

Abstract
The amelogenesis imperfectas (AIs) are a clinically and genetically diverse group of conditions that are caused by mutations in a variety of genes that are critical for normal enamel formation. To date, mutations have been identified in four genes (AMELX, ENAM, KLK4, MMP20) known to be involved in enamel formation. Additional yet to be identified genes also are implicated in the etiology of AI based on linkage studies. The diverse and often unique phenotypes resulting from the different allelic and non-allelic mutations in these genes provide an opportunity to better understand the role of these genes and their related proteins in enamel formation. Understanding the AI phenotypes also provides an aid to clinicians in directing molecular studies aimed at delineating the genetic basis underlying these diverse clinical conditions. Our current knowledge of the known mutations and associated phenotypes of the different AI subtypes are reviewed.
Keywords: enamel, amelogen, kalikrien, matrix metalloproteinase, AMELX, ENAM, KLK4, MMP20, DLX3
INTRODUCTION
While many hereditary and environmental conditions are associated with abnormal enamel formation, amelogenesis imperfecta (AI) is a term used for hereditary conditions that affect enamel formation not associated with marked developmental defects in other tissues. Insightful evaluation of the AI phenotypes led to a variety of classifications of these conditions and even proposed mechanisms of the underlying pathology despite a lack of molecular evidence [Witkop and Sauk, 1976]. Since discovery of the amelogenin gene (AMELX) [Snead et al., 1989], the molecular basis of these conditions has progressed, albeit at a rather slow pace due to the conditions’ genetic diversity, relative rarity, and difficulty studying a tissue where mRNA and cell lines are not readily available. Despite these challenges, four genes have been identified as causative of these conditions and are associated with phenotypes that in many cases confirm developmental mechanisms previously hypothesized as responsible for the different AI subtypes. The most commonly accepted classification of AI recognizes four major AI types that were proposed by Carl Witkop, Jr. based on their phenotype and their proposed mechanism of development [Witkop, 1989].
Current molecular evidence supports many of Witkop’s proposed developmental mechanisms and their associated phenotypes [Witkop and Sauk, 1976], although the genetic heterogeneity that has emerged is more complex than many investigators expected. This is not altogether surprising given our current understanding that amelogenesis (the process of enamel formation) is an exquisitely orchestrated process involving strict genetic regulation of a variety of diverse processes. The ameloblasts (enamel producing cells) secrete a unique extracellular matrix that is processed by proteinases in a highly controlled fashion. They control the critical ionic and fluid concentration of the micro-environment, regulate the pH during mineralization, are motile and undergo tremendous morphological changes during their life cycle to accommodate these diverse physiological functions [Simmer and Fincham, 1995; Smith, 1998].
We now know that a variety of genetic mutations are associated with distinct disturbances in these critical processes that lead to the conditions referred to as AI. The most abundant extracellular matrix protein in developing enamel is amelogenin [Fincham and Simmer, 1997] and mutations in the gene encoding this protein are responsible for a variety of phenotypes ranging from deficiencies in the amount of enamel (hypoplastic AI) to defects in the mineralization of the enamel due to aberrant processing of the matrix (hypomaturation AI) [Wright et al., 2003]. Mutations in the ENAM gene that encodes another extracellular matrix protein result in a variety of hypoplastic phenotypes depending on the specific mutation and its effect on the protein [Rajpar et al., 2001; Mardh et al., 2002]. More recently, mutations in the genes (KLK4 and MMP20) encoding two known proteinases that are considered critical for processing of the extracellular matrix have been shown to cause defects in the final crystallite mineralization or maturation of the enamel (autosomal recessive pigmented hypomaturation AI; OMIM 204700) [Hart et al., 2004; Kim et al., 2005a]. The most common form of AI in North America (autosomal dominant hypocalcified AI) has not been associated with a specific gene and indeed evidence suggests that it is not caused by any of the known candidate AI genes [Hart et al., 2003]. Finally, AI type IV (OMIM 104510), that is associated with an unusual phenotype including hypoplastic enamel and elongation of the dental pulp or taurodontism, remains controversial as being a true AI subtype [Crawford and Aldred, 1990] due to its close and often misdiagnosed association with tricho-dento-osseous syndrome (OMIM 190320).
The best approach for correlating our current molecular understanding of AI and their associated phenotypes in a robust and meaningful nosology has yet to be developed [Aldred et al., 2003]. Ideally, a nosology will emerge that takes into consideration the mode of inheritance and phenotypic features that are essential for clinical delineation of each AI type, while also coupling these critical clinical features to our understanding of the molecular basis of these conditions. This manuscript reviews our current knowledge about the molecular basis of each of the AI types and their associated phenotypes. The different AI types are presented based on their mode of inheritance as opposed to a phenotype based approach as is traditionally used for AI as it is felt that this is clinically more relevant and is critical for patient counseling and investigative approaches directed at identifying the molecular basis of these conditions.
X-LINKED AMELOGENESIS IMPERFECTA
All of the X-linked forms of AI (OMIM 300391) with a known molecular basis are associated with mutations in the AMELX gene (located at chromosome Xp22) that codes for amelogenin. One family has shown significant linkage to another X linked interval (Xq22-28) [Aldred et al., 1992a] and there are other X-linked conditions that have significant enamel involvement making it likely there are other genes important in enamel formation on the X chromosome [Landy and Donnai, 1993; Mikkola and Thesleff, 2003; Balmer et al., 2004]. Amelogenin is the predominant extracellular matrix protein in developing enamel and is thought to form an organic scaffold through self assembly that is essential for controlling the highly ordered and directional growth of enamel crystallites [Robinson et al., 1990; Fincham and Simmer, 1997]. Although the exact functions of the amelogenin protein have not been fully established, its critical role in enamel development is verified by the association of enamel defects with AMELX mutations in humans, and the presence of severe enamel hypoplasia in the amelogenin knock-out mouse [Gibson et al., 2001]. Human amelogenin has a 16 amino acid signal peptide, and is secreted primarily as a 175 amino acid protein [Salido et al., 1992]. It is known that there are alternatively spliced amelogenin proteins formed by deleting or adding certain exon products [Simmer et al., 1994; Baba et al., 2002]. During normal enamel formation the amelogenin protein is processed in a controlled fashion thereby generating the regulatory mechanism considered essential for orderly crystallite growth [Hu et al., 2002]. The acidic amelogenin C-terminus has a high mineral affinity compared with the remaining amelogenin molecule and is rapidly cleaved after secretion of the parent protein from the ameloblasts [Shaw et al., 2004]. During normal enamel development the amelogenin proteins are almost completely removed allowing the enamel crystallites to grow and produce mature enamel that is greater than 95% mineral by weight [Smith, 1998].
AMELX Mutations are Diverse and Include Deletion, Missense, and Nonsense Mutations
These coding errors result in essentially two different and sometimes overlapping phenotypes including a deficiency in the amount of enamel (hypoplasia; OMIM 301200) and defects in enamel mineralization (hypomaturation) [Wright et al., 2003] The enamel phenotype appears to be related to the changes in amelogenin that vary from loss of the protein due to large deletions, signal peptide mutations, or alteration of specific functional domains. Because affected males will express only the mutant allele, with females showing a mosaic pattern of expression due to X chromosome inactivation (Lyonization), the male and female phenotypes vary markedly in severity and appearance [Witkop, 1967]. Females with AMELX mutations typically have either discolorations or bands of hypoplasia that run vertically on the teeth due to clusters of ameloblasts expressing either the normal or mutant AMELX allele [Hart et al., 2002]. There are 14 AMELX mutations reported to date (Table I) which are reviewed in the following section.
TABLE I.
AMELX Mutations
| Genomic DNA | cDNA | Protein | Male phenotype | Reference |
|---|---|---|---|---|
| g.2T >C | c.2T >C | p.M1T | Smooth hypoplastic (normal mineralization) | Kim et al. [2004] |
| g.11G >C | c.11G >C | p.W4S | Smooth hypoplastic (normal mineralization) | Kim et al. [2004] |
| g.11G >A | c.11G >A | p.W4X | Smooth hypoplastic (normal mineralization) | Sekiguchi et al. [2001] |
| g.14_22del | c.14_22del | p.I5_A8delinsT | Smooth hypoplastic (normal mineralization) | Lagerström-Fermer and Landegren [1995] |
| g.1148_54del | c.55_54del | p.18del | Hypomaturation (some hypoplasia) | Lagerström et al. [1991] |
| g.3455C >T | c.152C >T | p.T51I | Hypomaturation (some hypoplasia) | Lench and Winter [1995] |
| g.3458delC | c.155delC | p.P52fsX53 | Hypomaturation (some hypoplasia, variable) | Aldred et al. [1992b]; Lench et al. [1994] |
| g.3781C >A | c.208C >A | p.P70T | Hypomaturation (some hypoplasia) | Collier et al. [1997]; Hart et al. [2000] |
| g.3803A >T | c.230A >T | p.H77L | Hypomaturation | Hart et al. [2002] |
| g.3958delC | c.385delC | p.H129fsX187 | Smooth hypoplastic | Sekiguchi et al. [2001] |
| g.3993delC | c.420delC | p.Y141fsX187 | Smooth hypoplastic | Greene et al. [2002] |
| g.4046delC | c.473delC | p.P158fsX187 | Smooth hypoplastic | Lench and Winter [1995] |
| g.4114delC | c.541delC | p.L181fsX187 | Smooth hypoplastic (some hypomineralization) | Kindelan et al. [2000]; Hart et al. [2001] |
| g.4144G >T | c.571G >T | p.E191X | Smooth hypoplastic | Lench and Winter [1995] |
AMELX Major Deletions and Mutations in the Signal Peptide Coding Region
A 5 Kb deletion, including all genomic DNA from the beginning of exon 3 through part of exon 7 (g.1148_47del), was the first AI associated mutation discovered [Lagerström et al., 1991]. This represents what amounts to a human knockout of the AMELX gene with only two amino acids beyond the signal peptide potentially being coded by the mutant gene. The associated phenotype reported in the two Swedish families having this mutation is described as being predominantly a hypomineralization/hypomaturation defect [Lagerström-Fermer and Landegren, 1995]. The enamel has a mottled white opaque coloration and microradiographic studies show that it is hypomineralized [Backman and Anneroth, 1989]. There also is variable enamel hypoplasia or generalized thinning of the enamel which is what one would predict for hemizygous affected males [Backman, 1988].
Four mutations in the 16 amino acid signal peptide have been identified. These mutations are all thought to cause a loss of protein or cause the protein not to be secreted from the cell. All signal peptide mutations identified to date result in a severe reduction in enamel thickness and a clinical phenotype of smooth hypoplastic AI. The little enamel that is present is described as hard and well-mineralized (Fig. 1).
Fig. 1.
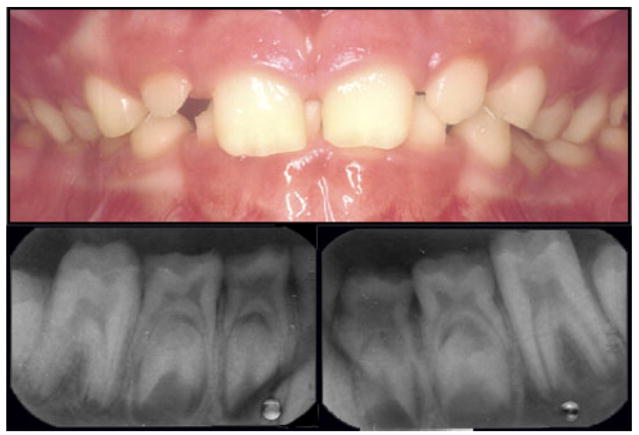
Loss of amelogenin due to signal peptide mutations results in a marked reduction in enamel thickness as seen in this male with a p.M1T mutation. The teeth are relatively normal in color and slightly reduced in size and the extremely thin enamel can be more readily visualized on the dental radiographs [Kim et al., 2004]. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
AMELX Mutations in the N Terminus Coding Region
Four AMELX mutations involving part of the N-terminus of the amelogenin protein have been described. The first of these was a frameshift mutation [Aldred et al., 1992b; Lench et al., 1994] resulting from a single nucleotide substitution in exon 5 and the introduction of a premature stop codon. This mutation is predicted to result in an amelogenin protein that is 36 amino acids in length. Both families described with this mutation show primarily a hypomineralization/hypomaturation phenotype but also varying degrees of hypoplasia. There was marked phenotypic variability in the severity between same gender individuals even within the same family [Aldred et al., 1992b; Lench et al., 1994].
Three missense mutations affecting the N-terminal region of the amelogenin protein involve single nucleotide substitutions that cause single amino acid changes. One of these mutations occurs in exon 5 (g.3455C >T) and produces a Threonine to Isoleucine change at amino acid 51 (p.T51I) [Lench and Winter, 1995]. The phenotype resulting from this one amino acid change is described as hypomineralization/hypomaturation with brown discoloration of the enamel. Similarly, two other mutations (P70T and H77L) that occur in exon 6 produce an enamel phenotype classified as hypomaturation with brown discolored enamel (Fig. 2) [Collier et al., 1997; Ravassipour et al., 2000; Hart et al., 2002]. All of these mutations cause changes in the protein at or in close proximity to a region that has specific proteinase cleavage sites. Studies have verified that the P70T change in amelogenin does cause a reduction in the ability of MMP20 (the primary proteinase for processing amelogenin) to cleave the mutant protein compared with wild type protein [Li et al., 2001].
Fig. 2.
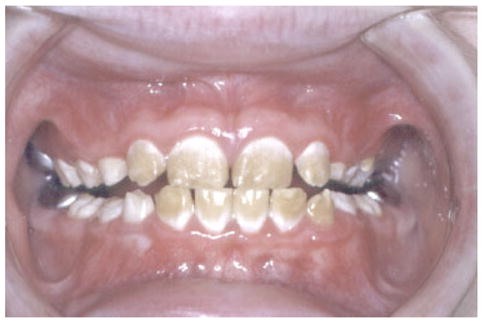
Point AMELX mutations altering the tyrosine rich region of the amelogenin protein cause hypomaturation of the enamel producing the characteristic white cervical opaque and coronal brown discoloration of the enamel, as seen in this male with a P70T mutation. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
AMELX Mutations in the C Terminus Coding Region
There are five different mutations that introduce a premature stop codon and truncate the C-terminus of the amelogenin protein. Four of these mutations are single nucleotide deletions at various locations in exon 6. All introduce a premature stop codon at amino acid position 187 [Lench and Winter, 1995; Kindelan et al., 2000; Sekiguchi et al., 2001; Hart et al., 2002]. The amelogenin proteins produced by these four mutations are similar in that all lack the C-terminal 18 amino acids. However, they all differ in the extent and type of amino acid changes occurring between the point of the deletion and the stop codon. One AMELX mutation introduces a single nucleotide change (c.571G >T) and a premature stop codon late in exon 6 (p.E191X), causing truncation of the protein at position 191. This results in a secreted amelogenin protein 14 amino acids shorter than the wild type [Lench and Winter, 1995]. Despite these differences, all mutations evaluated to date that affect the C-terminus of the amelogenin protein are associated with a generalized thinning of the enamel and a smooth hypoplastic phenotype (Fig. 3). The C-terminus is known to have an affinity for forming enamel crystallites [Shaw et al., 2004] and likely plays a critical role in amelogenin scaffold assembly during enamel development.
Fig. 3.
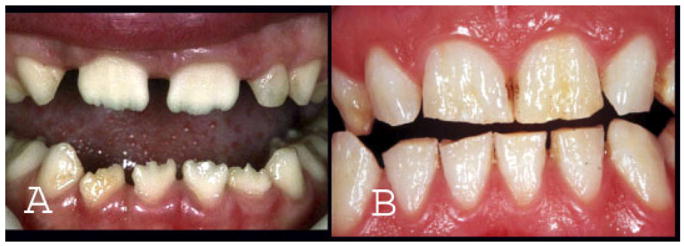
AMELX mutations affecting the C-terminus of the amelogenin protein cause a marked generalized decrease in enamel thickness in males (A) and produces vertical grooves in females due to Lyonization, as seen in the dentition of this heterozygous female (B). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
AUTOSOMAL DOMINANT AMELOGENESIS IMPERFECTA
ENAM Mutations: Genotype and Phenotype
In addition to amelogenin there are numerous other extracellular matrix components in developing enamel, including proteins such as ameloblastin, enamelin and proteinases that are required to process the matrix proteins during mineralization and are coded by genes on autosomes [Krebsbach et al., 1996; Smith, 1998; Hu et al., 2000]. The first molecular defect associated with an autosomal AI type was identified as a mutation in the enamelin coding gene (ENAM) located on chromosome 4q21 [Rajpar et al., 2001]. Enamelin is considered to be an enamel-specific protein secreted by ameloblasts and is localized at the secretory face of the cell [Fukae et al., 1993]. Enamelin is a glycosylated protein secreted in relatively low amounts (1–5% of matrix) [Yamakoshi et al., 1998; Hu et al., 2001]. While the specific role of enamelin in amelogenesis is unknown, it is thought to be involved in crystallite growth regulation and crystallite elongation.
Two clinically distinct forms of autosomal dominant AI, smooth hypoplastic AI and local hypoplastic AI (OMIM 104500), are associated with multiple allelic ENAM mutations (Table II) [Rajpar et al., 2001; Mardh et al., 2002]. The clinically distinct phenotypes resulting from these allelic mutations are hypothesized to result from haploinsufficiency in the local hypoplastic type and from a dominant negative effect in the more severe generalized hypoplastic type [Mardh et al., 2002]. The local hypoplastic phenotype resulting from ENAM mutations that essentially stop protein production from one allel is characterized by horizontal bands of hypoplastic pits that encompass the tooth (Fig. 4). In contrast, mutations that result in a secreted but altered protein are associated with a generalized thin hypoplastic phenotype (Fig. 5) where close evaluation often reveals fine horizontal bands and pitting on the surface of the enamel.
TABLE II.
ENAM Mutations
| Genomic DNA | cDNA | Protein | Phenotype | Reference |
|---|---|---|---|---|
| g.2382A >T | c.157A >T | p.K53X | Local hypoplastic | Mardh et al. [2002] |
| g.6395G >A | IVS7 +1G >A; c.534 +1G >A | p.A158_Q178del | Generalized thin hypoplastic | Rajpar et al. [2001] |
| g.8344delG | IVS8 +1delG; c.588 +1delG | p.N197fsX277 | Generalized thin hypoplastic | Kida et al. [2002]; Hart et al. [2003] |
| g.13185_13186insAG | c/1258_1259insAG | p.P422fsX448 | Generalized thin hypoplastic | Hart et al. [2004] |
Fig. 4.
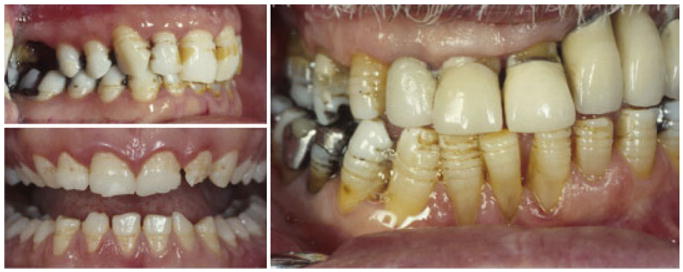
Autosomal dominant ENAM mutations associated with haploinsufficiency produce a local hypoplastic phenotype characterized by horizontal bands of pits that occur at the same height on the crowns of all teeth despite their marked difference in chronological development as seen in the teeth of this affected individual [Mardh et al., 2002]. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Fig. 5.
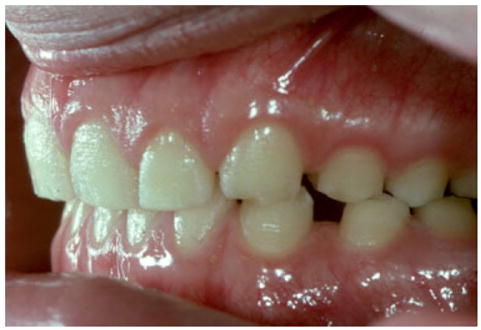
The generalized thin enamel in this affected individual has a rough pitted surface that results from an ENAM mutation producing a dominant negative affect. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Autosomal Dominant Hypocalcified AI
The most common form of AI in North America is thought to be the autosomal dominant hypocalcified type although current epidemiological studies are lacking [Witkop, 1957]. The molecular etiology for this AI type remains unknown at this time. However linkage studies in two large kindreds have excluded all known AI candidate genes as causative in these two families (i.e., tuftelin, enamelin, ameloblastin, kallikrien 4, and enamelysin) [Hart et al., 2003].
This indicates that there are additional, as yet undiscovered genes associated with autosomal dominant AI.
Autosomal Dominant Hypoplastic Hypomaturation AI With Taurodontism
Whether or not autosomal dominant hypoplastic hypomaturation AI with taurodontism (ADHHAI) is truly a distinct entity has been controversial [Crawford and Aldred, 1990; Seow, 1993]. Many of the cases reported as ADHHAI were later found to be the autosomal dominant condition named for its three principally affected tissues (hair, teeth, and bones), tricho-dento-osseous syndrome (TDO, OMIM 190320) [Crawford and Aldred, 1990; Wright et al., 1997]. The molecular etiology of TDO was discovered to involve a four base pair deletion in the distal-less 3 homeobox gene (DLX3 located at chromosome 17q21) [Price et al., 1998]. One family having the ADDHHAI with taurodontism phenotype was evaluated and showed no mutations in the coding or splice site regions of the DLX3 gene suggesting ADDHHAI with taurodontism is indeed a distinct entity [Price et al., 1999]. Recently a new DLX3 mutation involving a two base pair deletion in the homeobox region of the gene was reported as being associated ADDHHAI with taurodontism [Dong et al., 2005]. While the family members clearly showed the characteristic dental phenotype of thin discolored enamel and enlarged pulp chambers, they lacked a hair or bone phenotype. Investigation of an additional family having the same DLX3 CT deletion revealed, through careful phenotype analysis, that while not having the characteristic kinky curly hair associated with the DLX3 4 base pair deletion, all affected members did have a distinct hair phenotype. Hair of individuals having the DLX3 CT deletion was unmanageable, had a significantly smaller shaft diameter, and frequently displayed vertical grooves similar to those observed in hair shafts of individuals with the DLX3 four base pair deletion. The bone phenotype associated with the DLX3 CT deletion mutation is either not present or appears much milder compared with the four base pair deletion affected individuals. Careful bone phenotypic studies have not yet been completed. The presence of a hair and tooth phenotype associated with the DLX3 CT deletion indicates this mutation is associated with a mild expression of TDO and not ADDHHAI. The molecular defect for ADDHHAI with taurodontism remains to be discovered.
AUTOSOMAL RECESSIVE AMELOGENESIS IMPERFECTA
Two well characterized forms of AI are associated with the major proteinases responsible for processing the enamel extracellular matrix during enamel development [Hu et al., 2002]. Enamelysin, a matrix metalloproteinase (coded by the MMP20 gene), is secreted most abundantly during the secretory stage of development and processes amelogenin in a highly controlled fashion [Bartlett and Simmer, 1999]. Kalikrien four (formally known as enamel matrix serine proteinase 1, EMSP1), is a serine proteinase (coded by the KLK4 gene) and is secreted most abundantly during the maturation stage. KLK4 can cleave a variety of proteins and is thought to be responsible for removing the matrix proteins almost entirely to allow final growth of the enamel crystallites [Simmer and Hu, 2002]. Mutations in both of these proteinases are associated with pigmented hypomaturation AI (OMIM 204700) and enamel that is not mineralized to its full extent and shows increased protein retention (Table III). One kindred has been described with an autosomal recessive mode of inheritance for an ENAM mutation [Kim et al., 2005b] that is associated with a phenotype similar to the dominant negative ENAM mutation phenotypes.
TABLE III.
MMP20 and KLK4 Mutations
| Genomic DNA | cDNA | Protein | Phenotype | Reference |
|---|---|---|---|---|
| MMP20 Mutations | ||||
| g.30561A >T | c.954-2A >T or c.IVS6-2A-T | p.I319Fs338X or p.I319X | Pigmented hypomaturation decreased mineral | Kim et al. [2005] |
| g.16250T >A | c.678T >A | p.H226Q | Hypomaturation | Ozdemir et al. [2005] |
| KLK4 Mutations | ||||
| g.2142G >A | c.458G >A | p.W153X | Pigmented hypomaturation decreased mineral | Hart et al. [2004] |
Enamelysin (MMP20) Mutations: Genotype and Phenotype
Enamelysin (coded by the MMP20 gene) is considered to be a tooth specific matrix metalloproteinase. Two mutations have been identified in this gene that purportedly would cause loss of function. Both MMP20 mutations are associated with the pigmented hypomaturation AI phenotype characterized by enamel that has a reduced mineral content [Kim et al., 2005b]. The teeth have an orange–brown coloration, have a normal enamel thickness, and the enamel lacks its normal radioopacity showing little contrast when compared with dentin (Fig. 6).
Fig. 6.
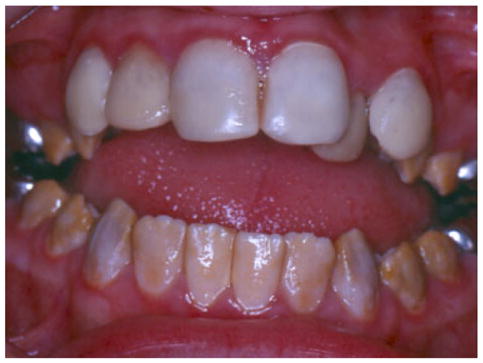
The autosomal recessive pigmented hypomaturation phenotype produced by a MMP20 mutation characterized by teeth that have reduced mineral content and have a white-to-brown discoloration [Kim et al., 2005b]. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Kalikrein 4 Mutations: Genotype and Phenotype
The enamel phenotype observed with KLK4 mutation [Hart et al., 2004] is consistent with a loss of protein function critical for its purported role in enamel mineralization and its known temporal and spatial expression [Simmer and Hu, 2002]. The transcript produced by the g.2142G >A mutation would result in a 153 amino acid protein that lacks the S207 of the catalytic triad that is essential for proteolytic activity [Hart et al., 2004]. The truncated product would also lack the conserved asparagine and glycine encoded by exon 6 that characterize the binding pocket that defines substrate specificity [Komatsu et al., 2003].
Teeth from the affected individuals have a marked orange–brown coloration in both the primary and permanent dentitions (Fig. 7). Radiographically, the teeth have normal morphology with enamel that has a decreased radiodensity compared with normal teeth. The enamel crystallites appear to have increased space between them and the enamel has an increased retention of amelogenin-like protein.
Fig. 7.
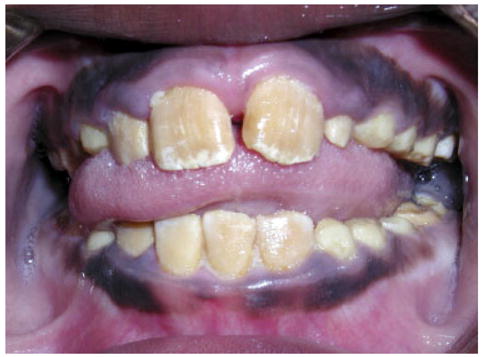
Autosomal recessive pigmented hypomaturation resulting from a KLK4 mutation is characterized by enamel of normal thickness that has a marked orange–brown discoloration, as seen in this affected female. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
DISCUSSION
Evaluation of the phenotype and genotype relationship in AI suggests there is clustering of phenotypes depending on the specific gene involved, the type of mutation and its effect on the translated protein, and the protein functional properties. The demarcation between phenotypes is not always exact with some overlap occurring between hypoplastic and hypomineralization defects [Wright et al., 2003]. This is not surprising given that AI genes and their associated proteins are involved in related pathways critical to the secretion, organization, processing, and/or mineralization of the developing enamel.
Four genes (AMELX, ENAM, KLK4, and MMP20) and 23 different mutations have been reported in the literature to date. Most of the identified allelic mutations are in the AMELX gene, although the two proteinase gene mutations were discovered only recently. While four genes have been associated with different AI types, there are other candidate genes proposed that have not, as yet, been related to an AI phenotype. Ameloblastin, the second most abundant protein in the enamel matrix is coded for by the AMBN gene [Krebasbach et al., 1996]. No human mutations in the AMBN gene have been associated with AI. However, based on the marked hypoplastic enamel phenotype in Ambn null mice [Fukumoto et al., 2004] it appears clear that this protein is critical for normal enamel formation. Negative linkage studies of kindreds with autosomal dominant hypocalcified AI indicate that genes other than the candidates proposed to date are also involved in AI. Collectively, these studies continue to indicate that genetic heterogeneity and phenotypic diversity are greater than one might have predicted based on classifications recognizing four main types and 14 subtypes.
Clinicians involved in diagnosing and managing these diverse conditions should be familiar with the different phenotypes and associated inheritance patterns. This information is critical for helping to narrow the search for a candidate gene so that a definitive molecular etiology can be established. There are some phenotypes that are so distinct, such as the ENAM associated local hypoplastic phenotype (horizontal bands of enamel pits), that the gene of major effect can often be effectively predicted. This also has proven true for the AMELX P70T mutation which produces a distinctly discolored hypomaturation phenotype where the cervical areas of the teeth are opaque white and the coronal portions of the teeth are brown.
However, in many cases having knowledge of the phenotype without information of the likely mode of inheritance pattern will often be insufficient to pursue a meaningful and informative molecular study. For example, a hypomaturation phenotype can be associated with multiple modes of inheritance and known genetic mutations such as an autosomal recessive trait (KLK4 or MMP20 mutation) or X-linked hypomaturation AI (AMELX mutations). A hypoplastic phenotype could be caused by X-linked (AMELX 5 prime, major deletion or 3 prime mutations), autosomal recessive (ENAM), or dominant (ENAM) mutations. As new genes are identified there is a high probability that they will have phenotypes that overlap the known clinical phenotypes making molecular identification even more complicated.
Since AI was first described there have been numerous nomenclatures proposed and in recent years much discussion about a molecular based nosology [Winter and Brook, 1975; Witkop, 1989; Aldred et al., 2003]. Although this would appear to be an obvious direction for future delineation of these clinically and genetically diverse conditions, it is as yet unclear how merging mode of inheritance, phenotypic information, and molecular basis of the condition can be best achieved. Future nomenclatures must be robust enough for effective communication between clinicians, geneticists, and researchers. Given the need for information about the mode of inheritance and phenotype necessary to pursue meaningful molecular studies, the adoption of a nomenclature that is predicated on the inheritance and phenotype, with further delineated of the molecular basis, appears to be the most useful at this time [Nussier et al., 2004]. This is in contrast to the nomenclature proposed by Witkop that is predicated on phenotype followed by mode of inheritance [Witkop, 1989]. The emerging complexity and heterogeneity of AI will likely require a consortium of experts to establish a more useful and meaningful nosology as our knowledge of these diverse hereditary defects of enamel continues to expand.
Footnotes
Grant sponsor: NIDCR; Grant number: 12879.
References
- Aldred MJ, Crawford PJM, Roberts E, Gillespie CM, Thomas NT, Fenton I, Sandkuijl LA, Harper PS. Genetic heterogeneity in X-linked amelogenesis imperfecta. Genomics. 1992a;14:567–573. doi: 10.1016/s0888-7543(05)80153-3. [DOI] [PubMed] [Google Scholar]
- Aldred MJ, Crawford PJM, Roberts E, Thomas NST. Identification of a nonsense mutation in the amelogenin gene (AMELX) in a family with X-linked amelogenesis imperfecta (AIH1) Hum Genet. 1992b;90:413–416. doi: 10.1007/BF00220469. [DOI] [PubMed] [Google Scholar]
- Aldred MJ, Savarirayan R, Crawford PJM. Amelogenesis imperfecta: A classification and catalogue for the 21st century. Oral Disease. 2003;9:19–23. doi: 10.1034/j.1601-0825.2003.00843.x. [DOI] [PubMed] [Google Scholar]
- Baba O, Takahashi N, Terashima T, Li W, DenBesten PK, Takano Y. Expression of alternatively spliced RNA transcripts of amelogenin gene exons 8 and 9 and its end products in the rat incisor. J Histochem Cytochem. 2002;50:1229–1236. doi: 10.1177/002215540205000910. [DOI] [PubMed] [Google Scholar]
- Backman B. Amelogenesis imperfecta-clinical manifestations in 51 families in a northern Swedish country. Scand J Dent Res. 1988;96:505–516. doi: 10.1111/j.1600-0722.1988.tb01590.x. [DOI] [PubMed] [Google Scholar]
- Backman B, Anneroth G. Microradiographic study of amelogenesis imperfecta. Scand J Dent Res. 1989;97:316–329. doi: 10.1111/j.1600-0722.1989.tb01619.x. [DOI] [PubMed] [Google Scholar]
- Balmer R, Cameron AC, Ades L, Aldred MJ. Enamel defects and Lyonization in focal dermal hypoplasia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;98:686–691. doi: 10.1016/j.tripleo.2004.02.061. [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Simmer JP. Proteinases in developing dental enamel. Crit Rev Oral Biol Med. 1999;10:425–441. doi: 10.1177/10454411990100040101. [DOI] [PubMed] [Google Scholar]
- Collier PM, Sauk JJ, Rosenbloom J, Yuan ZA, Gibson CW. An amelogenin gene defect associated with human x-linked amelogenesis imperfecta. Archs Oral Biol. 1997;42:235–242. doi: 10.1016/s0003-9969(96)00099-4. [DOI] [PubMed] [Google Scholar]
- Crawford PJM, Aldred MJ. Amelogenesis imperfecta with taurodontism and the tricho-dento-osseous syndrome: Separate conditions or a spectrum of disease? Clin Genet. 1990;38:44–50. doi: 10.1111/j.1399-0004.1990.tb03546.x. [DOI] [PubMed] [Google Scholar]
- Dong J, Amor D, Aldred MJ, Gu T, Escamilla M, MacDougall M. DLX3 mutation associated with autosomal dominant amelogenesis imperfecta with taurodontism. Am J Med Genet Part A. 2005;133A:138–141. doi: 10.1002/ajmg.a.30521. [DOI] [PubMed] [Google Scholar]
- Fincham AG, Simmer JP. Dental Enamel. Chichester: John Wiley & Sons; 1997. Amelogenin Proteins of Developing Dental Enamel; pp. 118–134. [DOI] [PubMed] [Google Scholar]
- Fukae M, Tanabe T, Uchida T, Yamakoshi Y, Shimizu M. Enamelins in the newly formed bovine enamel. Calcif Tissue Int. 1993;53:257–261. doi: 10.1007/BF01320911. [DOI] [PubMed] [Google Scholar]
- Fukumoto S, Kiba T, Hall B, Iehara N, Nakamura T, Longenecker G, Krebsbach PH, Nanci A, Kulkarni AB, Yamada Y. Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J Cell Biol. 2004;167:973–983. doi: 10.1083/jcb.200409077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson CW, Yuan ZA, Hall B, Longenecker G, Chen E, Thyagarajan T, Sreenath T, Wright JT, Decker S, Piddington R, et al. Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem. 2001;276:31871–31875. doi: 10.1074/jbc.M104624200. [DOI] [PubMed] [Google Scholar]
- Greene SR, Yuan ZA, Wright JT, Amjad AH, Abrams WR, Buchanan JA, Gibson GW. A gene deletion resulting in amelogenin with nine cysteine residues leads to amelogenesis imperfecta. Archs Oral Biol. 2002;47:211–217. doi: 10.1016/s0003-9969(01)00111-x. [DOI] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Gibson C, Wright JT. Mutational analysis of X-linked amelogenesis imperfecta in multiple families. Archs Oral Biol. 2000;45:79–86. doi: 10.1016/s0003-9969(99)00106-5. [DOI] [PubMed] [Google Scholar]
- Hart PS, Aldred MA, Crawford PJM, Wright NJ, Hart TC, Wright JT. Two amelogenin gene mutations cause different amelogenesis imperfecta phenotypes. Archs Oral Biol. 2002;47:255–260. doi: 10.1016/s0003-9969(02)00003-1. [DOI] [PubMed] [Google Scholar]
- Hart PS, Wright JT, Savage M, Kang G, Bensen JT, Gorry MC, Hart TC. Exclusion of candidate genes in two families with autosomal dominant hypocalcified amelogenesis imperfecta. Eur J Oral Sci. 2003;111:326–331. doi: 10.1034/j.1600-0722.2003.00046.x. [DOI] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons DG, Hong SP, Wright JT. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet. 2004;41:545–549. doi: 10.1136/jmg.2003.017657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Hart T, Dupont B, Chen J, Jiang H, Sun X, Qian Q, Zhang C, Wright T, Simmer J. Cloning human enamelin cDNA, chromosomal localization and analysis of expression during tooth development. J Dent Res. 2000;79:912–919. doi: 10.1177/00220345000790040501. [DOI] [PubMed] [Google Scholar]
- Hu JC-C, Sun X, Zhang C, Simmer JP. A comparison of enamelin and amelogenin expression in developing mouse molars. Eur J Oral Sci. 2001;109:125–132. doi: 10.1034/j.1600-0722.2001.00998.x. [DOI] [PubMed] [Google Scholar]
- Hu JCC, Sun XL, Zhang CH, Liu SX, Bartlett JD, Simmer JP. Enamelysin and kallikrein-4 mRNA expression in developing mouse molars. Eur J Oral Sci. 2002;110:307–315. doi: 10.1034/j.1600-0722.2002.21301.x. [DOI] [PubMed] [Google Scholar]
- Kida M, Ariga T, Shirakawa T, Oguchi H, Sakiyama Y. Autsomal-dominant hypoplastic form of amelogenesis imperfecta caused by an enamelin gene mutation at the exon-intron boundary. J Dent Res. 2002;81:738–742. doi: 10.1177/0810738. [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hu YY, Lin BP-L, Yamada CJM, Wright JT, Boyd C, Rayes SK, Feigal RJ, Hu JC-C. Amelogenin p.MIT and p.W4S mutations underlying hypoplastic X-linked amelogenesis imperfecta. J Dent Res. 2004;83:378–383. doi: 10.1177/154405910408300505. [DOI] [PubMed] [Google Scholar]
- Kim JW, Seymen F, Lin BP, Kiziltan B, Gencay K, Simmer JP, Hu JC. ENAM mutations in autosomal-dominant amelogenesis imperfecta. J Dent Res. 2005a;84:278–282. doi: 10.1177/154405910508400314. [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hart TC, Hart PS, Ramaswami MD, Bartlett JD, Hu JC. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet. 2005b;42:271–275. doi: 10.1136/jmg.2004.024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindelan S, Brook A, Gangemi L, Lench N, Wong F, Fearne J, Jackson Z, Foster G, BMJ S. Detection of a novel mutation in X-linked amelogenesis imperfecta. J Dent Res. 2000;79:1978–1982. doi: 10.1177/00220345000790120901. [DOI] [PubMed] [Google Scholar]
- Komatsu N, Takata M, Otsuki N, Toyama T, Ohka R, Takehara K, Saijoh K. Expression and localization of tissue kallikrein mRNAs in human epidermis and appendages. J Invest Dermatol. 2003;121:542–549. doi: 10.1046/j.1523-1747.2003.12363.x. [DOI] [PubMed] [Google Scholar]
- Krebasbach PH, Lee SK, Matsuki Y, Kozak CA, Yamada KM, Yamada Y. Full-length sequence, localization, and chromosomal mapping of ameloblastin. J Biol Chem. 1996;271:4431–4435. doi: 10.1074/jbc.271.8.4431. [DOI] [PubMed] [Google Scholar]
- Lagerström M, Dahl N, Nakahori Y, Nakagome Y, Backman B, Landegren U, Pettersson U. A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1) Genomics. 1991;10:971–975. doi: 10.1016/0888-7543(91)90187-j. [DOI] [PubMed] [Google Scholar]
- Lagerström-Fermer M, Landegren U. Understanding enamel formation from mutations causing X-linked amelogensis imperfecta. Con Tis Res. 1995;32:241–246. doi: 10.3109/03008209509013729. [DOI] [PubMed] [Google Scholar]
- Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome) J Med Genet. 1993;30:53–59. doi: 10.1136/jmg.30.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lench NJ, Winter GB. Characterisation of molecular defects in X-linked amelogenesis imperfecta (AIH1) Hum Mut. 1995;5:251–259. doi: 10.1002/humu.1380050310. [DOI] [PubMed] [Google Scholar]
- Lench NJ, Brook AH, Winter GB. SSCP detection of a nonsense mutation in exon 5 of the amelogenin gene (AMGX) causing X-linked amelogenesis imperfecta. Hum Mol Genet. 1994;3:827–828. doi: 10.1093/hmg/3.5.827. [DOI] [PubMed] [Google Scholar]
- Li W, Gibson C, Abrams W, Andrews D, DenBesten P. Reduced hydrolysis of amelogenin may result in X-linked amelogenesis imperfecta. Matrix Biol. 2001;19:7555–7760. doi: 10.1016/s0945-053x(00)00121-9. [DOI] [PubMed] [Google Scholar]
- Mardh KC, Backman B, Holgren G, Hu JC-C, Simmer JP, Forsman-Semb K. A nonsense mutation in the enamelin gene causes local hypoplastic autosomal dominant amelogenesis imperfecta (AIH2) Human Mol Genet. 2002;11:1069–1074. doi: 10.1093/hmg/11.9.1069. [DOI] [PubMed] [Google Scholar]
- Mikkola ML, Thesleff I. Ectodysplasin signaling in development. Cytokine Growth Factor Rev. 2003;14:211–224. doi: 10.1016/s1359-6101(03)00020-0. [DOI] [PubMed] [Google Scholar]
- Nussier M, Yassin O, Hart TC, Samimi A, Wright JT. Phenotypic diversity and revision of the nomenclature for autosomal recessive amelogenesis imperfecta. Oral Surg Oral Pathol Oral Med. 2004;97:220–230. doi: 10.1016/j.tripleo.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Ozdemir D, Hart PS, Ryu OH, Choi SJ, Ozdemir-Karatas M, Firatli E, Piesco N, Hart TC. MMP20 active-site mutation in hypomaturation amelogenesis imperfecta. J Dent Res. 2005;84:1031–1035. doi: 10.1177/154405910508401112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JA, Bowden DW, Wright JT, Pettenati MJ, Hart TC. Identification of a mutation in DLX3 associated with tricho-dento-osseous (TDO) syndrome. Hum Mol Genet. 1998;7:563–569. doi: 10.1093/hmg/7.3.563. [DOI] [PubMed] [Google Scholar]
- Price JA, Wright JT, Crawford PC, Aldred MJ, Hart TC. Tricho-dento-osseous syndrome and amelogenesis imperfecta with taurodontism are genetically distinct conditions. Clinical Genet. 1999;56:35–40. doi: 10.1034/j.1399-0004.1999.550105.x. [DOI] [PubMed] [Google Scholar]
- Rajpar MH, Harley K, Laing C, Davies DM, Dixon MJ. Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Human Mol Genet. 2001;10:1673–1677. doi: 10.1093/hmg/10.16.1673. [DOI] [PubMed] [Google Scholar]
- Ravassipour DB, Hart S, Hart TC, Ritter AV, Yamauchi RM, Gibson C, Wright JT. Unique enamel phenotype associated with amelogenin gene (AMELX) codon 41 point mutation. J Dent Res. 2000;79:1476–1481. doi: 10.1177/00220345000790070801. [DOI] [PubMed] [Google Scholar]
- Robinson C, Shore RC, Kirkham J, Stonehouse NJ. Extracellular processing of enamel matrix proteins and the control of crystal growth. J Biol Buccale. 1990;18:355–361. [PubMed] [Google Scholar]
- Salido E, Yen P, Koprivnikar K, Yu L-C, Shapiro L. The human enamel protein gene amelogenin is expressed from both the X and the Y chromosomes. Am J Hum Genet. 1992;50:303–316. [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi H, Alaluusua S, Minaguchi K, Yakushui M. A new mutation in the amelogenin gene causes X-linked amelogenesis imperfecta. J Dent Res. 2001;80:617. (Abstract 0722). [Google Scholar]
- Seow WK. Trichodentooseous (TDO) syndrome: Case report and literature review. J Pediatr Dent. 1993;15:355–361. [PubMed] [Google Scholar]
- Shaw WJ, Campbell AA, Paine ML, Snead ML. The COOH terminus of the amelogenin, LRAP, is oriented next to the hydroxyapatite surface. J Biol Chem. 2004;27:40263–40266. doi: 10.1074/jbc.C400322200. [DOI] [PubMed] [Google Scholar]
- Simmer JP, Fincham AG. Molecular mechanisms of dental enamel formation. Crit Rev Oral Biol Med. 1995;6:84–108. doi: 10.1177/10454411950060020701. [DOI] [PubMed] [Google Scholar]
- Simmer JP, Hu JC. Expression, structure, and function of enamel proteinases. Connect Tissue Res. 2002;43:441–449. doi: 10.1080/03008200290001159. [DOI] [PubMed] [Google Scholar]
- Simmer JP, Hu C-C, Lau EC, Sarte P, Slavkin HC, Fincham AG. Alternative splicing of the mouse amelogenin primary RNA transcript. Calcif Tissue Int. 1994;55:302–310. doi: 10.1007/BF00310410. [DOI] [PubMed] [Google Scholar]
- Smith CE. Cellular and chemical events during enamel maturation. Crit Rev Oral Biol Med. 1998;9:128–161. doi: 10.1177/10454411980090020101. [DOI] [PubMed] [Google Scholar]
- Snead ML, Lau EC, Fincham AG, Zeichner-David M, Davis C, Slavkin HC. Of mice and men: Anatomy of the amelogenin gene. Connect Tissue Res. 1989;22:101–109. [PubMed] [Google Scholar]
- Winter GB, Brook AH. Enamel hypoplasia and anomalies of the enamel. Dent Clin N Amer. 1975;19:3–24. [PubMed] [Google Scholar]
- Witkop CJ. Hereditary defects in enamel and dentin. Acta Genet. 1957;7:236–239. doi: 10.1159/000150974. [DOI] [PubMed] [Google Scholar]
- Witkop CJJ. Partial expression of sex-linked amelogenesis imperfecta in females compatible with the Lyon hypothesis. Oral Surg Oral Med Oral Pathology. 1967;23:174–182. doi: 10.1016/0030-4220(67)90092-8. [DOI] [PubMed] [Google Scholar]
- Witkop CJJ. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited, problems in classification. J Oral Pathol. 1989;17:547–553. doi: 10.1111/j.1600-0714.1988.tb01332.x. [DOI] [PubMed] [Google Scholar]
- Witkop CJ, Sauk JJ. Heritable defects of enamel. In: Stewart R, Prescott G, editors. Oral Facial Genetics. St. Louis: C.V. Mosby Company; 1976. pp. 151–226. [Google Scholar]
- Wright JT, Kula K, Hall KI, Simmons JH, Hart TC. Analysis of the tricho-dento-osseous syndrome genotype and phenotype. Am J Med Genet. 1997;72:197–204. doi: 10.1002/(sici)1096-8628(19971017)72:2<197::aid-ajmg14>3.3.co;2-0. [DOI] [PubMed] [Google Scholar]
- Wright JT, Hart PS, Aldred MJ, Seow WK, Crawford PJM, Hong SP, Gibson C, Hart TC. Relationship of phenotype and genotype in X-linked amelogenesis imperfecta. Connect Tissue Res. 2003;44:72–78. [PubMed] [Google Scholar]
- Yamakoshi Y, Pinheiro F, Tanabe T, Fukae M, Shimizu M. Sites of asparagine-linked oligosaccharides in porcine 32 kDa enamelin. Connect Tissue Res. 1998;39:39–46. doi: 10.3109/03008209809023910. [DOI] [PubMed] [Google Scholar]