

SUMMARY
We utilized the budding yeast Saccharomyces cerevisiae as a model to systematically explore physiological roles for yeast and mammalian aldo-keto reductases. Six open reading frames encoding putative aldo-keto reductases were identified when the yeast genome was queried against the sequence for human aldose reductase, the prototypical mammalian aldo-keto reductase. Recombinant proteins produced from five of these yeast open reading frames demonstrated NADPH-dependent reductase activity with a variety of aldehyde and ketone substrates. A triple aldo-keto reductase null mutant strain demonstrated a glucose-dependent heat shock phenotype which could be rescued by ectopic expression of human aldose reductase. Catalytically-inactive mutants of human or yeast aldo-keto reductases failed to effect a rescue of the heat shock phenotype, suggesting that the phenotype results from either an accumulation of one or more unmetabolized aldo-keto reductase substrates or a synthetic deficiency of aldo-keto reductase products generated in response to heat shock stress. These results suggest that multiple aldo-keto reductases fulfill functionally redundant roles in the stress response in yeast.
Keywords: aldo-keto reductase, aldose reductase, Saccharomyces cerevisiae, mutagenesis, heat shock
1. Introduction
Aldo-keto reductases (AKR)1 comprise a superfamily of structurally-similar proteins that catalyze the NADPH-dependent conversion of various carbonyl compounds into their corresponding alcohol products. In humans, the AKRs are implicated in diseases involving disposition of excess aldoses. Aldose reductase (AR), a prototypical AKR, catalyzes the conversion of glucose into sorbitol, which represents the first step in the polyol pathway. In humans with diabetes mellitus, increased activity of the polyol pathway due to chronic hyperglycemia has been associated with complications such as cataract and retinopathy [1], neuropathy [2], and nephropathy [3]. Beneficial roles for AKRs have been difficult to establish because tissues typically contain many similar enzymes with overlapping substrate and inhibitor specificities. Genetics-based approaches to explore physiological function using the mouse model have also been inconclusive, primarily due to probable redundancies between aldose reductase and similar aldo-keto reductases such as MVDP [major vas deferens protein, ref. 4] and FR-1 [fibroblast growth factor regulated-1, ref. 5]. Disruption of the aldose reductase gene in mice demonstrate a relatively benign phenotype resembling that of diabetes insipidis, which may be related to altered osmotic regulation in the kidneys of mutant animals [6,7].
The purpose of the current study was to utilize a simpler and more manageable eukaryotic model system to explore AKR gene function. By BLAST analysis of the Saccharomyces cerevisiae genome, we previously identified six open reading frames (YHR104w, YOR120W, YDR368W, YBR149W, YJR096W, YDL124W) which encode proteins with high predicted sequence similarity to human aldose reductase [8]. Three of these ORFs (YHR104W, YDR368W, YOR120W) have been previously shown to encode proteins with robust activity as NADPH-dependent carbonyl reductases [9–11], whereas one (YBR149W) functions primarily in the direction of alcohol (arabinose) oxidation [12]. In preliminary studies, we previously demonstrated that a triple null strain produced by disruption of ORFs encoding the three known AKRs exhibits a heat shock phenotype [13]. In the current study, we sought to evaluate the remaining two ORFs identified in our BLAST analysis (YJR096W and YDR368W) with respect to the kinetic properties of their encoded proteins as well as to probe for phenotypes of a mutant strain deficient in the five targeted aldo-keto reductases.
2. Materials and Methods
Expression and purification of recombinant yeast AKRs
Slightly different methods were used for production of recombinant yeast AKRs. The ORF for YJR096W (AKR5F), obtained from The Institute for Genome Research (TIGR, Rockville, MD), was subcloned into the prokaryotic expression plasmid pET23D (Novagen, Madison, WI) for expression of the recombinant protein in Escherichia coli strain BL21. The ORFs for YDL124W (AKR5G) and YOR120W (AKR3A1), obtained by PCR amplification from genomic DNA purified from S. cerevisiae strain S288c, were cloned into pET16B and pET23D (Novagen), respectively. Coding sequences in all plasmids were confirmed against the genome sequence of wild type S. cerevisiae strain S288c [14] prior to expression studies.
Recombinant proteins were purified from cultures grown in baffled shake flasks. In all cases, log phase cultures were grown for 4–16 hours following treatment with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) to induce expression of recombinant proteins. Cells were harvested by centrifugation (15,000 x g, 15 min, 4 °C) and were lysed after treatment with DNase (Sigma, St. Louis, MO) and lysozyme as described previously [15]. Extraction buffers contained the protease inhibitors (each at 1 μg/ml) antipain, bestatin, chymotrypsin, leupeptin, and pepstatin (Roche, Indianapolis, IN).
All extraction and purification steps were carried out at 4°C. As a group, yeast AKRs were readily prepared as recombinant proteins in E. coli host cultures. All except AKR5G (Ydl124w) were recovered as buffer-soluble proteins following extraction from host cells. In preliminary studies of AKR5G, expression of the encoded protein with an amino terminal histidine tag (7 residues in length) was associated with poor aldo-keto reductase activity [8]. For this reason, all yeast AKRs used in this study were expressed without fusion with affinity epitopes.
Methods and conditions for purification of each of the individual recombinant yeast AKRs are given below. In all cases, our methods routinely yielded in excess of 30 mg of highly purified protein from each liter of culture.
AKR3A1 (Yor120w)
Initial purification of AKR3A1 from host cell lysates was achieved by IEC using a carboxymethyl exchange resin (Macro-Prep CM, Bio-Rad). Lysate material from host cultures was first extensively dialyzed against a solution of 20 mM (2-[N-morpholinoethanesulfonic acid, MES), pH 6.0, containing 1 mM each of DTT and EDTA. After a brief centrifugation to remove insoluble materials, the dialyzed lysate was applied at 1 ml/min to a 2.5 x 10 cm column packed with the Macro-Prep CM resin. Unadsorbed material was flushed from the column with approximately 80 ml loading buffer. Bound materials were then eluted with a linear gradient of NaCl (0–0.5 M), contained in the loading buffer, over a volume of 600 ml. The eluate was continuously monitored by absorbance at 280 nm and was collected into 10 ml fractions for measurement of NADPH-dependent reductase activity using 5 mM DL-glyceraldehyde as substrate. Column fractions with reductase activity were pooled and dialyzed against 10 mM potassium phosphate buffer, pH 7.0, containing 1 mM DTT and 1 mM EDTA. Yor120w was further purified by dye-affinity chromatography using Affi-Gel Blue® resin (Bio-Rad, Hercules, CA). After loading to the column (2.5 x 5 cm) and a thorough wash with the loading buffer, bound material was eluted with a linear gradient of NaCl (0–1.4 M) contained in 400 ml loading buffer supplemented with 1mM each of DTT and EDTA. The eluate was continuously monitored by absorbance at 280 nm and was collected into 10 ml fractions. Fractions containing reductase activity were pooled and concentrated by ultrafiltration to approximately 1 mg/ml. The purified material was stored at 4°C and used within 2 weeks or stored at −70°C.
Expression and purification of the Y55F mutant of AKR3A1 was carried out exactly as for the wild type protein. Purification of the mutant protein was monitored by the presence of a highly enriched~36 kDa protein band on SDS-PAGE analysis of purification fractions. Materials extracted from induced cultures of the E. coli strain BL21 host transformed with a nonrecombinant (empty) pET expression plasmid were analyzed in a like manner.
AKR5F (Yjr096w)
Purification of recombinant AKR5F (Yjr096w) was achieved by anion exchange chromatography using DEAE-Sepharose (Amersham Biosciences, Piscataway, NJ). After overnight dialysis against 50 mM Tris-HCl, pH 8.0 containing 1 mM EDTA and 1 mM DTT, the cell-free extract was centrifuged briefly and the resulting supernatant applied to a 2.5x10 cm column of the ion exchange chromatography (IEC) resin. Bound proteins were eluted with a linear gradient of NaCl (0–0.4 M) contained in 600 ml of equilibrating buffer. Presence of AKR5F in column fractions was estimated by measuring NADPH-dependent reductase activity using 2 mM p-nitrobenzaldehyde as the aldehyde substrate. Proteins in column fractions were also visualized after separation by SDS-PAGE and Coomassie staining. Fractions containing reductase activity were pooled, dialyzed into 10 mM potassium phosphate buffer, pH 7.0, and applied to a 2.5 x 5 cm chromatography column packed with Affi-Gel Blue® (Bio-Rad) previously equilibrated with the same buffer. After a wash to remove unadsorbed material, bound proteins were eluted with a linear gradient of sodium chloride (0–2.5 M) contained in the equilibration buffer. Reductase assays on column fractions identified an activity peak that was coincident with a peak defined by A280. Fractions containing reductase activity were pooled, concentrated to ~ 1 mg/ml, and stored at 4 °C.
AKR5G (Ydl124w)
Preliminary studies showed that AKR5G expressed as a His-tagged fusion protein had very low AKR activity [16, data not shown]. In the present study, the enzyme was expressed without an amino-terminal His-tag epitope. The majority of recombinant AKR5G was found in the insoluble cell pellet following DNAse-lysozyme extraction (not shown), and was recovered by treatment for 30 minutes at 4 °C with a cracking buffer consisting of 10 mM sodium phosphate, pH 7.2, 6 M urea, 142 mM β-mercaptoethanol, and protease inhibitors essentially as described previously [17]. Materials solubilized in cracking buffer were dialyzed (Mr 12,000–14,000 cut-off tubing) against 100 volumes of DEAE column buffer (10 mM TrisHCl, pH 7.4, 0.5 mM DTT, 0.5 mM EDTA. After centrifugation to remove insoluble materials, lysates were applied at 1 ml/min to a chromatography column (2.5 x 10 cm) packed with DEAE ion exchange resin previously equilibrated with column buffer noted above. After flushing out unadsorbed materials with a 500 ml buffer wash, bound proteins were eluted with a linear gradient (0–200 mM) of sodium chloride prepared in column buffer. Eluate fractions (10 mL) were assayed for the presence of NADPH-dependent aldo-keto reductase activity using 5 mM DL-glyceraldehyde as a carbonyl substrate. Fractions with reductase activity were pooled and further purified by dye affinity chromatography essentially as described above for purification of AKR5F, with the exception that bound materials were eluted first with a step increase from 0 to 1.5 M NaCl in the column buffer (200 mL) followed by a linear NaCl gradient (1.5–2.5 M) contained in 300 ml column buffer. Fractions containing reductase activity were pooled, dialyzed against 40 volumes of 10 mM potassium phosphate, pH 7.2, containing 0.5 mM DTT and 0.5 mM EDTA. Purified materials were stored at 4 °C for up to 2 weeks or for indefinite periods at −70 °C.
Enzyme assays
Reductase activity was measured at 23 °C using a Cary 1E spectrophotometer interfaced with a peltier temperature-controlling device. Assay mixtures of 1.0 ml contained 0.15 mM NADPH and aldehyde substrate in a triple buffer system consisting of 25 mM 2-(N-morpholino)ethanesulfonic acid, 25 mM potassium phosphate, and 90 mM Tris, at a pH titrated to correspond to the pH optimum for the enzyme being tested (see below). Reactions were initiated by addition of enzyme, and initial rates calculated from the decrease in absorbance at 340 nm upon oxidation of NADPH. To determine steady state kinetic constants, initial velocities observed as a function of substrate concentration (ranging from approximately 0.1- to 5-times Km) were fitted to the Michaelis-Menten equation using Graphpad Prizm (San Diego, CA).
Strains, Gene Deletion, and Growth Conditions
Parental wild-type yeast strain (BY4742) and isogenic single deletion mutants, which were made available through the Yeast Deletion Project (ref PMID: 12140549) see http://www.stanford.edu/group/yeastdeletionproject/deletions3.html), were obtained from Research Genetics (Carlsbad, CA). A list of yeast strains used in this study is summarized in Table I. Generation of AKR gene deletion mutants was performed by the PCR-mediated gene disruption approach [18]. Targeting primers for disruption of open reading frames used in conjunction with several different selectable markers carried in the pRS4XX series of plasmids are shown in Table II. PCR amplimers for targeted gene disruption were used to transform recipient cells using the lithium acetate method. After growth of transformants on selective media, deletion of the targeted gene sequences was routinely confirmed by PCR using primer sets directed toward sites internal as well as flanking the targeted gene of interest (Table III).
Table I.
Strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Research Genetics |
| ARD10 | BY4742 gre3Δ::HIS3 ypr1Δ::kanMX4 gcy1::URA3 | This study |
| ARD11 | BY4742 gre3Δ::HIS3 ypr1Δ::kanMX4 gcy1::URA3 ydl124w Δ::LEU2 | This study |
| ARD12 | BY4742 gre3Δ::HIS3 ypr1Δ::kanMX4 gcy1::URA3 ydl124w Δ::LEU2 yjr096w Δ::bler | This study |
Table II.
Oligonucleotides used for construction of deletion strains
| Targeted Gene/ORF | Primer sequence 5′ to 3′ | Template |
|---|---|---|
| GCY1 | Sense:
ATATGCCTATCAGGCATTCACCCGTGTGACGAATCGCACA//AGATTGTACTGAGAGTGCAC Anti-sense: TGCCTTATACACCACCTATTCTGGCATCTGCGGGATTTCG//CTGTGCGGTATTTCACACCG |
pRS406 (URA3) |
| YDL124W | Sense
5′ACACATGTAAACGGTTGTGTTACCCTAAAGAAACGGAGGTCAGTTAACAACACTTTTACAGATGCAGCTGAAGCTTCGTACGC3′ Anti-sense 5′AAGTCGATTAATAAAGTTATACAAAACAAATATGACTCGTACATAAATTATCCGGTATTTAGCATAGGCCACTAGTGGATCTG3′ |
pUG73(LEU2) |
| YJR096W | Sense
5′ATATAAGAGCATAAGCAACTGATCTTACTTTAGTAATTAACTTAGCATACCTAGCCCGAAGGACAGCTGAAGCTTCGTACGC3′ Anti-sense 5′AAAAAGCTACATGTAATATTAATACTTTGTGAAACGTAAACTAATTATATGTTCTATATACGTGCATAGGCCACTAGTGGATCTG |
pUG66(bler) |
Table III.
Verification oligonucleotides.
| Gene/ORF | Confirmation primers |
|---|---|
| YPR1 | A: 5′-CATTCTCACTGATGCTATTGTC-3′
B: 5′-ATCAATGTGTCTGTATCCAGC-3′ C: 5′-ATGGCACTGTTGACATCG-3′ D: 5′-GATGACGACGGTCCGAA-3′ B′ (HIS3): 5′-ACGCACTCTCACTACG-3′ C′: (HIS3): 5′-CCTAGCGATAGAGCACT-3′ |
| GRE3 | A: 5′-CTCCTGGTACACTGAAGTAT-3′
B: 5′-TGAGACCTTCTTGATTACAT-3′ C: 5′-GTGCTTGCGACTACGG-3′ D: 5′-TCAACCATACAAGAGATGAA-3′ B′: (KanMX) 5′-TGTACGGGCGACAGTCACAT-3′ C′: (KanMX) 5′-CCTCGACATCATCTGCCCAGAT-3′ |
| GCY1 | A: 5′-CCTATCAGGCATTCACC-3′
B: 5′-GACGTGCCAGCTAATAAC-3′ C: 5′-ATGGCTACCGACACATTG-3′ D: 5′-CTTGCCTTATACACCACC-3′ B′: (URA3) 5′-AGTTATCCTTGGATTTGG-3′ C′: (URA3) 5′-ATCTCATGGATGATATCC-3′ |
| YDL124W | A: 5′-CGAAAGCGCGGAGAATTAA-3′
B: 5′-TGCCTTCCCAACTTCTGGATA-3′ C: 5′-TTGGTAGAAGCATACTCGCCA-3′ D: 5′-TAGCAACACATGATGCGCGT-3′ B′: (LEU2) 5′-AGTTATCCTTGGATTTGG-3′ [see ref.42] C′: (LEU2) 5′-ATCTCATGGATGATATCC-3′ [see ref.42] |
| YJR096W | A: 5′-TGAAGTAGTTATCATACATATTCCGTCG-3′
B: 5′-GAATCAAAAGAAGATCGATGTATTGC-3′ C: 5′-GGTCAACCAAATCGAGATATCACCT-3′ D: 5′-GGTAAGACTAGTAACGTCACATCGAAG-3′ B′: (bler) 5′-GGATGTATGGGCTAAATG-3′ [see ref.42] C′: (bler) 5′-CCTCGACATCATCTGCCC-3′ [see ref.42] |
| |
Cloning and mutagenesis of AKR3A1 (Gcy1) and AKR1B1 (human aldose reductase) for expression in yeast
While in the context of pBLUESCRIPT, codons for tyrosine-48 of AKR1B1 (human AR) [19], and the homologous site (tyrosine-55) of AKR3A1 (Gcy1), were mutated to phenylalanine using a QuikChange site-directed mutagenesis kit (Stratagene). For preparation of yeast expression constructs, the mutant genes were released by endonuclease treatment (SpeI and HindIII for AKR1B1, and BamHI and PstI for AKR3A1) and were ligated into the corresponding sites of the episomal GPD425 expression vector as described above. DNA sequencing across ligation sites and open reading frames was carried out to confirm the fidelity of all plasmid constructs.
3. Results
Characterization of yeast aldo-keto reductases
Yeast AKRs may be identified according to a gene name, an ORF association, and according to a systematic AKR nomenclature [20]. A cross-referenced list of genes, ORFs, and AKR designations is shown in Table IV. By BLAST search, we previously identified six ORFs (YDR368W, YHR104W, YOR120W, YBR149W, YJR096W, YDR124W) in the S. cerevisiae genome sequence which showed clear amino acid sequence similarity (30–44% identity) to human aldose reductase (AKR1B1) [8]. Alignment of the deduced sequences against the primary structure of human aldose reductase indicates that amino acid side chains known to facilitate catalysis in mammalian AKRs are strictly conserved across these yeast proteins [19,21,22]. As a group, these proteins share approximately 30–40% sequence identity, excepting AKR3A1 (Gcy1) and AKR3A2 (Ypr1), which are approximately 65% identical. To evaluate the ability of the encoded proteins to function as an NADPH-dependent aldo-keto reductase, ORFs for putative AKRs were cloned into expression plasmids and over-expressed in Escherichia coli host cultures. Studies of AKR3C (Ara1) were not undertaken because, unlike AR, this enzyme functions preferentially in the direction of NADP-dependent aldose oxidation [12]. Various structural and kinetic properties of some yeast AKRs examined in our study, including, AKR3A2 (Ypr1) and AKR2B6 (Gre3), were reported previously by us [8] and other investigators [9–11,23] using recombinant and native enzymes purified from yeast cultures, respectively. In the present work, we carried out a systematic evaluation of the remaining putative AKRs including AKR5F (Yjr096w), AKR5G (Ydl124w) and AKR3A1 (Gcy1) so as to further our understanding of these proteins as functional aldo-keto reductases.
Table IV.
Aldo-keto reductases in Saccharyomyces cerevisiae
| ORF Designation | YHR104W | YOR120W | YDR368W | YBR149W | YJR096W | YDL124W |
| Gene Name | GRE3 | GCY1 | YPR1 | ARA1 | ||
| AKR Designationa | AKR2B6 | AKR3A1 | AKR3A2 | AKR3C | AKR5F | AKR5G |
| Reported | Aldo-keto | Aldo-keto | Xylose | Arabinose | Xylose/arabino | Xylose/arabino |
| Function | reductase | reductase | reductase | Dehydrogenase | se reductase | se reductase |
| References | [26,30,36,43] | [27,30,41] | [10,25] | [12] | [30] | [30] |
Nomenclature for the AKR Superfamily is available at http://www.med.upenn.edu/akr/
Since only limited data was previously published on the putative reductase activities of these putative AKRs [8,11,11,16], we initiated a screen for aldo-keto reductase activity using 150 μM NADPH as cofactor and either DL-glyceraldehyde or p-nitrobenzaldehyde as aldehyde substrate. These enzymes all showed robust activity, with optima at pH 7.5-8.5 for AKR5F and pH 6.0–7.5 for AKR3A1 (Figure 2). Reductase activity for AKR5G showed no distinct optimum over a pH range of pH 5 to pH 8.5, but dropped sharply above pH 8.5 (Figure 1). The reductase activity level of each protein was not above background when assays were conducted using DL-glyceraldehyde or p-nitrobenzaldehyde as the aldehyde substrate and NADH instead of NADPH. Using reaction conditions optimized for each enzyme, catalytic constants were determined with a variety of aldehyde substrates. As shown in Table V, the catalytic efficiency of AKR3A1 was generally higher than AKR5F or AKR5G with almost all substrates examined. No measurable activity was observed when each of these enzymes was assayed using aldose substrates such as D-glucose and D-xylose. As a group, the enzymes showed highest catalytic efficiencies with aromatic aldehydes such as benzaldehydes and phenylglyoxal.
Fig. 2.
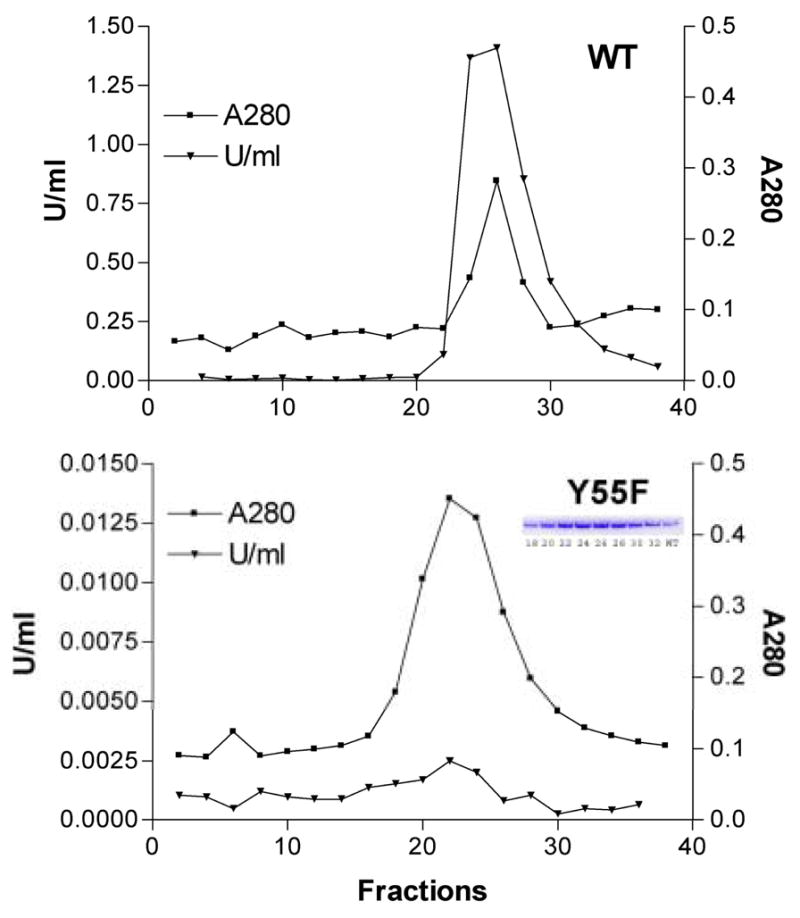
Purification of AKR3A1 (Gcy1). Wild type (WT) and mutant (Y55F) AKR3A1 were purified by dye-affinity chromatography using Affi-Gel Blue (Bio-Rad). Column fractions were measured for protein by absorbance at 280 nm (▪) and for NADPH-linked reductase activity using 5 mM DL-glyceraldehyde as substrate (▾). Note 100-fold difference in scale on the ordinate axes showing enzyme activities. (Inset: SDS-PAGE of material from the indicated column fractions; WT, wild type AKR3A1).
Fig. 1.
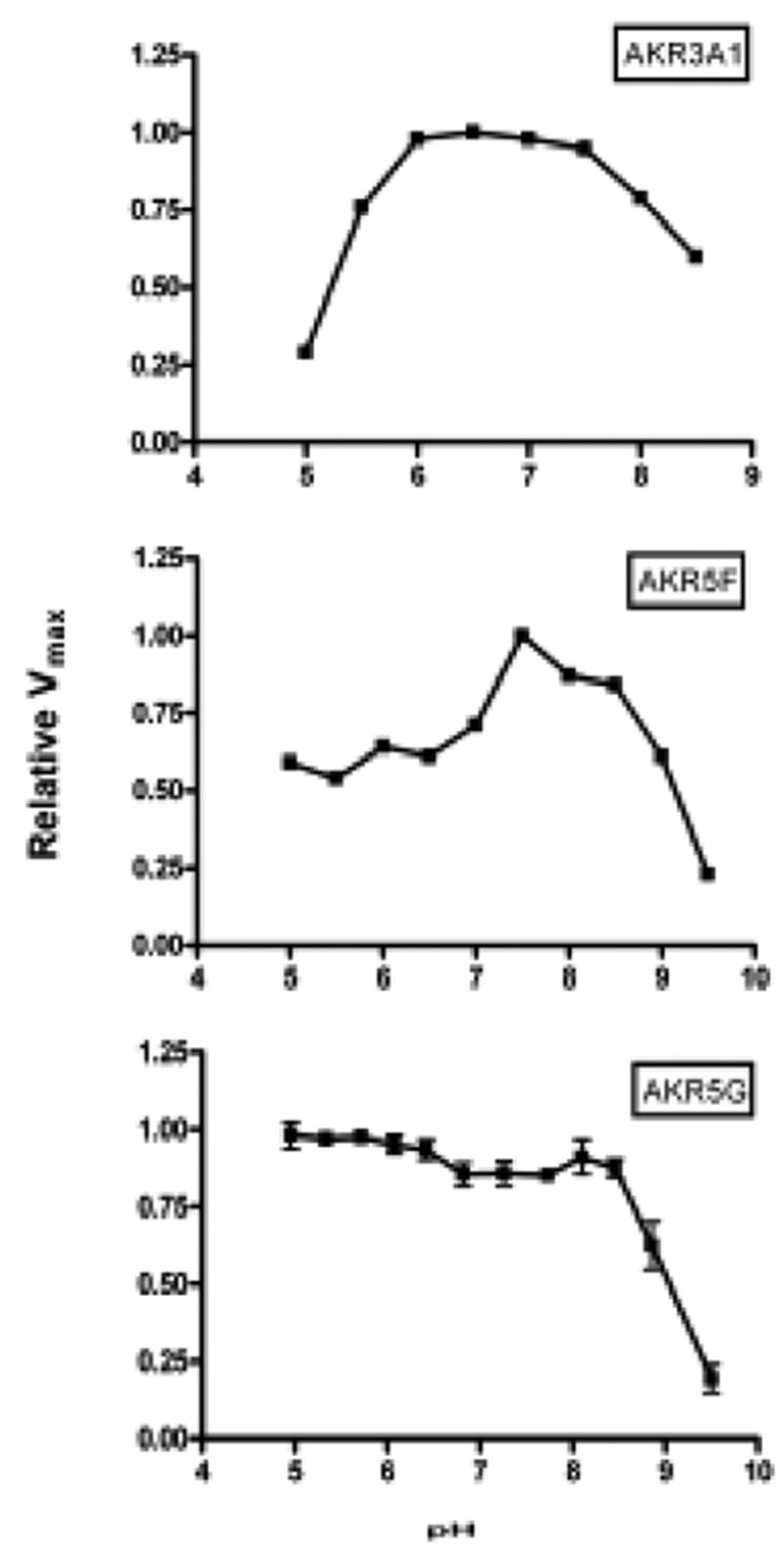
pH Optima of yeast AKRs. Reductase activities were measured in a 1.0 ml assay solution containing enzyme, aldehyde substrate and 0.15 mM NADPH in a triple buffer system titrated to the indicated pH values.
Table V.
Kinetic constants for yeast aldo-keto reductases
| AKR3A1 | AKR5F | AKR5G | |
|---|---|---|---|
| Yor120w Gcy1 | Yjr096w | Ydl124w | |
| DL-Glyceraldehyde | |||
| kcat (min−1) | 539 | 6.6 | 4.1 |
| Km (mM) | 0.97 ± 0.1 | 54 ± 2 | 0.24 ± 0.03 |
| kcat/Km (min−1mM−1) | 556 | 0.12 | 17 |
| p-Nitrobenzaldehyde | |||
| kcat (min−1) | 71 | 88 | 3.3 |
| Km (mM) | 0.13 ± 0.002 | 0.5 ± 0.06 | 0.03 ± 0.004 |
| kcat/Km (min−1mM−1) | 546 | 176 | 110 |
| Benzaldehyde | |||
| kcat (min−1) | 58 | 4 | 4 |
| Km (mM) | 5.2 ± 0.6 | 47 ± 6 | 0.24 ± 0.03 |
| kcat/Km (min−1mM−1) | 11 | 0.09 | 17 |
| Phenylglyoxal | |||
| kcat (min−1) | 111 | 2.4 | 985 |
| Km (mM) | 8.7 ± 0.19 | 35 ± 0.1 | 4.2 ± 0.3 |
| kcat/Km (min−1mM−1) | 13 | 0.07 | 234 |
| Acrolein | |||
| kcat (min−1) | 30 | 1.0 | 3.4 |
| Km (mM) | 50 ± 12.8 | 22 ± 5.2 | 125 ± 88 |
| kcat/Km (min−1mM−1) | 0.6 | 0.05 | 0.03 |
| Butyraldehyde | |||
| kcat (min−1) | 81 | 0.5 | 14 |
| Km (mM) | 54 ± 2.8 | 18 ± 8 | 210 ± 145 |
| kcat/Km (min−1mM−1) | 1.5 | 0.03 | 0.07 |
| D-glucose | |||
| kcat (min−1) | NMA | NMA | NMA |
| D-Xylose | |||
| kcat (min−1) | NMA | NMA | NMA |
| NADPH | |||
| kcat (min−1) | 438 | 123 | 7.2 |
| Km (mM) | 0.012 ± 0.003 | 0.37 ± 0.1 | 0.023 ± .005 |
| kcat/Km (min−1mM−1) | 36500 | 332 | 313 |
Activity measurements with Yjr096w were performed at pH 8.0 and at pH 6.5 for Ydl124w and Gcy1. In all cases, NADPH was held constant at 0.15 mM. Apparent Km and kcat values were determined by fitting initial rate data to a general Michaelis-Menten equation using Graphpad Prizm (San Diego). For each enzyme-substrate pair, n≥10. NMA, no measurable activity. Apparent Km for NADPH was determined in the presence of fixed concentrations of aldehyde substrate including 0.4 mM p-nitrobenzaldehyde for AKR3A1 (Yor120w), 2 mM p-nitrobenzaldehyde for AKR5F (Yjr096w), and 25 mM phenylglyoxal for AKR5G (Ydl124w).
Alignment of amino acid sequences demonstrates that a tyrosine at the position corresponding to tyrosine-48 in human aldose reductase is conserved among all yeast AKRs examined in this study [8]. We hypothesized that substitution of phenylalanine for tyrosine-55 would result in loss of AKR activity in AKR3A1 (Gcy1), similar to that observed with aldose reductase [19] and 3α-hydroxysteroid dehydrogenase [24]. As with wild type AKR3A1, the Y55F mutant was expressed to high levels in IPTG-induced cultures and was obtained in virtually homogeneous form by a two-step column purification procedure. Although reductase activity in the Y55F mutant was virtually undetectable, purification of the mutant was easily followed by the appearance of a protein band of approximately Mr 36000 on SDS-PAGE of material from column fractions (Figure 2). High reductase activity levels were found in the corresponding fractions from preparations starting with wild type AKR3A1. A protein band corresponding in size and abundance to AKR3A1 was not observed in whole cell extracts or in purification fractions using material from induced E.coli expression cultures transformed with an empty pET21d expression plasmid (not shown).
Consistent with results from other aldo-keto reductases, the substitution of Phe for Tyr-55 caused a dramatic reduction in catalytic efficiency. Using DL-glyceraldehyde as a typical carbonyl substrate, the catalytic efficiency of wild type AKR3A1 was reduced from 553 mmol−1min−1 to 0.16 mmol−1min−1 for the Y55F mutant. Virtually all of this change results from a reduction in kcat, as there was a relatively small change in Km DL-glyceraldehyde. The Km was 0.97±0.1 mM for wild type and 1.5±0.1 mM for the Y55F mutant.
Phenotypic Characteristics of AKR deletion mutants
Based on this and previous work, it is clear that two of the yeast AKRS examined, AKR5F (Yjr096w)and AKR5G (Ydl124w), have a relatively poor ability to catalyze reduction of carbonyls represented in our battery of substrates. Given that AKR3A1 (Gcy1), AKR3A2 (Ypr1) and AKR2B6 (Gre3) demonstrate more robust activity as aldo-keto reductases, we hypothesized that these enzymes would be excellent candidates to test for functional conservation with mammalian aldo-keto reductases [8]. Accordingly, GCY1, YPR1, and GRE3 were disrupted in a series of mutant strains to test the hypothesis that human aldose reductase can functionally compensate for one or more of the yeast AKR genes.
Single gene deletion mutants of YPR1, GRE3 or GCY1 do not display an obvious phenotype [25–27]. This is not surprising since these genes may be functionally redundant. We examined double and triple AKR gene deletion mutants with a variety of phenotypic screens including growth rate at normal (30°C) or elevated temperatures (37°C) in YPD, cell morphology/budding pattern, growth sensitivity to various stress inducible agents, growth auxotrophy for different carbon sources and nutrients, and growth sensitivity to screening drugs selected to probe major signaling transduction pathways and nucleic acid metabolism (Table VI). In preliminary studies, we observed that mutants missing any two of the three AKR genes were not distinguishable from the wild type strain, but deletion of all three AKR genes (ypr1Δ/gre3Δ/gcy1Δ; strain ARD10) resulted in a marked heat shock sensitivity [13]. In the current study, we show that further disruption of ORFs that encode the two additional putative AKRs resulted in an enhancement of the heat shock phenotype (Figure 3A).
Table VI.
Phenotypic screens for yeast AKR mutants
| Phenotype | Screening agent/parameter | References |
|---|---|---|
| Heat shock | Loss of viability following heat shock up to 55°C | [44,45] |
| Oxidative stress | YPD + 0.88 mM hydrogen peroxide | [46,47] |
| YPD + 1.5 mM diamide | [48] | |
| YPD + 10 μM paraquat | [49] | |
| Osmotic stress | YPD + 1 M sorbitol
YPD + 1 M NaCl |
|
| Temperature sensitivity | Growth at 37°C, YPD | |
| Growth/morphology | Growth rate (doubling time) | |
| Viability (exclusion of propidium iodide) | ||
| Budding pattern (light microscopy) | ||
| Calcofluor sensitivity (YPD + 0.1% calcofluor) | [50] | |
| Carbon auxotrophy | YP + 2% glucose
YP + 2% galactose YP + 2% raffinose YP + 2% maltose YP + 2% sucrose YP + 3% glycerol YP + 2% potassium Acetate |
|
| Nitrogen starvation | Loss of growth on nitrogen-free medium | [45] |
| Signal transduction | YPD + 10 mM caffeine | [51] |
| YPD + 0.1 μg/ml staurosporine | [52] | |
| Nucleic acid metabolism | YPD + 0.05% methyl methanesulfonate | [53] |
| YPD + 100 mM hydroxyurea | [54] | |
| YPD + 60 μg/ml 6-azauracil | [55] |
Fig. 3.
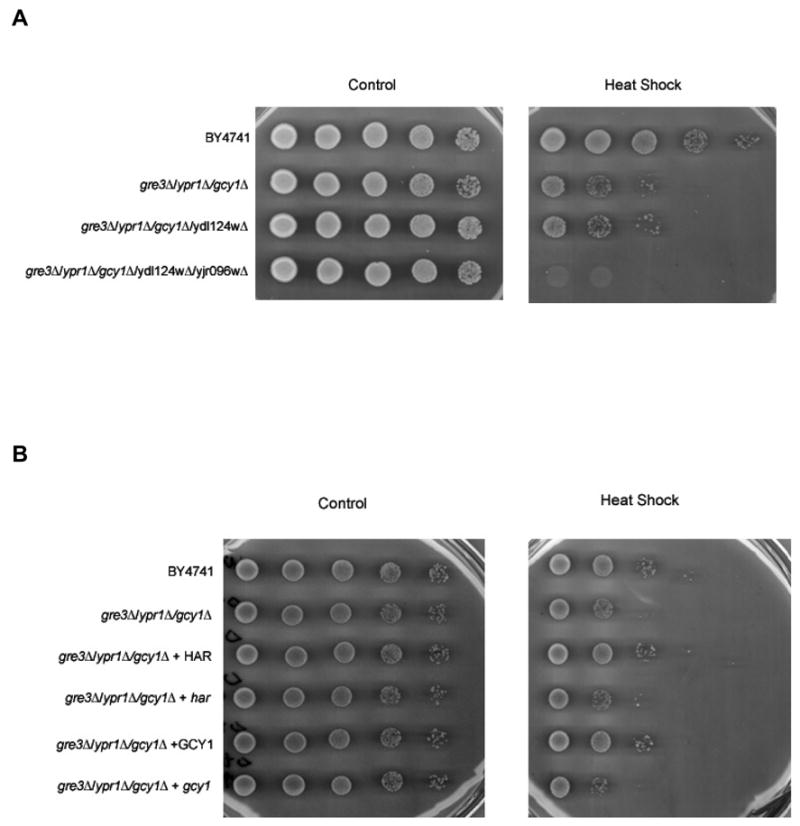
Heat shock phenotype of AKR mutant strains. After heat shock treatment, cell suspensions were serially diluted into water and spotted on selective plates (−Leu). After incubation for ~72 h at 30°, plates were imaged on a flat bed scanner to document recovery of growth. (A) Growth recovery following heat shock in the wild type parental strain (BY4741) and isogenic strains containing 3 (ARD10), 4 (ARD11), and 5 (ARD12) disrupted AKR genes. Genotypes of relevant strains are ARD10 (BY4742 gre3Δ::HIS3 ypr1Δ::kanMX4 gcy1::URA3), ARD11 (BY4742 gre3Δ::HIS3 ypr1Δ::kanMX4 gcy1::URA3 ydl124wΔ::LEU2), and ARD12 (BY4742 gre3Δ::HIS3 ypr1Δ::kanMX4 gcy1::URA3 ydl124wΔ::LEU2 yjr096wΔ::bler). (B) Heat shock rescue experiments. Strains included the wild type parental strain (BY4741), and the triple AKR null strain ARD10 (BY4742 gre3Δ::HIS3 ypr1Δ::kanMX4 gcy1::URA3). Heat shock rescue experiments were carried out by transforming strains BY4741 and ARD10 with expression plasmids encoding either human aldose reductase (HAR, AKR1B1) or Gcy1 (AKR3A1). Dependence of phenotypic rescue on reductase activity was assessed by transforming heat shock sensitive strains with functionally inactive point mutants of human aldose reductase (har) or Gcy1 (gcy1).
Transformation of strain ARD10 with a plasmid carrying GCY1, but not gcy1 Y55F, partially rescued the heat shock sensitivity. (Figure 3B). The human aldose reductase cDNA expressed from the GPD promoter partially rescued heat shock sensitivity, but not if it carried a mutation of the active site tyrosine-48 residue (Y48F) to create a catalytically inactive enzyme (Figure 3B). These results indicate that either human or yeast AKRs is capable of rescuing the heat shock phenotype in triple AKR deficient mutant strains, suggesting some degree of functional conservation among AKRs in yeast and humans.
4. Discussion
The budding yeast Saccharomyces cerevisiae has six ORFs that encode putative aldo-keto reductases (Table IV). Products of YHR104W (Gre3) and YDR368W (Ypr1) were initially identified as stress response proteins on the basis of their upregulation following growth in the presence of hypertonic media [26,28]. The product of YOR120W (Gcy1) was initially identified as a galactose-inducible crystallin-like yeast protein [27]. Subsequent studies demonstrated that each of these gene products had properties typical of classical aldo-keto reductase, i.e., catalytic activity with various classes of carbonyl-containing substrates, preferentially in the direction of carbonyl reduction, using NADPH as the reducing coenzyme [9–11]. Current evidence suggests that YBR149W (Ara1) encodes a dehydrogenase that functions preferentially in the direction of arabinose oxidation rather than reduction [12]. The functions of the remaining ORFs identified in our BLAST analysis, including YDL124W and YJR096W, have not been extensively studied. Ishihara recently reported evidence that a yeast protein, identical to that encoded by YDL124W, catalyzes the NADPH-dependent reduction of aromatic α-keto amides and α-keto esters [29]. Recent studies also indicate that the proteins encoded by YJR096W and YDL124W and may play a role in arabinose and xylose metabolism [30].
In preliminary studies, we showed that Gre3, Ypr1, Gcy1, and human AR, purified as recombinant proteins, have NADPH-dependent aldo-keto reductase activity using aldehyde substrates such as DL-glyceraldehyde and p-nitrobenzaldehyde [8]. This suggested to us that the functions of AKR genes between yeasts and humans may be similar. Our observation that ectopic expression of human aldose reductase rescues the heat shock phenotype of the triple AKR null strain suggests that human and yeast AKRs fulfill similar catalytic functions that are necessary for recovery from stress associated with heat shock. Since the catalytically-inactive mutants of human aldose reductase or Gcy1 failed to show a rescue effect, it is possible that the heat shock phenotype results from either an accumulation of one or more unmetabolized AKR substrates or a synthetic deficiency of AKR products generated in response to stress or culture conditions. Further studies are ongoing to identify pathways and processes that are influenced by the action of AKRs.
Previous studies have shown that 4-hydroxynonenal (4-HNE), a lipid-derived aldehyde produced during oxidative stress, is a high affinity substrate for aldose reductase in mammalian cells [31]. In addition, aldose reductase gene expression is induced in response to osmotic stress [32,33]. Accordingly, we expected that deletion of the three cognate AKR genes would induce phenotypes representative of oxidative and osmotic stress. However, this is not the case, as essentially no difference was observed in the growth of this mutant when compared to wild type yeast under various oxidative and osmotic stress conditions. We hypothesize that this outcome may result from at least two possibilities. First, lipid-derived reactive aldehydes, such as 4-HNE, are not produced in yeast due to low amounts of polyunsaturated fatty acids, the obligate precursors to lipid aldehydes generated by oxidative stress [34]. Although the heat shock phenotype was glucose-dependent, we consider it unlikely that any of the three disrupted AKRs play roles in the response to osmotic stress through production of an osmolyte such as sorbitol. Unlike human AR, none of the yeast AKRs are capable of converting glucose to sorbitol, an active osmolyte known to be involved in renal response to hypertonicity [35]. However, at least two of the yeast AKRs, namely GRE3 and YPR1, were identified as osmoregulated genes in previous studies [26,26,28]. Similar to findings reported here, previous investigators observed no measurable difference from wild type control when gre3 or ypr1 mutants were subjected to osmotic stress [26,36].
Although AKR3A2 (Ypr1) and AKR2B6 (Gre3) have been studied in the context of their activity as NADPH-dependent carbonyl reductases, very little is known regarding the catalytic activities of other yeast enzymes such as AKR3A1 (Gcy1), AKR5F (Yjr096w) and AKR5G (Ydl124w). Our results demonstrate each of these gene products has easily measured activity as an NADPH-dependent aldehyde reductase. Human and yeast AKRs appear to have similar activities when assays were conducted with substrates such as aliphatic and aromatic aldehydes. These results confirm that yeast, like mammalian tissues, contain multiple AR-like enzymes with activity against a broad range of aromatic and aliphatic aldehydes [37].
Crystallography studies of mammalian aldose reductases indicate that the proteins adopt a (β/α)8 barrel structural fold with the active site buried deep within the hydrophobic barrel core. Functional and structural studies with human aldose reductase (AKR1B1) suggest the importance of Asp-48, Lys-77, Tyr-48 and His-110 as key residues in the catalytic sequence [19,38,39]. Comparison with AKR1B1 shows that all of these key residues are conserved at homologous sites throughout the yeast AKRs. Similar to that observed with human aldose reductase [19], mutagenesis of the presumptive hydrogen donor to create the Y55F mutant of AKR3A1 (Gcy1) resulted in essentially complete loss of AKR activity. Structural studies have shown that, unlike the case with Tyr-48 in human aldose reductase [19,40], the orientation of the Tyr-55 side chain is not favorably positioned to function as an acid in the reduction reaction [41]. Further study will be required to establish whether AKR3A1 utilizes a different catalytic mechanism than hypothesized for mammalian AKRs.
Since phenotypic abnormalities did not appear until ORFs corresponding to at least three yeast AKRs were interrupted, we hypothesize that these enzymes fulfill functionally redundant roles even though their expression may fall under different transcriptional control. Some differences were noted between human and yeast enzymes with respect to catalytic efficiency with some physiologically relevant substrates such as glucose. However, the observation that human aldose reductase can rescue the heat shock phenotype of the triple AKR null mutant suggests that human and yeast AKRs share overlapping physiological roles. Future studies will be needed to identify pathways and structures that may be affected by deficiencies in AKR gene expression in S cerevisiae. Such studies may provide valuable clues in our ongoing studies to discover physiological roles of this enzyme family in human tissues.
Acknowledgments
We wish to thank Dr. Mark Johnston for advice and help with the manuscript, his associate Linda Riles for her technical assistance with targeted gene disruption procedures, and Loryn Rikimaru, B.S.N. Murthy, Kent Morris and Michael Young for their assistance with cloning and expression of recombinant yeast proteins. This work was supported in part by NIH grants EY05856, EY02687, DK20579 and by awards to the Department of Ophthalmology and Visual Sciences from Research to Prevent Blindness, Inc. Support from the Kilo Diabetes and Vascular Research Foundation is also gratefully acknowledged.
The abbreviations used are
- AKR
aldo-keto reductase
- AR
aldose reductase
- GPD
glyceraldehyde 3-phosphate dehydrogenase
- IEC
ion exchange chromatography
- ORF
open reading frame
- YPD
yeast peptone dextrose, synthetic complete
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kinoshita JH, Nishimura C. The involvement of aldose reductase in diabetic complications. Diabetes-Metabolism Rev. 1988;4:323. doi: 10.1002/dmr.5610040403. [DOI] [PubMed] [Google Scholar]
- 2.Oates PJ. Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol. 2002;50:325. doi: 10.1016/s0074-7742(02)50082-9. [DOI] [PubMed] [Google Scholar]
- 3.Raptis AE, Viberti G. Pathogenesis of diabetic nephropathy. Exp Clin Endocrinol Diabetes. 2001;109(Suppl 2):S424–S437. doi: 10.1055/s-2001-18600. [DOI] [PubMed] [Google Scholar]
- 4.Pailhoux EA, Martinez A, Veyssiere GM, Jean CG. Androgen-dependent protein from mouse vas deferens. cDNA cloning and protein homology with the aldo-keto reductase superfamily. J Biol Chem. 1990;265:19932. [PubMed] [Google Scholar]
- 5.Donohue PJ, Alberts GF, Hampton BS, Winkles JA. A delayed-early gene activated by fibroblast growth factor-1 encodes a protein related to aldose reductase. J Biol Chem. 1994;269:8604. [PubMed] [Google Scholar]
- 6.Ho HT, Chung SK, Law JW, Ko BC, Tam SC, Brooks HL, Knepper MA, Chung SS. Aldose reductase-deficient mice develop nephrogenic diabetes insipidus. Mol Cell Biol. 2000;20:5840. doi: 10.1128/mcb.20.16.5840-5846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aida K, Ikegishi Y, Chen J, Tawata M, Ito S, Maeda S, Onaya T. Disruption of aldose reductase gene (Akr1b1) causes defect in urinary concentrating ability and divalent cation homeostasis. Biochem Biophys Res Commun. 2000;277:281. doi: 10.1006/bbrc.2000.3648. [DOI] [PubMed] [Google Scholar]
- 8.Petrash JM, Murthy BS, Young M, Morris K, Rikimaru L, Griest TA, Harter T. Functional genomic studies of aldo-keto reductases. Chem Biol Interact. 2001;130–132:673. doi: 10.1016/s0009-2797(00)00258-1. [DOI] [PubMed] [Google Scholar]
- 9.Kuhn A, van Zyl C, van Tonder A, Prior BA. Purification and partial characterization of an aldo-keto reductase from Saccharomyces cerevisiae. Applied & Environmental Microbiology. 1995;61:1580. doi: 10.1128/aem.61.4.1580-1585.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura K, Kondo S, Kawai Y, Nakajima N, Ohno A. Amino acid sequence and characterization of aldo-keto reductase from bakers' yeast. Biosci Biotechnol Biochem. 1997;61:375. doi: 10.1271/bbb.61.375. [DOI] [PubMed] [Google Scholar]
- 11.Hur E, Wilson DK. Crystallization and aldo-keto reductase activity of Gcy1p from Saccharomyces cerevisiae. Acta Crystallogr D Biol Crystallogr. 2000;56( Pt 6):763. doi: 10.1107/s0907444900004704. [DOI] [PubMed] [Google Scholar]
- 12.Kim ST, Huh WK, Lee BH, Kang SO. D-arabinose dehydrogenase and its gene from Saccharomyces cerevisiae. Biochim Biophys Acta. 1998;1429:29. doi: 10.1016/s0167-4838(98)00217-9. [DOI] [PubMed] [Google Scholar]
- 13.Chang Q, Harter TM, rikimaru LT, Petrash JM. Aldo-keto reductases as modulators of stress response. Chem Biol Interact. 2003;143–144:325. doi: 10.1016/s0009-2797(02)00216-8. [DOI] [PubMed] [Google Scholar]
- 14.Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M, Weng S, Botstein D. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merck KB, De Haard Hoekman WA, Oude Essink BB, Bloemendal H, de Jong WW. Expression and aggregation of recombinant alpha A-crystallin and its two domains. Biochim Biophys Acta. 1992;1130:267. doi: 10.1016/0167-4781(92)90439-7. [DOI] [PubMed] [Google Scholar]
- 16.Chang Q, Harter TM, Griest TA, Murthy BSN, Petrash JM. Aldo-Keto Reductases in the Stress Response of the Budding Yeast Saccharomyces Cerevisiae. In: Penning TM, Petrash JM, editors. Aldo-Keto Reductases and Toxicant Metabolism, ACS Symposium Series. American Chemical Society; Washington, D.C.: 2004. pp. 225–238. [Google Scholar]
- 17.Andley UP, Mathur S, Griest TA, Petrash JM. Cloning, expression, and chaperone-like activity of human alphaA- crystallin. J Biol Chem. 1996;271:31973. doi: 10.1074/jbc.271.50.31973. [DOI] [PubMed] [Google Scholar]
- 18.Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 19.Tarle I, Borhani DW, Wilson DK, Quiocho FA, Petrash JM. Probing the active site of human aldose reductase. Site-directed mutagenesis of Asp-43, Tyr-48, Lys-77, and His-110. J Biol Chem. 1993;268:25687. [PubMed] [Google Scholar]
- 20.Jez JM, Penning TM. The aldo-keto reductase (AKR) superfamily: an update. Chem Biol Interact. 2001;130–132:499. doi: 10.1016/s0009-2797(00)00295-7. [DOI] [PubMed] [Google Scholar]
- 21.Wilson DK, Nakano T, Petrash JM, Quiocho FA. 1.7 A structure of FR-1, a fibroblast growth factor-induced member of the aldo-keto reductase family, complexed with coenzyme and inhibitor. Biochemistry. 1995;34:14323. doi: 10.1021/bi00044a009. [DOI] [PubMed] [Google Scholar]
- 22.Bennett MJ, Schlegel BP, Jez JM, Penning TM, Lewis M. Structure of 3 alpha-hydroxysteroid/dihydrodiol dehydrogenase complexed with NADP+ Biochemistry. 1996;35:10702. doi: 10.1021/bi9604688. [DOI] [PubMed] [Google Scholar]
- 23.Kim ST, Huh WK, Kim JY, Hwang SW, Kang SO. D-arabinose dehydrogenase and biosynthesis of erythroascorbic acid in Candida albicans. Biochim Biophys Acta. 1996;1297:1. doi: 10.1016/0167-4838(96)00077-5. [DOI] [PubMed] [Google Scholar]
- 24.Schlegel BP, Jez JM, Penning TM. Mutagenesis of 3 alpha-hydroxysteroid dehydrogenase reveals a "push-pull" mechanism for proton transfer in aldo-keto reductases. Biochemistry. 1998;37:3538. doi: 10.1021/bi9723055. [DOI] [PubMed] [Google Scholar]
- 25.Ford G, Ellis EM. Characterization of Ypr1p from Saccharomyces cerevisiae as a 2- methylbutyraldehyde reductase. Yeast. 2002;19:1087. doi: 10.1002/yea.899. [DOI] [PubMed] [Google Scholar]
- 26.Garay-Arroyo A, Covarrubias AA. Three genes whose expression is induced by stress in Saccharomyces cerevisiae. Yeast. 1999;15:879. doi: 10.1002/(SICI)1097-0061(199907)15:10A<879::AID-YEA428>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 27.Oechsner U, Magdolen V, Bandlow W. A nuclear yeast gene (GCY) encodes a polypeptide with high homology to a vertebrate eye lens protein. FEBS Lett. 1988;238:123. doi: 10.1016/0014-5793(88)80240-0. [DOI] [PubMed] [Google Scholar]
- 28.Norbeck J, Blomberg A. Metabolic and regulatory changes associated with growth of Saccharomyces cerevisiae in 1.4 M NaCl. Evidence for osmotic induction of glycerol dissimilation via the dihydroxyacetone pathway. J Biol Chem. 1997;272:5544. doi: 10.1074/jbc.272.9.5544. [DOI] [PubMed] [Google Scholar]
- 29.Ishihara K, Yamamoto H, Mitsuhashi K, Nishikawa K, Tsuboi S, Tsuji H, Nakajima N. Purification and characterization of alpha-keto amide reductase from Saccharomyces cerevisiae. Biosci Biotechnol Biochem. 2004;68:2306. doi: 10.1271/bbb.68.2306. [DOI] [PubMed] [Google Scholar]
- 30.Traff KL, Jonsson LJ, Hahn-Hagerdal B. Putative xylose and arabinose reductases in Saccharomyces cerevisiae. Yeast. 2002;19:1233. doi: 10.1002/yea.913. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava S, Chandra A, Bhatnagar A, Srivastava SK, Ansari NH. Lipid peroxidation product, 4-hydroxynonenal and its conjugate with GSH are excellent substrates of bovine lens aldose reductase. Biochem Biophys Res Commun. 1995;217:741. doi: 10.1006/bbrc.1995.2835. [DOI] [PubMed] [Google Scholar]
- 32.Petrash JM, Flath M, Sens D, Bylander J. Effects of osmotic stress and hyperglycemia on aldose reductase gene expression in human renal proximal tubule cells. Biochem Biophys Res Commun. 1992;187:201. doi: 10.1016/s0006-291x(05)81479-2. [DOI] [PubMed] [Google Scholar]
- 33.Cowley BD, Jr, Ferraris JD, Carper D, Burg MB. In vivo osmoregulation of aldose reductase mRNA, protein, and sorbitol in renal medulla. Am J Physiol. 1990;258:F154–F161. doi: 10.1152/ajprenal.1990.258.1.F154. [DOI] [PubMed] [Google Scholar]
- 34.Wonisch W, Hayn M, Schaur RJ, Tatzber F, Kranner I, Grill D, Winkler R, Bilinski T, Kohlwein SD, Esterbauer H. Increased stress parameter synthesis in the yeast Saccharomyces cerevisiae after treatment with 4-hydroxy-2-nonenal. FEBS Lett. 1997;405:11. doi: 10.1016/s0014-5793(97)00123-3. [DOI] [PubMed] [Google Scholar]
- 35.Burg MB. Role of aldose reductase and sorbitol in maintaining the medullary intracellular milieu. Kidney Int. 1988;33:635. doi: 10.1038/ki.1988.46. [DOI] [PubMed] [Google Scholar]
- 36.Ford G, Ellis EM. Three aldo-keto reductases of the yeast Saccharomyces cerevisiae. Chem Biol Interact. 2001;130–132:685. doi: 10.1016/s0009-2797(00)00259-3. [DOI] [PubMed] [Google Scholar]
- 37.Petrash JM. All in the family: aldose reductase and closely related aldo-keto reductases. Cell Mol Life Sci. 2004;61:737. doi: 10.1007/s00018-003-3402-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bohren KM, Grimshaw CE, Lai CJ, Harrison DH, Ringe D, Petsko GA, Gabbay KH. Tyrosine-48 is the proton donor and histidine-110 directs substrate stereochemical selectivity in the reduction reaction of human aldose reductase: Enzyme kinetics and crystal structure of the Y48H mutant enzyme. Biochemistry. 1994;33:2021. doi: 10.1021/bi00174a007. [DOI] [PubMed] [Google Scholar]
- 39.Liu SQ, Bhatnagar A, Srivastava SK. Bovine lens aldose reductase. pH-dependence of steady-state kinetic parameters and nucleotide binding. J Biol Chem. 1993;268:25494. [PubMed] [Google Scholar]
- 40.Wilson DK, Bohren KM, Gabbay KH, Quiocho FA. An unlikely sugar substrate site in the 1.65 A structure of the human aldose reductase holoenzyme implicated in diabetic complications. Science. 1992;257:81. doi: 10.1126/science.1621098. [DOI] [PubMed] [Google Scholar]
- 41.Hur E, Wilson DK. The crystal structure of the GCY1 protein from S. cerevisiae suggests a divergent aldo-keto reductase catalytic mechanism. Chem Biol Interact. 2001;130–132:527. doi: 10.1016/s0009-2797(00)00296-9. [DOI] [PubMed] [Google Scholar]
- 42.Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002;30:e23. doi: 10.1093/nar/30.6.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeong EY, Sopher C, Kim IS, Lee H. Mutational study of the role of tyrosine-49 in the Saccharomyces cerevisiae xylose reductase. Yeast. 2001;18:1081. [PubMed] [Google Scholar]
- 44.Choder M, Young RA. A portion of RNA polymerase II molecules has a component essential for stress responses and stress survival. Mol Cell Biol. 1993;13:6984. doi: 10.1128/mcb.13.11.6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sass P, Field J, Nikawa J, Toda T, Wigler M. Cloning and characterization of the high-affinity cAMP phosphodiesterase of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1986;83:9303. doi: 10.1073/pnas.83.24.9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krems B, Charizanis C, Entian KD. Mutants of Saccharomyces cerevisiae sensitive to oxidative and osmotic stress. Current Genetics. 1995;27:427. doi: 10.1007/BF00311211. [DOI] [PubMed] [Google Scholar]
- 47.Krems B, Charizanis C, Entian KD. The response regulator-like protein Pos9/Skn7 of Saccharomyces cerevisiae is involved in oxidative stress resistance. Current Genetics. 1996;29:327. doi: 10.1007/BF02208613. [DOI] [PubMed] [Google Scholar]
- 48.Kushnir S, Babiychuk E, Kampfenkel K, Belles-Boix E, Van Montagu M, Inze D. Characterization of Arabidopsis thaliana cDNAs that render yeasts tolerant toward the thiol-oxidizing drug diamide. Proc Natl Acad Sci U S A. 1995;92:10580. doi: 10.1073/pnas.92.23.10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin SJ, Culotta VC. The ATX1 gene of Saccharomyces cerevisiae encodes a small metal homeostasis factor that protects cells against reactive oxygen toxicity. Proc Natl Acad Sci U S A. 1995;92:3784. doi: 10.1073/pnas.92.9.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roncero C, Valdivieso MH, Ribas JC, Duran A. Isolation and characterization of Saccharomyces cerevisiae mutants resistant to Calcofluor white. J Bacteriol. 1988;170:1950. doi: 10.1128/jb.170.4.1950-1954.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frederick DL, Tatchell K. The REG2 gene of Saccharomyces cerevisiae encodes a type 1 protein phosphatase-binding protein that functions with Reg1p and the Snf1 protein kinase to regulate growth. Molecular & Cellular Biology. 1996;16:2922. doi: 10.1128/mcb.16.6.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishizuka Y. Studies and perspectives of protein kinase C. Science. 1986;233:305. doi: 10.1126/science.3014651. [DOI] [PubMed] [Google Scholar]
- 53.Masson JY, Ramotar D. The Saccharomyces cerevisiae IMP2 gene encodes a transcriptional activator that mediates protection against DNA damage caused by bleomycin and other oxidants. Mol Cell Biol. 1996;16:2091. doi: 10.1128/mcb.16.5.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Z, Elledge SJ. Isolation of crt mutants constitutive for transcription of the DNA damage inducible gene RNR3 in Saccharomyces cerevisiae. Genetics. 1992;131:851. doi: 10.1093/genetics/131.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Exinger F, Lacroute F. 6-Azauracil inhibition of GTP biosynthesis in Saccharomyces cerevisiae. Curr Genet. 1992;22:9. doi: 10.1007/BF00351735. [DOI] [PubMed] [Google Scholar]