

Abstract
Interleukin (IL)-18 is a cardiotropic proinflammatory cytokine chronically elevated in the serum of patients with cardiac hypertrophy (LVH). The purpose of this study was to examine the role of IL-18 in pressure-overload hypertrophy using wild type (WT) and IL-18 −/− (null) mice. Adult male C57Bl/6 mice underwent transaortic constriction (TAC) for 7 days or sham surgery. Heart weight/body weight ratios showed blunted hypertrophy in IL-18 null TAC mice compared to WT TAC animals. Microarray analyses indicated differential expression of hypertrophy-related genes in WT versus IL-18 nulls. Northern, Western, and EMSA analyses showed Akt and GATA4 were increased in WT but unchanged in IL-18 null mice. Our results demonstrate blunted hypertrophy with reduced expression of contractile-, hypertrophy-, and remodeling-associated genes following pressure overload in IL-18 null mice, and suggest that IL-18 plays a critical role in the hypertrophic response.
Keywords: Interleukins, myocardial hypertrophy, Akt, signal transduction, mouse, pressure overload
1. Introduction
Cardiac hypertrophy is an adaptive response of the heart to prolonged increases in hemodynamic workload. Although this compensatory process is initially beneficial in normalizing wall stress and oxygen consumption, sustained left ventricular hypertrophy (LVH) significantly increases the risk of developing heart failure and of sudden death [1]. In addition to functional alterations, chronic pressure overload leads to cytoarchitectual changes in the heart characterized by structural remodeling of the muscular, vascular, and extracellular matrix components of the myocardium[2]. Despite recent progress in elucidating the pathological processes involved in myocardial hypertrophy, the molecular mechanisms that underlie LVH remain unclear.
Sustained production of inflammatory cytokines and elevated adrenergic activation play critical roles in the progression of LVH[3, 4]. While low levels of cytokines may be protective for myocytes, persistently high levels are detrimental[5, 6]. Interleukin (IL)-18 is chronically induced during various immune, infectious, and inflammatory conditions[7–9]. Mature IL-18 exerts its biological effects through binding to its cognate receptor (IL-18R) composed of an α and β subunit. The expression of IL-18 serves to amplify the inflammatory response by inducing cytokines, chemokines, and adhesion molecules in both immune and non-immune cells[10, 11]. Recently it was shown that daily systemic IL-18 administration led to rapid LVH in the absence of hemodynamic stress demonstrating a role for IL-18 in cardiac growth[12]. In addition, IL-18 was shown to depress myocardial function in vivo and cardiomyocyte contractility in vitro[12, 13]. Finally, IL-18 has been demonstrated to be elevated in the serum of patients with LVH. These observations demonstrate a role for IL-18 in the progression of inflammatory heart disease and suggest a potent role for this cytokine in cardiac growth. Despite recent progress in our understanding of IL-18, studies defining the signaling pathways and mechanisms responsible for IL-18-dependent cardiac growth in the pressure-overloaded heart are lacking.
We previously reported a direct pro-hypertrophic effect of IL-18 on cardiomyocytes in vitro involves the activations of Akt and GATA4 [14]. The purpose of this study was to determine whether this pro-hypertrophic signaling pathway is blunted in IL-18 gene knockout mice following trans-aortic constriction (TAC). We demonstrate here, for the first time, that IL-18 plays a key role in compensatory cardiac growth in response to pressure overload.
2. Materials and Methods
2.1. Mouse Model
The protocol conformed to the “Guidelines for the Care and Use of Laboratory Animals” (NIH publication No. 86-23, revised in 1985). Homozygous IL-18 −/− (null) mice[15] were purchased from The Jackson Laboratory (Bar Harbor, ME). Pressure overload was achieved as described[16, 17]. Briefly, age (3–4 month old) and weight-matched (25–30g) male WT C57Bl/6 and IL-18 null C57Bl/6 mice (n=5–9/group) underwent surgery for the placement of a constricting suture around the aorta (TAC) or sham surgery (controls). The suture was tightened around the aorta and a 27-gauge needle between the brachiocephalic trunk and left common carotid artery, which after removal provided a consistent degree of constriction. The mice were recovered and after 7 days, the hearts were removed, weighed, snap frozen, and stored at −80°C. Additional groups of animals underwent identical procedures for the purpose of providing adequate heart tissues for EMSA and Western analyses, and invasive LV pressure-volume relations.
2.2. Hemodynamic Measurements
Briefly, mice were anesthetized, intubated and subjected to anterior thoracotomy. The apex of the heart was stabbed with a 30-gauge needle and a miniaturized conductance catheter (Millar Instruments, Houston, TX) was advanced retrograde into the LV along the long axis, with the proximal electrode just within the myocardial wall of the apex. The inferior vena cava (IVC) was isolated immediately below the diaphragm. Baseline pressure-conductance relations at 10 and 100 kHz were acquired and stored for offline conversion to PV relations as previously described[18]. Data were acquired during transient occlusion of the IVC.
2.3. Microarray Analysis
RNA samples from individual mice (n=4–5/group) were pooled into the following 4 experimental groups: sham-operated WT, banded WT, sham-operated IL-18 null, and banded IL-18 null. For each group, the mRNA expression levels of >14,000 well-characterized murine genes were measured using four Mouse Genome 430A 2.0 high-density oligonucleotide arrays (Affymetrix, Santa Clara, CA). Preparation of target RNA, array hybridization, washing, and scanning was performed by standard Affymetrix protocols at the UTHSCSA Microarray Core Facility using 10 μg of input RNA and 20 μg biotin-labeled cRNA. Arrays were scanned using an Affymetrix GeneChip® Scanner 3000, and the image files were converted to probe-level data using Microarray Suite Expression Analysis (MAS 5.0) software (Affymetrix). Gene expression data were background adjusted and normalized using GCRMA. Contrast parameterizations of normalized expression data sets were performed using the affylmGUI and limma (v1.8.14) statistical packages for R from Bioconductor with fitting to a general linear model. Probe sets were deemed differentially expressed at the >2-fold absolute ratio of signal intensities. Annotation of differentially expressed probe sets was provided by the NetAffx bioinformatics center[19], and classification of genes according to biological process, cellular component, and biochemical function provided by gene ontogeny (GO) consortium. The normalized data were subjected to hierarchical and k-means clustering to visualize distinct patterns of gene expression. Identification of over-represented functional classes among the differentially expressed genes was determined using the GenMapp bioinformatics tool[20]. The microarray data has been submitted to the Gene Expression Omnibus (GEO), accession number GSE5129.
2.4. Northern Blot Analysis, Western blot analysis, and electrophoretic mobility shift assay (EMSA)
Northern Blot Analysis, Western blot analysis, and electrophoretic mobility shift assay (EMSA) were carried out as described[14, 21–23].
2.5. Statistical Analyses
The LV PV relations were analyzed with software developed in our laboratory and licensed to and modified by Millar Instruments (PVAN, Conductance Technologies, San Antonio, TX). The algorithms used for dual frequency were developed by us and published previously[18]. Absolute volume measurements from the conductance catheter were calibrated with a correction for α , which was defined as the ratio of flow probe stroke volume to conductance stroke volume. Each mouse was analyzed with its individual α.
Comparisons between sham-operated controls and TAC mice were performed for measures of GATA4 DNA binding activity, gene expression by Northern blotting and protein levels by Western blotting by ANOVA with post hoc Dunnett’s t tests. F tests and Dunnett’s t tests with values of p<0.05 were considered statistically significant.
3. Results
3.1. TAC Phenotypes
Pressure overload (TAC) stimulated increased heart weight/body weight ratios in WT mice (6.3±1.0 vs. 4.3±0.5 mg/g, p<0.05) with blunted hypertrophy in the IL-18 null mice (5.1±0.7, 4.12 ±0.47 mg/g) as compared to their respective sham operated controls. The impact of IL-18 gene knockout on LV geometry and function following TAC is demonstrated in Fig. 1 which shows representative LV pressure/volume relations from sham and TAC WT and IL-18 null mice during transient vena caval occlusion. As indicated, LV contractility (curvilinear end systolic pressure/volume relation; E’max) increased in WT TAC hearts while it decreased in the IL-18 null TAC mice as compared to respective shams. End-diastolic volumes (EDV) decreased from 31±4 to 29±3 mmHg following TAC in WT animals whereas EDV increased from 27±4 to 35±2 mmHg in response to TAC in IL-18 null mice.
Fig. 1.
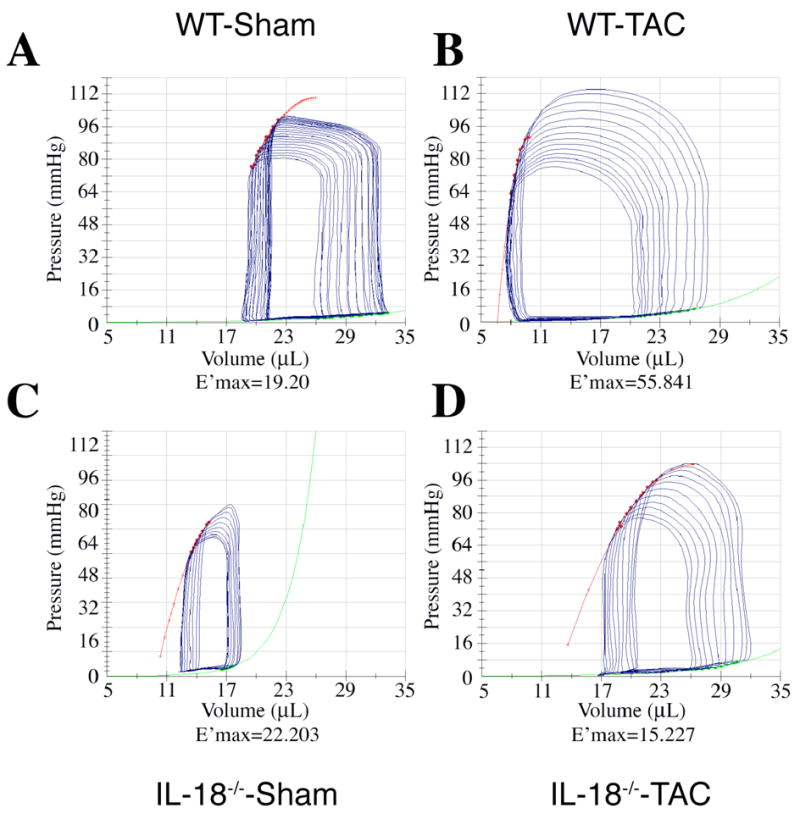
Representative LVPV relations in WT sham (A), WT TAC (B), IL-18 null sham (C), and null TAC (D). E’max indicates curvilinear end-systolic elastance (red line). LV contractility increased in WT mice after 1 week of TAC whereas IL-18 null animals exhibited depressed contractility following TAC.
3.2. Microarray Gene Analyses
Pressure overload altered the expression of 2729 genes common to the hearts of both WT and IL-18 null mice. 1835 transcripts were up regulated and 894 were downregulated. Interestingly, only 286 additional genes were altered in IL-18 null TAC hearts, whereas pressure overload induced changes in another 2470 transcripts in WT hearts. Table 1 shows fold-changes in hypertrophy-related transcript levels between sham and TAC WT and IL-18 null mice.
Table 1.
Selected cardiac growth/hypertrophy-related genes are differentially expressed at 1 week in WT and IL-18 null TAC mice. Fold-changes from corresponding sham-operated controls are shown for each phenotype.
| Gene I.D. (Symbol) | Gene Description | Wild Type | IL-18 Null |
|---|---|---|---|
| Cytostructural/Contractile | |||
| MYH6 | α -myosin heavy chain | 39.9 | −1.1 |
| MYH7 | β -myosin heavy chain | 132.6 | 1.0 |
| DES | desmin | 841.9 | 1.2 |
| ACTB | β -actin | 113.7 | 1.2 |
| TUBA8 | α -tubulin | 65.5 | −1.3 |
| Substrate/Metabolic/Signaling | |||
| PPARA | peroxisome proliferative activated receptor α | 4.3 | −1.7 |
| CPT1B | carnitine palmitoyltransferase | 131.6 | −3.3 |
| SLC2A1 | glucose transporter (Glut1) | −2.5 | 2.0 |
| SLC2A4 | Insulin-sensitive glucose transporter (Glut4) | 8.9 | −2.0 |
| PYGM | muscle glycogen phosphorylase | 185.8 | −1.3 |
| PDK2 | pyruvate dehydrogenase kinase 2 | 91.4 | −2.0 |
| PPP3CB | calcineurin A β | 17.6 | −2.5 |
| GSK3B | glycogen synthase kinase 3β | 2.5 | 1.0 |
| DSCR1/MCIP1 | myocyte- enriched calcineurin-interacting protein 1 | 7.9 | 1.0 |
| SOCS3 | suppressor of cytokine signaling 3 | −12.5 | 30.9 |
| GATA4 | GATA binding protein 4 | 56.0 | −2.0 |
| MYOCD | Myocardin | 18.3 | 1.0 |
| Nkx2-5 | Nkx2.5 | 3.8 | −0.7 |
| NPPA | ANP, atrial natriuretic peptide | 2.7 | 5.7 |
| AKT1 | v-akt murine thymoma viral oncogene homolog 1 | 9.4 | 1.0 |
| AKT2 | v-akt murine thymoma viral oncogene homolog 2 | 23.3 | 1.3 |
| Extracellular Matrix/Remodeling | |||
| MMP2 | matrix metalloproteinase 2 | 23.0 | 4.1 |
| TIMP1 | tissue inhibitor of metalloproteinase 1 | 7.4 | 324.1 |
| TIMP2 | tissue inhibitor of metalloproteinase 2 | 6.3 | −2.5 |
| TIMP3 | tissue inhibitor of metalloproteinase 3 | 11.0 | −1.3 |
| TIMP4 | tissue inhibitor of metalloproteinase 4 | 7.0 | −1.4 |
3.3. Transcription Factors
GATA4 was upregulated in WT TAC mice as compared to IL-18 null TAC animals, where there was no change. Northern blotting confirmed the microarray results demonstrating significant upregulation of GATA4 transcripts in the WT TAC mice (Fig. 2A and 2B). Similarly, the hypertrophy-related myocardin gene was upregulated in WT TAC mice and was confirmed by Northern blotting (Fig. 2A and 2C). GATA4 protein levels (Fig. 3A and 3B) were increased significantly in the hearts of WT mice though not in IL-18 null animals after TAC. As shown in Fig. 3C and 3D EMSA analyses indicated GATA4 DNA binding activity was significantly upregulated in the hearts of WT TAC mice compared to WT shams (260.3±21.8 vs. 157.7±15.9; p<0.01). Pressure overload did not induce GATA4 DNA binding in IL-18 null TAC animals. No difference in GATA4 DNA binding was observed between WT and IL-18 null shams (158.7±17.5 vs. 157.7±15.9).
Fig. 2.
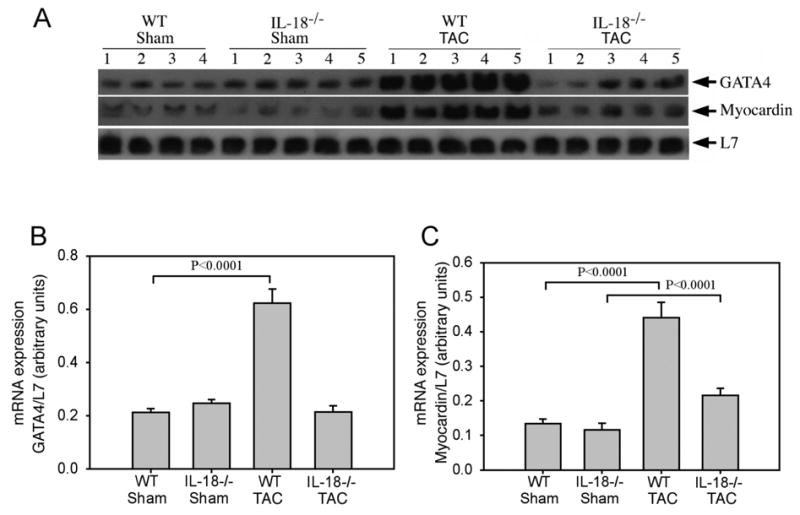
TAC enhances pro-hypertrophic gene expression in wild type, but not in IL-18 null mice.A, GATA4 and myocardin expression by Northern blot (20 μg total RNA/lane). B, Autoradiographic signal intensity for GATA4 and myocardin mRNA is expressed in arbitrary units relative to L7 ribosomal RNA (B, GATA4; C, Myocardin).
Fig. 3.
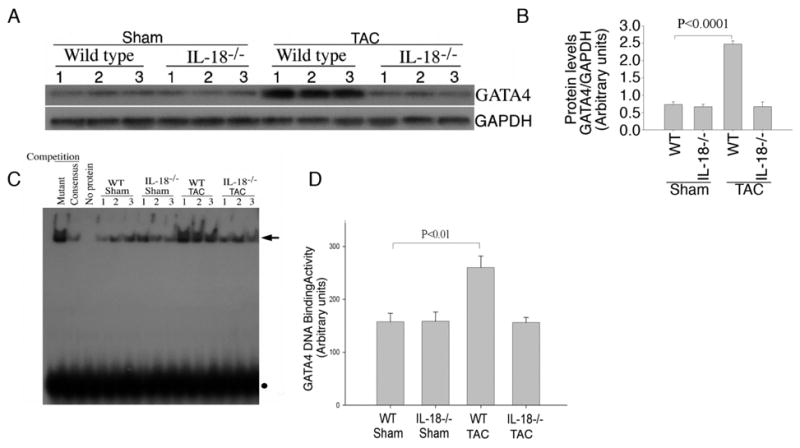
A, TAC enhances GATA4 protein levels in WT, but not in IL-18 null mice. GAPDH was used as an internal control. The autoradiographic signals shown in panel A were semi-quantified, and the signal intensity for GATA4 protein is expressed in arbitrary units relative to that of corresponding GAPDH levels (B). TAC enhances GATA4 DNA binding activity in WT, but not in IL-18 null mice. C, GATA4 DNA binding activity in nuclear protein extracts from myocardium of sham and TAC WT and IL-18 null mice. Each lane contains 10 μg of nuclear protein extract. In competition experiments with mutant or consensus GATA4 oligonucleotides, nuclear protein extracts from WT mice were preincubated with 75-fold molar excess of either unlabelled double-stranded mutant or consensus GATA4 oligonucleotide respectively, followed by the addition of32P-labelled consensus GATA4 probe. Arrow indicates GATA4-specific DNA–protein complexes. Solid circle shows unincorporated labeled probe. D, Densitometric analysis of the autoradiographic signals shown in C was semi-quantified by video image analysis.
Activation of Akt by various stimuli has been shown to be required for myocyte hypertrophy[24]. We observed upregulation of Akt1 (9.4-fold) and Akt2 (23.3-fold) transcripts in the hearts of WT TAC mice (Table 1). As shown in Fig. 4A and 4B, western blotting revealed significantly increased phospho-Akt levels in WT TAC animals (p<0.0001) whereas IL-18 null TAC mice exhibited minimal Akt activation (p<0.05). These data indicate IL-18 may be required for full Akt activation in the pressure-overloaded heart.
Fig. 4.
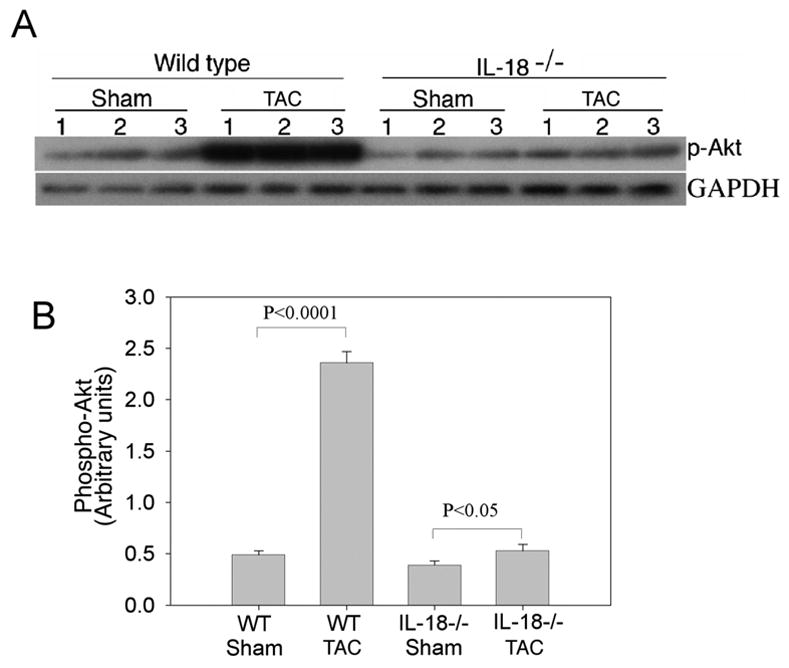
A, TAC enhances phospho-Akt levels in wild type, but not in IL-18 null mice. Phospho-Akt levels in tissue homogenates were quantified by Western blotting using activation-specific antibodies (Ser473). GAPDH was used as an internal control. B, The autoradiographic signals obtained in A were semi-quantified, and the signal intensity for phospho-Akt is expressed in arbitrary units relative to that of corresponding GAPDH levels.
4. Discussion
This study expands on several in vitro reports from our group demonstrating the important role of IL-18 in cardiac pathophysiology[14, 21–23]. The novel and significant finding of this study is that IL-18 is required for full hypertrophic response in an in vivo murine model of chronic pressure overload. We found that the presence or absence of IL-18 resulted in differential regulation of hypertrophy-related genes, suggesting a key role for IL-18 in the early hypertrophic response to pressure overload. Our results support a putative mechanism for IL-18-dependent cardiac growth in vivo whereby stimulated Akt phosphorylation leads to nuclear localization and DNA binding of GATA4 resulting in the expression of hypertrophy-related genes. This scheme is further supported by the results obtained from the hearts of IL-18 null TAC mice where Akt phosphorylation was prevented, GATA4 nuclear transport and DNA binding activity were impaired in association with dysregulation of various genes including those coding for cytoarchitectial, metabolic, and remodeling proteins. These gene alterations resulted in a phenotype of blunted cardiac hypertrophy in response to TAC. Thus, we demonstrate, for the first time, that IL-18 plays a critical role in the cardiac growth response to pressure overload in the intact beating heart.
These observations are in accord with a previous study from our laboratory showing IL-18 stimulates cardiac myocytes hypertrophy through a pathway involving sequential activation of PI3K, Akt, and GATA4[14]. A role for Akt in hypertrophic signaling is now well established and supported by numerous reports[24, 25]. Once activated by hypertrophic signals, Akt participates in cellular growth, proliferation[14], survival[26], and regulation of glucose metabolism[24]. The results of our microarray analysis show that in addition to increased phosphorylation of Akt, both Akt1 and Akt2 transcripts were upregulated in the hearts of WT TAC but not IL-18 null TAC mice, suggesting Akt may be regulated by IL-18 at multiple levels, transcriptional and post-translational. Akt may also regulate IL-18 itself providing a positive feedback mechanism resulting in enhanced myocardial cell survival and sustained cardiac growth signaling.
We have previously shown IL-18-dependent Akt activation stimulates GATA4 protein expression and DNA binding activity in cardiac myocytes[14]. The cardiac-specific transcription factor GATA4 regulates the expression of many genes essential for heart development as well as regulating myocardial genes in response to various stressful stimuli including pressure overload. Garg and colleagues demonstrated that significant knockdown of GATA4 protein in cardiac myocytes in vivo attenuated hypertrophy induced by either pressure overload or exercise[27]. They found decreased cardiac hypertrophy and earlier functional decompensation correlated with the extent of GATA4 knockdown. In our study, microarray and Northern blot analyses indicated a robust induction of GATA4 transcripts in the hearts of WT TAC mice while IL-18 null TAC animals exhibited significant downregulation. In addition, both GATA4 protein levels and DNA binding activity were stimulated in WT mice but not in IL-18 null animals after TAC. A plausible mechanism for this regulation could involve the phosphorylation and inactivation of glycogen synthase kinase (GSK)3β by Akt[24]. GSK3β phosphorylates GATA4 and its co-transcriptional activator NFAT. Phosphorylation of GATA4 leads to nuclear export and cessation of transcriptional activity[28]. Similarly, phosphorylation of NFAT by GSK3β results in the masking of nuclear import sequences leading to translocation to the cytosol such that inhibition of GSK3β by Akt would be expected to maintain the GATA4/NFAT complex in the nucleus.
Our study should be interpreted in light of certain limitations. We assessed the effects of TAC on heart weight/body weight ratios and gene expression after only 7 days, and as such we addressed mechanisms related to the compensatory (growth) phase of hypertrophy unrelated to a transition to heart failure. Using a similar murine pressure overload model, Zhao and coworkers[29] reported most genes were differentially regulated by 10 days, similar to the time period used in this study. Wagner and coworkers have shown that TAC resulted in stabilization of hypertrophy in as few as 20 days in mice[30]. We chose the time period for this study to illuminate the primary impact of IL-18 on pressure overload-induced cardiac growth while minimizing secondary alterations in gene expression to the extent possible. With regard to microarray gene analyses, although pooling RNA samples provides an advantage by effectively averaging biological variation[31], the lack of replicates in the microarray experiments limits the calculation of fundamental statistics and probabilities, leaving fold-change as the parameter on which to base differential gene expression. Nevertheless, 2-fold thresholds have been successfully used as a statistical threshold for eliminating the majority of false positives. The use of microarrays served as a screening tool to identify candidate genes that may be differentially expressed in our model. Thus, for confirmatory purposes, we verified several of the key genes by Northern blotting.
In summary, our results demonstrate blunted hypertrophy and reduced expression of contractile-, hypertrophy-, and remodeling-associated genes following pressure overload in IL-18 null mice, and suggest that IL-18 plays a critical role in the hypertrophic response.
Acknowledgments
This work was supported in part by AHA, Texas Affiliate BGIA 0565018Y (JTC), the Research Service of the Department of Veterans Affairs (BC, MDF), HL68020 and HL079926 from the NIH (BC, MDF, respectively), and the Janey Briscoe Center of Excellence in Cardiovascular Disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Vakili BA, Okin PM, Devereux RB. Prognostic implications of left ventricular hypertrophy. Am Heart J. 2001;141:334–341. doi: 10.1067/mhj.2001.113218. [DOI] [PubMed] [Google Scholar]
- 2.Wakatsuki T, Schlessinger J, Elson EL. The biochemical response of the heart to hypertension and exercise. Trends Biochem Sci. 2004;29:609–617. doi: 10.1016/j.tibs.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Prabhu SD. Cytokine-Induced Modulation of Cardiac Function. Circ Res. 2004;95:1140–1153. doi: 10.1161/01.RES.0000150734.79804.92. [DOI] [PubMed] [Google Scholar]
- 4.Mann DL. Inflammatory Mediators and the Failing Heart: Past, Present, and the Foreseeable Future. Circ Res. 2002;91:988–998. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- 5.Wong G, Goeddel D. Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science. 1988;242:941–944. doi: 10.1126/science.3263703. [DOI] [PubMed] [Google Scholar]
- 6.Nakano M, Knowlton AA, Dibbs Z, Mann DL. Tumor Necrosis Factor-{alpha} Confers Resistance to Hypoxic Injury in the Adult Mammalian Cardiac Myocyte. Circ. 1998;97:1392–1400. doi: 10.1161/01.cir.97.14.1392. [DOI] [PubMed] [Google Scholar]
- 7.Bazan J, Timans J, Kastelein R. A newly defined interleukin-1β [comment] Nature. 1996;379:15. doi: 10.1038/379591a0. [DOI] [PubMed] [Google Scholar]
- 8.Dinarello C, Novick D, Puren A, Fantuzzi G, Shapiro L, Muhl H, Yoon D, Reznikov L, Kim S, Rubinstein M. Overview of interleukin-18: more than an interferon-gamma inducing factor. J Leuk Biol. 1998;63:658–664. [PubMed] [Google Scholar]
- 9.Dinarello C, Fantuzzi G. Interleukin-18 and host defense against infection. J Infect Diseases. 2003;187:15. doi: 10.1086/374751. [DOI] [PubMed] [Google Scholar]
- 10.Jablonska E, Jablonski J. Effect of IL-18 on the release of IL-6 and its soluble receptors: sIL-6Ralpha and sgp130 by human neutrophils. Immunological Investigations. 2002;31:159–167. doi: 10.1081/imm-120016237. [DOI] [PubMed] [Google Scholar]
- 11.Jablonska E, Izycka A, Jablonska J, Wawrusiewicz N, Piecuch J. Role of IL-18 in the secretion of Il-1beta, sIL-1RII, and IL-1Ra by human neutrophils. Immunological Investigations. 2001;30:221–229. doi: 10.1081/imm-100105066. [DOI] [PubMed] [Google Scholar]
- 12.Woldbaek PR, Sande JB, Stromme TA, Lunde PK, Djurovic S, Lyberg T, Christensen G, Tonnessen T. Daily administration of interleukin-18 causes myocardial dysfunction in healthy mice. Am J Physiol Heart Circ Physiol. 2005;289:H708–714. doi: 10.1152/ajpheart.01179.2004. [DOI] [PubMed] [Google Scholar]
- 13.Pomerantz B, Reznikov L, Harken A, Dinarello C. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci USA. 2001;98:2871–2876. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandrasekar B, Mummidi S, Claycomb WC, Mestril R, Nemer M. Interleukin-18 Is a Pro-hypertrophic Cytokine That Acts through a Phosphatidylinositol 3-Kinase-Phosphoinositide-dependent Kinase-1-Akt-GATA4 Signaling Pathway in Cardiomyocytes. J Biol Chem. 2005;280:4553–4567. doi: 10.1074/jbc.M411787200. [DOI] [PubMed] [Google Scholar]
- 15.Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K, Akira S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 1998;8:383–390. doi: 10.1016/s1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura A, Rokosh DG, Paccanaro M, Yee RR, Simpson PC, Grossman W, Foster E. LV systolic performance improves with development of hypertrophy after transverse aortic constriction in mice. American Journal Of Physiology Heart And Circulatory Physiology. 2001;281:H1104–H1112. doi: 10.1152/ajpheart.2001.281.3.H1104. [DOI] [PubMed] [Google Scholar]
- 17.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J, Jr, Chien KR. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feldman MD, Mao Y, Valvano JW, Pearce JA, Freeman GL. Development of a multifrequency conductance catheter-based system to determine LV function in mice. Am J Physiol Heart Circ Physiol. 2000;279:H1411–1420. doi: 10.1152/ajpheart.2000.279.3.H1411. [DOI] [PubMed] [Google Scholar]
- 19.Liu G, Loraine A, Shigeta R, Cline M, Cheng J, Valmeekam V, Sun S, Kulp D, Siani-Rose M. NetAffx: Affymetrix probesets and annotations. Nucleic Acids Res. 2003;31:82–86. doi: 10.1093/nar/gkg121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doniger S, Salomonis N, Dahlquist K, Vranizan K, Lawlor S, Conklin B. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;4 doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandrasekar B, Valente A, Freeman G, Mahimainathan L, Mummidi S. Interleukin-18 induces human cardiac endothelial cell death via a novel signaling pathway involving NF-kappaB-dependent PTEN activation. Biochem Biophys Res Com. 2006;339:956–963. doi: 10.1016/j.bbrc.2005.11.100. [DOI] [PubMed] [Google Scholar]
- 22.Chandrasekar B, Colston JT, de la Rosa SD, Rao PP, Freeman GL. TNF-[alpha] and H2O2 induce IL-18 and IL-18R[beta] expression in cardiomyocytes via NF-[kappa]B activation. Biochem Biophys Res Com. 2003;303:1152–1158. doi: 10.1016/s0006-291x(03)00496-0. [DOI] [PubMed] [Google Scholar]
- 23.Chandrasekar B, Marelli-Berg F, Tone M, Bysani S, Prabhu S, Murray D. β-adrenergic stimulation induces interleukin-18 expression via beta2-AR, PI3K, Akt, IKK, and NF-κB. Biochem Biophys Res Com. 2004;319:304–311. doi: 10.1016/j.bbrc.2004.04.185. [DOI] [PubMed] [Google Scholar]
- 24.Latronico MVG, Costinean S, Lavitrano ML, Peschle C, Condorelli G. Regulation of Cell Size and Contractile Function by AKT in Cardiomyocytes. Ann NY Acad Sci. 2004;1015:250–260. doi: 10.1196/annals.1302.021. [DOI] [PubMed] [Google Scholar]
- 25.Shioi T, McMullen J, Kang P, Douglas P, Obata T, Franke T, Cantley L, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shao Z, Bhattacharya K, Hsich E, Park L, Walters B, Germann U, Wang Y, Kyriakis J, Mohanlal R, Kuida K, Namchuk M, Salituro F, Yao Y, Hou W, Chen X, Aronovitz M, Tsichlis P, Bhattacharya S, Force T, Kilter H. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ Res. 2006;98:111–118. doi: 10.1161/01.RES.0000197781.20524.b9. [DOI] [PubMed] [Google Scholar]
- 27.Garg V, Kathiriya I, Barnes R, Schluterman M, King I, Butler C, Rothrock C, Eapen R, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen J, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 28.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-Specific Deletion of Gata4 Reveals Its Requirement for Hypertrophy, Compensation, and Myocyte Viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 29.Zhao M, Chow A, Powers J, Fajardo G, Bernstein D. Microarray analysis of gene expression after transverse aortic constriction in mice. Physiol Genomics. 2004;19:93–105. doi: 10.1152/physiolgenomics.00040.2004. [DOI] [PubMed] [Google Scholar]
- 30.Wagner RA, Tabibiazar R, Powers J, Bernstein D, Quertermous T. Genome-wide expression profiling of a cardiac pressure overload model identifies major metabolic and signaling pathway responses. J Mol Cell Cardiol. 2004;37:1159–1170. doi: 10.1016/j.yjmcc.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Kendziorski C, Irizarry R, Chen K, Haag J, Gould M. On the utility of pooling biological samples in microarray experiments. Proc Natl Acad Sci USA. 2005;102:4252–4257. doi: 10.1073/pnas.0500607102. [DOI] [PMC free article] [PubMed] [Google Scholar]