA, McFarland L, Bachrach AF, Zackert WE, Roberts LJ, Morrow JD. Increased CSF F2-isoprostane concentration in probable AD. Neurology. 1999;52:562 – 565. doi: 10.1212/wnl.52.3.562. [DOI] [PubMed] [Google Scholar]
- 50.Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, 2nd, Morrow JD. Lipid peroxidation in aging brain and Alzheimer's disease. Free Radic Biol Med. 2002;33:620–626. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- 51.Montine TJ, Montine KS, McMahan W, Markesbery WR, Quinn JF, Morrow JD. F2-isoprostanes in Alzheimer and other neurodegenerative diseases. Antioxid Redox Signal. 2005;7:269 – 75. doi: 10.1089/ars.2005.7.269. [DOI] [PubMed] [Google Scholar]
- 52.Morrow JD, Robertsx LJ., 2nd Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Methods Enzymol. 1999;300:3 – 12. doi: 10.1016/s0076-6879(99)00106-8. [DOI] [PubMed] [Google Scholar]
- 53.Mundy WR, Freudenrich TM. Sensitivity of immature neurons in culture to metal-induced changes in reactive oxygen species and intracellular free calcium. Neurotoxicology. 2000;21:1135 – 1144. [PubMed] [Google Scholar]
- 54.Myhre O, Fonnum F. The effect of aliphatic, naphthenic, and aromatic hydrocarbons on production of reactive oxygen species and reactive nitrogen species in rat brain synaptosome fraction: the involvement of calcium, nitric oxide synthase, mitochondria, and phospholipase A. Biochem Pharmacol. 2001;62:119 – 128. doi: 10.1016/s0006-2952(01)00652-9. [DOI] [PubMed] [Google Scholar]
- 55.Palmeira CM, Serrano J, Kuehl DW, Wallace KB. Preferential oxidation of cardiac mitochondrial DNA following acute intoxication with doxorubicin. Biochim Biophys Acta. 1997;1321:101 – 106. doi: 10.1016/s0005-2728(97)00055-8. [DOI] [PubMed] [Google Scholar]
- 56.Patenaude A, Ven Murthy MR, Mirault ME. Mitochondrial thioredoxin system: effects of TrxR2 overexpression on redox balance, cell growth, and apoptosis. J Biol Chem. 2004;279:27302 – 27314. doi: 10.1074/jbc.M402496200. [DOI] [PubMed] [Google Scholar]
- 57.Roberts LJ, 2nd, Morrow JD. Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci. 2002;59:808 – 820. doi: 10.1007/s00018-002-8469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santos AC, Uyemura SA, Santos NA, Mingatto FE, Curti C. Hg(II)-induced renal cytotoxicity: in vitro and in vivo implications for the bioenergetic and oxidative status of mitochondria. Mol Cell Biochem. 1997;177:53 – 59. doi: 10.1023/a:1006861319378. [DOI] [PubMed] [Google Scholar]
- 59.Schousboe A, Westergaard N, Sonnewald U, Petersen SB, Yu AC, Hertz L. Regulatory role of astrocytes for neuronal biosynthesis and homeostasis of glutamate and GABA. Prog Brain Res. 1992;94:199 – 211. doi: 10.1016/s0079-6123(08)61751-3. [DOI] [PubMed] [Google Scholar]
- 60.Schousboe A, Westergaard N, Sonnewald U, Petersen SB, Huang R, Peng L, Hertz L. Glutamate and glutamine metabolism and compartmentation in astrocytes. Dev Neurosci. 1993;15:359 – 366. doi: 10.1159/000111356. [DOI] [PubMed] [Google Scholar]
- 61.Shanker G, Allen JW, Mutkus LA, Aschner M. Methylmercury inhibits cysteine uptake in cultured primary astrocytes, but not in neurons. Brain Res. 2001;914:159 – 165. doi: 10.1016/s0006-8993(01)02791-3. [DOI] [PubMed] [Google Scholar]
- 62.Shanker G, Aschner M. Identification and characterization of uptake systems for cystine and cysteine in cultured astrocytes and neurons: evidence for methylmercury-targeted disruption of astrocyte transport. J Neurosci Res. 2001;66:998 – 1002. doi: 10.1002/jnr.10066. [DOI] [PubMed] [Google Scholar]
- 63.Shanker G, Aschner M. Methylmercury-induced reactive oxygen species formation in neonatal cerebral astrocytic cultures is attenuated by antioxidants. Brain Res Mol Brain Res. 2003;110:85 – 91. doi: 10.1016/s0169-328x(02)00642-3. [DOI] [PubMed] [Google Scholar]
- 64.Shanker G, Syversen T, Aschner M. Astrocyte-mediated methylmercury neurotoxicity. Biol Trace Elem Res. 2003;95:1 – 10. doi: 10.1385/BTER:95:1:1. [DOI] [PubMed] [Google Scholar]
- 65.Shanker G, Aschner JL, Syversen T, Aschner M. Free radical formation in cerebral cortical astrocytes in culture in duced by Methylmercury. Molecular Brain Research. 2004;128:48 – 57. doi: 10.1016/j.molbrainres.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 66.Shanker G, Syversen T, Aschner JL, Aschner M. Modulatory effect of glutathione status and antioxidants on methylmercury-induced free radical formation in primary cultures of cerebral astrocytes. Brain Res Mol Brain Res. 2005;137:11 – 22. doi: 10.1016/j.molbrainres.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 67.Shenker BJ, Guo TL, Shapiro IM. Low-level methylmercury exposure causes human T-cells to undergo apoptosis: evidence of mitochondrial dysfunction. Environ Res. 1998;77:149 – 159. doi: 10.1006/enrs.1997.3816. [DOI] [PubMed] [Google Scholar]
- 68.Sonnewald U, Westergaard N, Schousboe A. Glutamate transport and metabolism in astrocytes. Glia. 1997;21:56 – 63. doi: 10.1002/(sici)1098-1136(199709)21:1<56::aid-glia6>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 69.Sorg O, Schilter B, Honegger P, Monnet-Tschudi F. Increased vulnerability of neurones and glial cells to low concentrations of methylmercury in a prooxidant situation. Acta Neuropathol (Berl) 1998;96:621 – 627. doi: 10.1007/s004010050943. [DOI] [PubMed] [Google Scholar]
- 70.Strubelt O, Kremer J, Tilse A, Keogh J, Pentz R, Younes M. Comparative studies on the toxicity of mercury, cadmium, and copper toward the isolated perfused rat liver. J Toxicol Environ Health. 1996;47:267 – 283. doi: 10.1080/009841096161780. [DOI] [PubMed] [Google Scholar]
- 71.Vitarella D, Kimelberg HK, Aschner M. Inhibition of regulatory volume decrease in swollen rat primary astrocyte cultures by Methylmercury is due to increased amiloride-sensitive Na+ uptake. Brain Res. 1996;732:169 – 178. doi: 10.1016/0006-8993(96)00518-5. [DOI] [PubMed] [Google Scholar]
- 72.Weinberg JM, Harding PG, Humes HD. Mitochondrial bioenergetics during the initiation of mercuric chloride-induced renal injury. II. Functional alterations of renal cortical mitochondria isolated after mercuric chloride treatment. J Biol Chem. 1982;257:68 – 74. [PubMed] [Google Scholar]
- 73.Wu Q, Sidoryk M, Mutkus L, Zielinska M, Albrecht J, Aschner M. Acrylamide stimulates glutamine uptake in Fischer 344 rat astrocytes by a mechanism involving upregulation of the amino acid transport system N. Ann N Y Acad Sci. 2005;1053:435 – 443. doi: 10.1111/j.1749-6632.2005.tb00052.x. [DOI] [PubMed] [Google Scholar]
- 74.Yee S, Choi H. Oxidative stress in neurotoxic effects of methylmercury poisoning. Neurotoxicology. 1996;17:17 – 26. [PubMed] [Google Scholar]
- 75.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139 – 176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]